

Abstract
The direct α-arylation of cyclic and acyclic ethers with heteroarenes has been accomplished via the design of a new photoredox mediated C–H functionalization pathway. Transiently generated α-oxyalkyl radicals, produced from a variety of widely available ethers via hydrogen atom transfer (HAT), were found to couple with a range of electron-deficient heteroarenes in a Minisci-type mechanism. This mild, visible light-driven protocol allows direct access to medicinal pharmacophores of broad utility using feedstock substrates and a commercial photocatalyst.
Keywords: photoredox catalysis, C–H functionalization, alkylation, ethers, heterocycles
Visible light photoredox catalysis has recently emerged as a powerful platform for the functionalization of C–H and C–X bonds via numerous single electron transfer (SET) pathways, a rapidly growing area of research that played a part in a large-scale renaissance of radical-based methodologies.[1] Recently, we sought to expand the types of organic molecules that can participate in photoredox mediated C–H functionalization[2] via the exploitation of physical properties that are predictable across a wide range of structural classes (e.g., bond dissociation energies (BDEs),[3] hydrogen atom transfer exchange constants[4] and oxidation potentials) (Figure 1). In previous work, we developed a new photoredox–organocatalytic C–H functionalization protocol that allows benzylic ethers to undergo selective α-oxy arylation (via radical-radical coupling) in the presence of functional groups that contain similar C–H bonds (e.g., α̃C–H amines, α̃C–H alcohols, and α̃C–H ethers).[5] In this manuscript, we report a new photocatalytic mechanism that allows for a complimentary class of organic molecules, namely dialkyl ethers, to formally undergo selective C–H bond functionalization and thereafter Minisci-type coupling[6] with electron-deficient heteroarenes. This mild, visible light-driven protocol enables the rapid conversion of feedstock ethers into α-oxy heteroarenes, an important and broadly employed pharmacophore within the realm of drug development.
Figure 1.

Photoredox mediated C–H functionalization.
Heteroaromatic moieties are undoubtedly among the most widespread constituents of pharmaceutical compounds.[7] Over the last decade, the direct C–H functionalization of heteroarenes has become a powerful and efficient transformation, with extensive applications in both medicinal and process chemistry.[8] Notably, the open-shell addition of alkyl groups to heteroarenes,[9] known as the Minisci reaction, has also enjoyed a broad scale re-adoption and application within drug discovery programs.[10] Recently, we questioned whether photoredox catalysis might be mechanistically coupled with the venerable Minisci reaction to create a new and selective C–H functionalization–heteroarylation reaction, a protocol that would convert cyclic and acyclic ethers to high value pharmacophore adducts [Eq. (1)]. While the use of unfunctionalized ethers in the Minisci transform was previously demonstrated in 1971, these exploratory studies require the use of metal salts or stoichiometric peroxides at high temperatures, a necessity which generally results in moderate or low yields, and the production of a variety of radical addition byproducts.[6] On this basis we sought to develop a broadly useful α-oxyalkylation reaction that employs photoredox catalysis in conjunction with low-boiling feedstock ethers and mild, room temperature conditions. Our hope was to develop a complementary Minisci-type mechanism that enables a general, selective and efficient C–H functionalization–heteroarylation reaction, and at the same time deliver a protocol that can convert a broad range of cyclic and acyclic ethers to high value pharmacophore adducts.
It has long been established that α-oxyalkyl radicals are relatively stable yet nucleophilic open-shell species that can readily react with electron-deficient substrates, such as olefins, imines, and heteroarenes.[11] However, the vast majority of photoredox-mediated C–H functionalization processes have been limited to α-alkyl and aryl substituted amines, while ethers and alcohols remain outside the scope of these SET–deprotonation pathways due to their comparatively high oxidation potentials (for THF, THP, and Et2O, E1/2ox > +2.4 V vs. SCE in 2:1 MeCN:H2O).[12] In an effort to expand the range of organic moieties that can participate in photocatalytic C–H functionalization (and to circumvent the limitations of an oxidation potential-gated electron-transfer mechanism) we sought to employ a hydrogen atom transfer (HAT) pathway, that would allow hydrogen abstraction via photoredox catalysis in lieu of direct substrate oxidation. Previous studies from our lab have shown that substrates which incorporate weak C–H BDEs, such as benzylic ethers, can be directly functionalized via the combination of an organocatalytic thiyl activation mode and photoredox catalysis.[5] In this study, we hypothesized that the intervention of a sulfate radical anion[13] should allow substrates that incorporate much stronger C–H BDEs, such as non-benzylic dialkyl ethers, to be directly activated and thereafter employed in Minisci-type chemistry. Moreover, the successful execution of these ideals would further demonstrate the utility of photoredox catalysis for medicinal chemistry.
The mechanistic details of our proposed α-arylation of ethers are outlined in Scheme 1. Irradiation of photoredox catalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1) using visible light from a household light bulb at room temperature will produce a long-lived (2.3 μs) photoexcited state, *IrIII[dF(CF3)ppy]2(dtbbpy)PF6 (2).[14] Given that the resulting *IrIII species is a strong reductant (E1/2IV/*III = −0.88 V vs. SCE in 2:1 MeCN:H2O),[12,15] we expected it to be capable of reducing the persulfate anion (3)[16] to afford IrIV[dF(CF3)ppy]2(dtbbpy)PF6 (4), sulfate dianion and the sulfate radical anion (5). We presumed that the desired α-oxyalkyl radical (7) could then be generated via a HAT event between the dialkyl ether (6) and sulfate radical anion.[17] At this stage, we expected that the resulting α-oxyalkyl radical (7) would be sufficiently nucleophilic to add to a protonated electron-deficient heteroarene (8) in a Minisci-type pathway to afford the amine radical cation (9). The cation (9), upon loss of a proton, would give the α-amino radical (10), which would undergo a second SET event with the oxidized photo-system (4) (E1/2IV/III = +1.70 V vs. SCE in 2:1 MeCN:H2O)[12] to regenerate the photocatalyst ground-state (1) and furnish the desired α-aryl ether product.
Scheme 1.

Proposed mechanism for α–arylation of ethers.
We recognize that α-oxyalkyl radical 7 might be generated via two distinct mechanisms: (1) hydrogen atom abstraction or (2) direct single-electron oxidation followed by deprotonation. While we cannot definitively rule out the latter possibility, we favor the proposed HAT pathway based on mechanistic evidence from the literature. Studies of the reaction of SO4•− and ethers via transient absorption spectroscopy,[18a] kinetic isotope effects,[18b] and correlation between BDEs and reaction rates[18c] are consistent with HAT as the operative mechanism. In the related case of alcohols, both ESR experiments[18b] and Minisci-type trapping reactions[13a] suggest that hydrogen abstraction, not SET–deprotonation, is the dominant pathway in the sulfate radical anion-mediated generation of α-oxyalkyl radicals.[X]
Our examination of the proposed α-arylation of ethers began with tetrahydropyran (THP), isoquinoline, and a series of photocatalysts and persulfates (Table 1). To our delight, the desired α-arylation product was obtained in 68% yield using the Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1) photocatalyst in combination with (NH4)2S2O8 as the oxidant, in MeCN under the irradiation of a 26 W household fluorescent light bulb (entry 1). The regioselectivity of addition at the isoquinoline C(1) position was fully expected based on the previously described polar effect.[19] Notably, the addition of water allowed an increase in yield (entry 2, 77%), presumably due in part to the dissolution of the persulfate salt to generate a transparent biphasic reaction mixture, a measure that allows enhanced photon penetration. Changing the oxidant to K2S2O8 improved the efficiency slightly, whereas the use of the more economical salt Na2S2O8 provided a superior 81% yield of the heteroarylation product (entries 3 and 4). An examination of several photocatalysts revealed that Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1) was the most effective in this α-arylation protocol (entries 5–7). It is intriguing to consider that in comparison to Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1), the less successful Ir(ppy)3 and Ir(ppy)2(dtbppy)PF6 catalysts are more reducing at the *IrIII excited state yet less oxidizing in the IrIV species, while Ru(bpy)3Cl2 exhibits a less reducing *RuII species yet more oxidizing RuIII species.[20–22] As such, we speculate that matched redox potentials are essential for both SET events to be efficient, a scenario that requires a highly tuned photocatalyst with respect to its intrinsic redox window. As a second issue, we were aware that the product ether α-C–H bonds are weaker than those of the ether starting materials, which could lead to multiple arylation or product decomposition events.[6b] Indeed, we did observe oxidative dimerization of the arylation products in a small number of cases, with isoquinoline substrates, however, the use of EtCN as the reaction medium alleviated this issue (providing the desired adduct in 88% yield, entry 8). Given that the reduction potential of the persulfate anion is low,[16] we expected that this reagent would be unable to react with the substrate ethers without further activation. Indeed, the critical roles of the photocatalyst and light in this α-arylation method were demonstrated using control experiments, wherein minimal amounts of desired product were detected in the absence of visible light photo-excitation (entries 9 and 10).
Table 1.
Preliminary studies toward α-heteroarylation of ethers.

| ||||
|---|---|---|---|---|
| Entry | Photocatalyst | Persulfate salt | Solvents | % Yield[a] |
| 1 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | (NH4)2S2O8 | MeCN | 68 |
| 2 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | (NH4)2S2O8 | MeCN/H2O | 77 |
| 3 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | K2S2O8 | MeCN/H2O | 79 |
| 4 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Na2S2O8 | MeCN/H2O | 81 |
| 5 | Ir(ppy)3 | Na2S2O8 | MeCN/H2O | 7 |
| 6 | Ir(ppy)2(dtbbpy)PF6 | Na2S2O8 | MeCN/H2O | 28 |
| 7 | Ru(bpy)3Cl2 | Na2S2O8 | MeCN/H2O | 22 |
| 8 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Na2S2O8 | EtCN/H2O | 88 |
| 9[b] | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Na2S2O8 | EtCN/H2O | <1 |
| 10 | none | Na2S2O8 | EtCN/H2O | 4 |
Yield determined by 1H NMR using 1,3-benzodioxole as the internal standard after work-up following the general procedure in Supporting Information.
Reaction performed in the absence of light.
TFA = trifluoroacetic acid. CFL = compact fluorescent light.
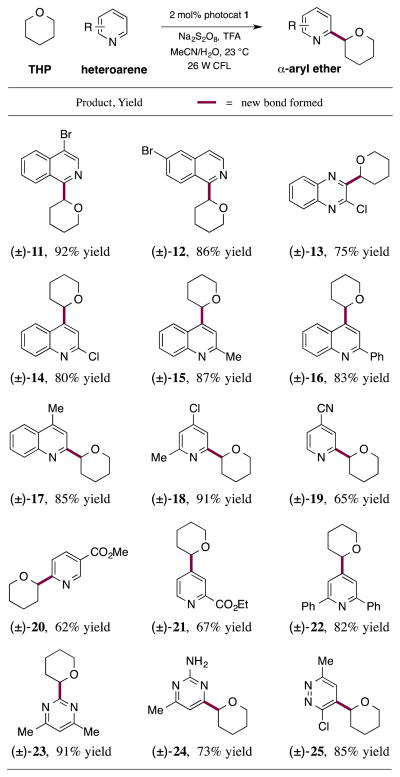
Having identified the optimal conditions for this α-ether arylation protocol, we next focused on defining the scope of the heteroarene component. As revealed in Table 2, a variety of electron-deficient heteroarenes will readily couple with THP in good to excellent yield at the most electrophilic site. Notably, bromo- or chloro-substituents at the C(4) and C(6) positions of isoquinoline are well tolerated, along with the C(2) positions of both quinoxaline and quinoline (products 11–14, 75–92% yields). Moreover, the use of 2-methyl or 2-phenylquinoline resulted in alkylation with THP selectively at the C(4) position while 4-methylquinoline underwent coupling exclusively at the C(2)-position (products 15–17, 83–87% yields). It is important to note that various pyridine derivatives with electron-withdrawing groups worked well in this ether-arylation protocol (products 18–22, 62–91% yields). Dimethylpyrimidine readily participated in this α-ethereal coupling with THP at the C(2) site in 91% yield (product 23). It should be noted that the use of free amine substituents on the aromatic ring resulted in diminished reactivity towards the addition of the nucleophilic α-oxyalkyl radical. More specifically, the coupling of 2-amino-4-methylpyrimidine with THP required 48 h to reach full conversion, albeit in a useful 73% yield (product 24). Again, high levels of regioselectivity were also obtained with pyridazine-based heteroarenes, providing the ortho-chloro alkylation adduct with almost complete site selectivity (product 25, 85% yield).
Table 2.
|
Yield of material isolated by column chromatography.
See Supporting Information for experimental details.
Next, we explored the scope of the ether component in this new α-heteroarylation protocol. As shown in Table 3, a range of cyclic and acyclic ethers were found to be suitable coupling partners. Besides tetrahydropyran (product 26, 88% yield), a series of tetrahydrofurans were found to be amenable to direct addition with isoquinoline (entries 27–29, 77–86% yields). It should be noted, however, for 2-methyltetrahydrofuran, that both the α-oxy methylene and α-oxy methine sites underwent arylation with a ~4:1 regioselectivity for the less hindered position (products 28, 81% yield). With this in mind it is intriguing to consider that the use of tetrahydrofurfuryl acetate led to α-methylene arylation with complete regioselectivity, a remarkable finding given the three available α-oxygen C–H containing positions (product 29, 77% yield). Moreover, the use of 3-oxotetrahydrofuran selectively gave the C(5)-arylation product in the presence of a β-carbonyl group, effectively leaving the α-position of the carbonyl untouched (product 30, 83% yield). A similar level of regiocontrol and efficiency was achieved with 4-oxotetrahydropyran (product 31, 86% yield). Recently oxetanyl moieties have emerged as a valuable new structural element in drug discovery.[23] However, at the present time, there are few synthetic methods that allow for the facile introduction of oxetane in a direct and routine fashion into medicinal agents.[23,24] We initially found that the use of this strained ring system led to an oxetanyl radical that underwent ring-opening polymerization under our optimal aqueous/acid conditions. Gratifyingly, when the coupling of oxetane and isoquinoline was carried out in MeCN (0.05 M) with the addition of (nBu)4NCl to help solubilize the persulfate anion, a useful 42% yield of the desired product was obtained (product 32). It should be noted that 1,4-dioxane is also amenable to this arylation protocol (product 33, 90% yield), while 1,3-dioxolane provided the masked formyl equivalent in 62% yield (product 34, 83% yield, r.r. 3:1). The use of acyclic dialkyl ethers was also demonstrated via the application of the diethyl and dibutyl variants, both providing the heteroarylation adducts in excellent yield (product 35 and 36, 93% and 88% yields, respectively). The coupling of diethyl ether is of particular interest, as its volatility (b.p. 35 °C) renders it incompatible with the temperatures required for thermal radical generation (80–90 °C).[6b,c] Moreover, dimethoxyethane underwent arylation at both the methylene and methyl sites (r.r. 2.3:1) with high levels of efficiency (product 37, 93% yield).
Table 3.

|
Yield of material isolated by column chromatography.
See Supporting Information for experimental details.
Regiomeric ratio (r.r.) determined by 1H NMR. 28: 3.5:1 r.r.; 34: 3:1 r.r.; 37: 2.3:1 r.r.
Diastereomeric ratio (d.r.) determined by 1H NMR. 28: 1.3:1 d.r.; 29: 2:1 d.r.
MeCN used as the only solvent; 2 equiv. of (nBu)4NCl added; no TFA added.
In summary, we have developed a photoredox catalytic approach to the direct α-arylation of ethers with electron-deficient heteroarenes. This visible light-promoted method shows a broad scope with regard to both the dialkyl ether and heteroarene substrates, providing consistently high yields of α-oxyalkylated arenes. Remarkably, this C(sp3)–C(sp2) bond construction was achieved efficiently via C–H functionalization of both the coupling partners at room temperature, thereby allowing the coupling of even highly volatile ether substrates. We anticipate this new photoredox-mediated C–H functionalization–Minisci addition will find broad application in both the academic and pharmaceutical sciences.
Supplementary Material
Footnotes
The authors are grateful for financial support provided by the NIH General Medical Sciences (Grant NIHGMS (R01 GM103558-03)) and gifts from Merck, Amgen, Abbvie, and BMS.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.
References
- 1.a) Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; b) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; c) Xuan J, Xiao WJ. Angew Chem. 2012;124:6934–6944. [Google Scholar]; Angew Chem Int Ed. 2012;51:6828–6838. doi: 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]; d) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Schultz DM, Yoon TP. Science. 2014;343:985. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) McNally A, Prier CK, MacMillan DWC. Science. 2011;334:1114–1117. doi: 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pirnot MT, Rankic DA, Martin DBC, MacMillan DWC. Science. 2013;339:1593–1596. doi: 10.1126/science.1232993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo Y-R. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton: 2007. [Google Scholar]
- 4.Mayer JM. Acc Chem Res. 2011;44:36–46. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qvortrup K, Rankic DA, MacMillan DWC. J Am Chem Soc. 2014;136:626–629. doi: 10.1021/ja411596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For a review of additions of nucleophilic radicals to heteroarenes, see: Minisci F, Fontana F, Vismara E. J Heterocycl Chem. 1990;27:79–96.For examples of Minisci reactions of oxyalkyl radicals generated via persulfate, see: Buratti W, Gardini GP, Minisci F, Bertini F, Galli R, Perchinunno M. Tetrahedron. 1971;27:3655–3668.Li X, Wang HY, Shi ZJ. New J Chem. 2013;37:1704–1706.For examples of Minisci reactions of oxyalkyl radicals generated via other methods, see: Minisci F, Bernardi R, Bertini F, Galli R, Perchinunno M. Tetrahedron. 1971;27:3575–3579.Minisci F, Citterio A, Vismara E. Tetrahedron. 1985;41:4157–4170.Fontana F, Minisci F, Yan YM, Zhao L. Tetrahedron Lett. 1993;34:2517–2520.
- 7.Lednicer D. Strategies for Organic Drug Synthesis and Design. John Wiley & Sons; Hoboken: 2008. [Google Scholar]
- 8.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174–238. doi: 10.1021/cr0509760.Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173–1193. doi: 10.1039/b606984n.Ackermann L, Vicente R, Kapdi AR. Angew Chem. 2009;121:9976–10011. doi: 10.1002/anie.200902996.Angew Chem Int Ed. 2009;48:9792–9826. doi: 10.1002/anie.200902996.Bellina F, Rossi R. Tetrahedron. 2009;65:10269–10310.Hari DP, König B. Angew Chem. 2013;125:4832–4842.Angew Chem Int Ed. 2013;52:4734–4743. doi: 10.1002/anie.201210276.For recent examples of additions of aryl radicals to heteroarenes, see: Xue D, Jia ZH, Zhao CJ, Zhang YY, Wang C, Xiao J. Chem Eur J. 2014;20:2960–2965. doi: 10.1002/chem.201304120.Zhang J, Chen J, Zhang X, Lei X. J Org Chem. 2014;79:10682–10688. doi: 10.1021/jo5020432.
- 9.a) Nagib DA, MacMillan DWC. Nature. 2011;480:224–228. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Molander GA, Colombel V, Braz VA. Org Lett. 2011;13:1852–1855. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fujiwara Y, Dixon JA, O’Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herlé B, Sach N, Collins MR, Ishihara Y, Baran PS. Nature. 2012;492:95–99. doi: 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Antonchick AP, Burgmann L. Angew Chem. 2013;125:3349–3353. doi: 10.1002/anie.201209584. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:3267–3271. doi: 10.1002/anie.201209584. [DOI] [PubMed] [Google Scholar]; e) Gui J, Zhou Q, Pan CM, Yabe Y, Burns AC, Collins MR, Ornelas MA, Ishihara Y, Baran PS. J Am Chem Soc. 2014;136:4853–4856. doi: 10.1021/ja5007838. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) DiRocco DA, Dykstra K, Krska S, Vachal P, Conway DV, Tudge M. Angew Chem. 2014;126:4902–4906. doi: 10.1002/anie.201402023. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2014;53:4802–4806. doi: 10.1002/anie.201402023. [DOI] [PubMed] [Google Scholar]
- 10.Doncton MAJ. Med Chem Commun. 2011;2:1135–1161. [Google Scholar]
- 11.Zhang SY, Zhang FM, Tu YQ. Chem Soc Rev. 2011;40:1937–1949. doi: 10.1039/c0cs00063a. [DOI] [PubMed] [Google Scholar]
- 12.Redox potentials measured by cyclic voltammetry. 2:1 MeCN:H2O was required to solubilize the electrolyte (nBu)4NPF6.
- 13.a) Caronna T, Citterio A, Grossi L, Minisci F, Ogawa K. Tetrahedron. 1976;32:2741–2745. [Google Scholar]; b) Dai C, Meschini F, Narayanam JMR, Stephenson CRJ. J Org Chem. 2012;77:4425–4431. doi: 10.1021/jo300162c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712–5719. [Google Scholar]; b) Cline ED, Bernhard S. Chimia. 2009;63:709–713. [Google Scholar]
- 15.Calculated from the corresponding ground state potential and emission maximum. See: Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617–1622. doi: 10.1021/jo202538x.b) Reference 14a.
- 16.While the oxidation potential for the S2O82−/SO4•− redox couple in MeCN could not be obtained by direct measurement or from the literature, the value has been estimated to be ≤ 0.35 V vs. SCE in aqueous solution. See: Memming R. J Electrochem Soc. 1969;116:785–790.b) Reference 13b.
- 17.HAT from ethers by sulfate radical anion has been reported previously. See: Huie RE, Clifton CL, Kafafi SA. J Phys Chem. 1991;95:9336–9340.Padmaja S, Alfassi ZB, Neta P, Huie RE. Int J Chem Kinet. 1993;25:193–198.
- 18.a) Wan LK, Peng J, Lin MZ, Muroya Y, Katsumura Y, Fu HY. Radiat Phys Chem. 2012;81:524–530. [Google Scholar]; b) Eibenberger H, Steenken S, O’Neill P, Schulte-Frohlinde D. J Phys Chem. 1978;82:749–750. [Google Scholar]; c) George C, El Rassy H, Chovelon J-M. Int J Chem Kinet. 2001;33:539–547. [Google Scholar]
- X.In order to rule out a radical chain reaction pathway via SET between 10 and 3, two identical reaction mixtures were irradiated with visible light for 30 minutes. At this point, one reaction was immediately worked up and analysed, while the other was allowed to proceed in the dark for an additional 3.5 h. The yields of these two reactions were found to be the same within experimental error, indicating that the reaction does not proceed in the absence of light and that a radical chain mechanism is unlikely to be operative. See Supporting Information for experimental details.
- 19.Minisci F, Vismara E, Fontana F, Morini G, Serravalle M, Giordano C. J Org Chem. 1987;52:730–736. [Google Scholar]
- 20.Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top Curr Chem. 2007;281:143–303. [Google Scholar]
- 21.Slinker JD, Gorodetsky AA, Lowry MS, Wang J, Parker S, Rohl R, Bernhard S, Malliaras GG. J Am Chem Soc. 2004;126:2763–2767. doi: 10.1021/ja0345221. [DOI] [PubMed] [Google Scholar]
- 22.a) Kalyanasundaram K. Coord Chem Rev. 1982;46:159–244. [Google Scholar]; b) Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, Von Zelewsky A. Coord Chem Rev. 1988;84:85–277. [Google Scholar]
- 23.Burkhard JA, Wuitschik G, Rogers-Evans M, Müller K, Carreira EM. Angew Chem. 2010;122:9236–9051. doi: 10.1002/anie.200907155. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:9052–9067. doi: 10.1002/anie.200907155. [DOI] [PubMed] [Google Scholar]
- 24.a) Coppi DI, Salomone A, Perna FM, Capriati V. Chem Commun. 2011;47:9918–9920. doi: 10.1039/c1cc13670d. [DOI] [PubMed] [Google Scholar]; b) Hashizume S, Oisaki K, Kanai M. Org Lett. 2011;13:4288–4291. doi: 10.1021/ol201629n. [DOI] [PubMed] [Google Scholar]; c) Ravelli D, Zoccolillo M, Mella M, Fagnoni M. Adv Syn Cat. 2014;356:2781–2786. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.