

Significance
Therapies for myocardial infarction consequent to ischemia/reperfusion injury (I/R) have been lacking. The netrin-1 receptor deleted in colorectal cancer (DCC) mediates netrin-1–dependent cardioprotective signaling. It is anticipated that any means of augmenting DCC signaling may induce cardioprotection on its own and enhance netrin-1/NO–dependent cardioprotection. The present study identifies a novel mechanism by which NO upregulates DCC abundance. NO inhibits seven in absentia homolog (SIAH), an E3 ubiquitin ligase that suppresses DCC. Inhibition of SIAH by in vivo RNA interference markedly reduces infarct size to improve cardiac function that was determined by echocardiography. Combined with netrin-1 perfusion, it further potentiated netrin-1’s cardioprotective effect. SIAH may therefore serve as a novel therapeutic target for myocardial infarction, particularly those suffering from cardiac I/R injury.
Keywords: SIAH, netrin-1, nitric oxide, DCC, cardioprotection
Abstract
Deleted in colorectal cancer (DCC), a large transmembrane receptor of netrin-1, is critical for mediating netrin-1’s cardioprotective function. In the present study we investigated novel mechanisms underlying netrin-1–induced, rapid, and feed-forward up-regulation of DCC, which is believed to sustain nitric oxide (NO) production to potentiate cardioprotection. Intriguingly, NO markedly reduced expression of the E3 ubiquitin ligase seven in absentia homolog (SIAH) that is specific for regulation of protesome-dependent DCC degradation, resulting in accumulation of DCC. The two SIAH isoforms compensate for each other when one is repressed; inhibition of both SIAH1 and SIAH2 using combined siRNAs significantly reduced infarct size while improving cardiac function after ischemia/reperfusion injury of the heart. This effect was absent in DCC-deficient mice. Moreover, in vivo RNAi inhibition of SIAH1/2 further augmented netrin-1’s cardioprotective function. In summary, these data identify a novel therapeutic target of SIAH in facilitating NO/netrin-1–dependent cardioprotection, using the DCC receptor. Combination of netrin-1 and SIAH RNAi may prove to be a substantially effective therapy for myocardial infarction.
Deleted in colorectal cancer (DCC), a single transmembrane receptor of netrin-1, was first discovered in 1990 during the search for candidate tumor suppressor genes in chromosome 18q (1). At present DCC is known to play important roles in the following biological processes: guidance of developing axons (2, 3), conditional control of apoptosis (4), tumorigenesis (5), and angiogenesis (6, 7). DCC-dependent production of nitric oxide (NO) mediates angiogenic responses of endothelial cells (6). We have recently identified a novel and robust effect of netrin-1 in cardioprotection (8, 9). Netrin-1 induces cardioprotection against ischemia/reperfusion (I/R) injury both ex vivo and in vivo, which is mediated by DCC/ERK1/2/eNOS/NO-dependent protection of cardiomyocytes from necrosis and apoptosis, mediated by NO-dependent attenuation of oxidative stress and preservation of mitochondrial function (8–10). A deficiency of DCC abolished the protective effects of netrin-1 against cardiac I/R injury, implying a crucial role of DCC in mediating netrin-1–induced cardioprotection (8, 10).
Although it is a large transmembrane protein, DCC protein abundance was significantly up-regulated by netrin-1 in I/R-injured heart within a very short time of 2 h (8). Intriguingly, the DCC accumulation induced by netrin-1 was attenuated by the NOS inhibitor l-NAME, implicating an NO-dependent mechanism (8). This NO-DCC feed-forward regulation, in addition to the initial DCC/ERK1/2/eNOS/NO pathway, may be extremely beneficial in potentiating netrin-1–dependent cardioprotection. It is possible that a proteasome dependent mechanism was involved in this quick action of protein regulation. Protein ubiquitination plays a key role in determining the half-life of proteins in mammalian cells. In ubiquitin-proteasome systems, E3 ubiquitin ligases recognize target substrates to induce transfer of ubiquitin from an E2 ubiquitin-conjugating enzyme to the substrates (11). Several specific E3 ligases play important roles in cardiac physiology and pathophysiology (12). We hypothesize that some specific E3 ligases might be involved in quickly regulating DCC in response to an increase in NO production, contributing to NO-mediated cardioprotection.
In the present study we fully characterized a novel role of an E3 ubiquitin ligase, SIAH (seven in absentia homolog), in mediating NO up-regulation of DCC to induce cardioprotection. Here we found that NO donor rapidly increased protein abundance of DCC by decreasing its level of ubiquitination, consequent to NO inhibition of SIAH. Inhibition of SIAH by in vivo RNAi significantly reduced infarct size to improve cardiac function, measured by echocardiography. Combined with netrin-1, SIAH inhibition further increased cardioprotective potency of netrin-1. Therefore, through accumulating DCC, SIAH inhibition can induce cardioprotection alone, or enhance netrin-1/NO–induced cardioprotection. Without doubt, SIAH can be developed as a novel target for the treatment of acute myocardial infarction, particularly for cardiac I/R injury induced by angioplasty.
Results
NO Donor Increases Protein Abundance of DCC in Endothelial Cells.
Our previous results have shown that DCC protein abundance was rapidly increased in netrin-1 perfused hearts during I/R injury, which is mediated by NO (8). Evidence from previous studies has also shown that endothelial cells can promote cardiomyocytes survival (13, 14). Indeed, activation of eNOS by netrin-1/DCC/ERK1/2 pathway increases NO production to result in cardioprotection. We hypothesize that NO-DCC feed-forward regulation occurs in endothelial cells to facilitate more NO production that is diffused to underneath cardiomyocytes to promote survival and preserve function. To test this hypothesis, we first examined the effect of NO donor on DCC protein abundance in endothelial cells. As shown in Fig. 1, exposure of bovine aortic endothelial cells (BAECs) to NO donor methylamine hexamethylene methylamine NONOate (MAHMA NONOate) resulted in a time-dependent increase in DCC protein expression, which occurred as quickly as 5 min after treatment (1.65 ± 0.16-fold vs. 0 min, P < 0.01). The response maximized at 30 min (2.15 ± 0.20-fold vs. 0 min, P < 0.001). These data implicate that NO can rapidly up-regulate protein abundance of DCC in endothelial cells.
Fig. 1.
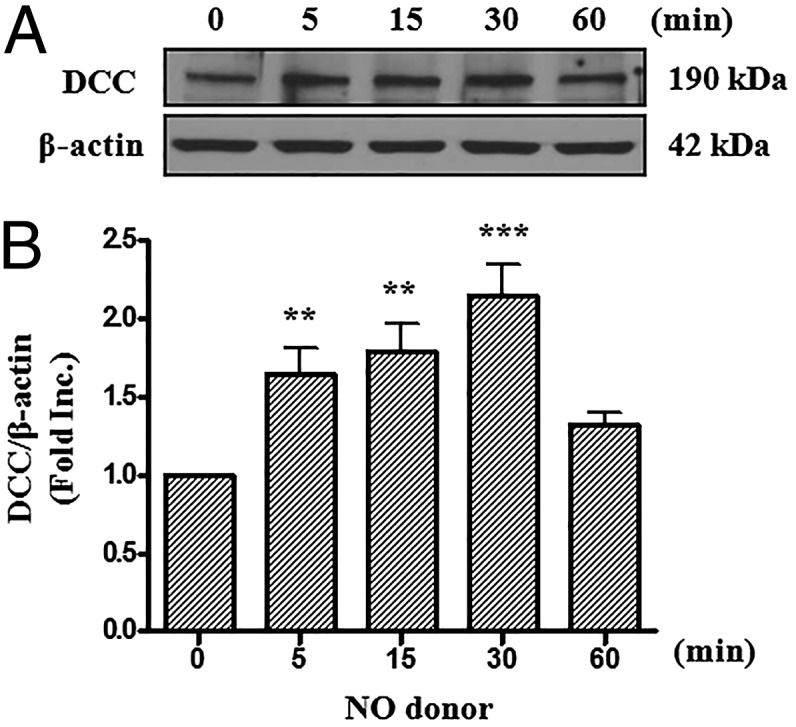
NO donor increases protein abundance of DCC. Bovine aortic endothelial cells were exposed to NO donor MAHMA-NONOate (6 mmol/L) and harvested at different time points (0, 5, 15, 30, or 60 min). Western blots were performed to detect DCC protein level. (A) Representative Western blots of DCC and β-actin. (B) Grouped densitometric data of DCC protein expression under NO donor treatment (mean ± SEM, n = 8). **P < 0.01, ***P < 0.001 vs. 0 min.
SIAH1 and SIAH2 Regulate DCC Stability via a Ubiquitin-Proteasome Pathway.
Remarkably, netrin-1 is able to feed-forwardly up-regulate its large transmembrane receptor DCC, which is required for NO production to mediate cardioprotection, as shown above and previously (8). NO donor increased protein abundance of DCC in endothelial cells within a very short time, which suggests a potential involvement of a proteasome-dependent degradation pathway. We then treated BAECs with the proteasome inhibitor MG132 for 0, 5, 15, 30, and 60 min and determined the protein levels of endogenous DCC by Western blots. Of note, MG132 treatment rapidly elevated DCC protein levels (Fig. 2A), suggesting that DCC stability is regulated by a proteasomal pathway.
Fig. 2.

SIAH1 and SIAH2 regulate DCC stability via the ubiquitin-proteasome pathway. (A) BAECs were exposed to MG132 (20 μmol/L) and harvested at different time points (0, 5, 15, 30, or 60 min). Western blots were performed to detect DCC protein level. Representative Western blots and grouped densitometric data of DCC protein expression are presented as mean ± SEM (n = 3). *P < 0.05, **P < 0.01 vs. 0 min. Inhibition of proteasome pathway with MG132 up-regulated DCC protein levels. (B) HA-DCC (500 ng) was cotransfected with different amount of pCMV-SIAH1, pCMV-SIAH1-DN, pCMV-SIAH2, or pCMV-SIAH2-DN (0, 10, or 100 ng) into HEK293T cells. The total amount of DNA was equalized by adding mock pCMV-Myc. Thirty hours later, protein abundance of DCC and SIAH1/2 were determined by Western blots with anti-HA antibody and anti-Myc antibody. Actin was used as an internal control. SIAH1 or -2 overexpression dose-dependently down-regulated DCC protein abundance. (C) HEK293T cells were cotransfected with HA-DCC and Myc-SIAH1 or Myc-SIAH2 constructs. Cell lysates were prepared and subjected to immunoprecipitation with anti-HA antibodies. The immunoprecipitates were analyzed by Western blots with anti-HA and anti-Myc antibodies. Both SIAH1 and SIAH2 can co-IP with DCC. (D) HA-DCC, Myc-ubiquitin, SIAH1 or -2 WT, and SIAH1 or -2-DN mutant constructs were cotransfected into HEK293T cells. DCC proteins were immunoprecipitated by anti-HA antibody. The polyubiquitinated forms of DCC were detected by Western blots with anti-Myc antibody. SIAH1 or -2 overexpression markedly increased polyubiquitination of DCC, which was blunted by SIAH1 or -2-DN mutant overexpression.
The E3 ubiquitin ligases SIAH1 and SIAH2 have been implicated in the regulation of DCC stability in mammalian cells. SIAH1 shares 77% amino acid identity with SIAH2 in humans (15–17). To examine a regulatory role of SIAH1 and SIAH2 in DCC degradation, we generated SIAH1 and SIAH2 dominant negative (DN) mutants (18). Cotransfection with SIAH1 or SIAH2, but not their DN mutants, dose-dependently diminished DCC protein abundance in HEK293T cells (Fig. 2B). Moreover, coimmunoprecipitation experiments indicated that both SIAH1 and SIAH2 can directly interact with DCC (Fig. 2C). To determine whether SIAH1 and SIAH2 affect DCC ubiquitination, we performed an in vivo ubiquitination assay. DCC was seen as a strong smear of bands when it was coexpressed with WT SIAH1 or SIAH2, but a much weaker smear of bands in the presence of SIAH1-DN or SIAH2-DN, which was similar to the control lane (Fig. 2D). Taken together, these data strongly suggest that SIAH1 and SIAH2 regulate DCC stability via a ubiquitin-proteasome pathway.
SIAH Inhibition Mediates NO Regulation of DCC.
Our data above confirmed a role of SIAH1 and SIAH2 in regulating DCC degradation. We next aimed to determine whether this pathway is involved in NO up-regulation of DCC. We examined the effect of NO donor on the ubiquitination level of endogenous DCC. After treatment with NO donor MAHMA NONOate, the poly-ubiquitinated forms of DCC were immunoprecipitated and detected by Western blots. Of note, NO donor markedly reduced ubiquitination of endogenous DCC (Fig. 3A). Furthermore, NO donor time-dependently abrogated SIAH1 expression, which corresponded well with an elevation in DCC abundance (Fig. 1 and Fig. 3 B and D). These data indicate that SIAH1-dependent ubiquitination of DCC was attenuated by NO donor to result in DCC up-regulation.
Fig. 3.

SIAH mediates NO regulation of DCC. (A) BAECs were transfected with Myc-ubiquitin for 48 h before being treated with MG132 (20 μmol/L) for 8 h and exposed to NO donor MAHMA-NONOate (6 mmol/L) for 20 min. DCC proteins were immunoprecipitated using anti-DCC antibody. The polyubiquitinated forms of DCC were detected by Western blots with anti-Myc antibody. NO donor induced a marked decrease in DCC polyubiquitination. (B and C) BAECs were treated with NO donor MAHMA-NONOate (6 mmol/L) and harvested at different time points (0, 5, 15, 30, or 60 min). Western blots were performed to detect SIAH1 (B) or SIAH2 (C) protein levels. Representative Western blots and grouped densitometric data are presented as mean ± SEM (n = 6). **P < 0.01 vs. 0 min. NO donor induced a significant reduction in SIAH1 protein expression. (D) Time-dependent responses of DCC, SIAH1, and SIAH2 protein levels under NO donor treatment. SIAH1 expression declined, whereas DCC protein levels increased in response to NO donor treatment. (E) Bovine aortic endothelial cells were transfected with empty Myc vector, Myc-SIAH1, or Myc-SIAH2 for 48 h, before exposure to NO donor MAHMA-NONOate (6 mmol/L, 20 min). DCC, SIAH1, and SIAH2 protein levels were determined using anti-DCC and anti-Myc antibodies. Actin was used as an internal control. Overexpression of SIAH1 or SIAH2 attenuated NO donor-induced increase in DCC expression. (F) Grouped densitometric data of DCC protein expression of E, mean ± SEM (n = 5). **P < 0.01.
However, SIAH2 protein level was not affected by NO donor stimulation (Fig. 3 C and D). It was puzzling at first, but our data showed that the SIAH isoforms compensate for each other (see below). To determine whether overexpression of SIAH can block NO up-regulation of DCC, we transfected Myc-SIAH1 or Myc-SIAH2 constructs into endothelial cells. Overexpression of either SIAH1 or SIAH2 prevented DCC up-regulation by NO donor (Fig. 3 E and F). These data reveal a central role of SIAH inhibition in mediating NO-dependent up-regulation of DCC.
SIAH1 and SIAH2 Compensate for Each Other.
To explore whether genetic inhibition of SIAH can promote DCC accumulation, we performed RNA interference experiments. SIAH1 and SIAH2 targeting siRNAs markedly decreased endothelial expression of SIAH1 and SIAH2 proteins to 39.3 ± 9.6% and 58.6 ± 3.0% of control, respectively (Fig. 4 A, B, D, and E), similar to what was shown in other cell lines (19, 20). Intriguingly, SIAH1 or SIAH2 was able to compensate for each other when one was suppressed, because RNAi for SIAH1 led to up-regulated SIAH2 by 1.37 ± 0.11-fold (Fig. 4C), and vice versa (1.34 ± 0.10-fold for SIAH1 when SIAH2 RNAi was applied; Fig. 4F). This observation was also confirmed in a mouse myoblast cell line C2C12 (Fig. S1). Cotransfection of both SIAH1 and SIAH2 siRNAs into endothelial cells increased protein abundance of DCC by 1.57 ± 0.06-fold (Fig. 5A). These data showed that SIAH1 and SIAH2 compensate for each other to synergistically regulate DCC protein abundance.
Fig. 4.

SIAH1 and SIAH2 compensate for each other. BAECs were transfected with 100 nmol/L scrambled siRNA or siRNA for SIAH1 (A–C), SIAH2 (D–F) for 48 h before being harvested and subjected to Western blot analyses. SIAH1 and SIAH2 were detected using anti-SIAH1 antibody and anti-SIAH2 antibody, and actin was used as an internal control. RNAi silencing of SIAH1 or SIAH2 resulted in compensatory up-regulation of SIAH2 or SIAH1, respectively. Grouped densitometric data of SIAH1 or SIAH2 protein levels are presented as mean ± SEM (n = 3). **P < 0.01, *P < 0.05.
Fig. 5.

SIAH1/2 RNAi up-regulates DCC protein abundance in endothelial cells and mouse heart. (A) BAECs were cotransfected with siRNAs for SIAH1 and SIAH2 together. DCC protein levels were determined by Western blots using anti-DCC antibody. Tubulin was used as an internal control. A combined treatment of SIAH1 and SIAH2 siRNAs resulted in an increase in DCC protein levels. (Lower) Grouped densitometric data of DCC protein levels, mean ± SEM (n = 4). **P < 0.01. (B) Mice were transfected with SIAH1/2 siRNA or their corresponding scrambled siRNAs (every 24 h, 7.5 nmol each time) via tail vain injection. Hearts were harvested for Western blot analysis of DCC protein expression, which showed up-regulation of DCC protein abundance in mice silenced of SIAH1/2 expression. Actin was used as an internal control. (Lower) Grouped densitometric data of DCC protein level, mean ± SEM (n = 3). *P < 0.05.
Inhibition of SIAH Induces and Potentiates Cardioprotection ex Vivo.
In view of the potent effect of SIAH in mediating NO regulation of DCC, we examined whether attenuation of SIAH leads to cardioprotection during I/R injury with or without netrin-1 perfusion. A combination of SIAH1 and SIAH2 siRNAs were delivered in vivo via tail vain injection, following our published protocol that demonstrates efficacy of in vivo RNAi (21). As shown in Fig. S2 A and B, results of immunofluorescent assay confirmed successful knockdown of corresponding SIAH isoforms in vivo in mouse heart. Intriguingly, in vivo RNAi of SIAH1/2 resulted in up-regulation of DCC protein abundance in I/R-injured hearts by 2.92 ± 0.39-fold (Fig. 5B). These data further demonstrate that SIAH1 and SIAH2 regulate DCC in mouse hearts.
I/R injury was performed in mouse hearts using a Langendorff system, as illustrated in Fig. 6A. In vivo RNAi inhibition of SIAH induced a significant reduction in infarct size compared with controls (40.6 ± 4.5% vs. 55.6 ± 1.4% for SIAH1/2 siRNA treated I/R vs. scrambled siRNA treated I/R, P < 0.01; Fig. 6 B and C). In additional experiments, a combinatory effect of netrin-1 and SIAH in vivo RNAi was examined. Our previous studies have established netrin-1’s cardioprotective role against I/R injury both ex vivo and in vivo (8–10). In scrambled siRNA treated group, netrin-1 reduced infarct size from 55.6 ± 1.4% to 27.7 ± 3.6% (P < 0.001). In vivo RNAi inhibition of SIAH further reduced infarct size of the netrin-1 treated group to 15.2 ± 1.2% (P < 0.01). These data demonstrate a clear additive effect of SIAH inhibition in affording augmented cardioprotection against I/R injury.
Fig. 6.

Inhibition of SIAH induces cardioprotection and potentiates netrin-1 induced cardioprotection. (A) Hearts isolated from mice injected with SIAH1/2 siRNA were subjected to I/R injury using a Langendorff perfusion system. Hearts were preperfused with Krebs–Henseleit buffer (KHB) for 30 min stabilization, followed by 45 min with or without netrin-1 perfusion. Then I/R injury was consistently produced by subjecting the hearts to 20 min of global ischemia, followed by reperfusion for 60 min with or without netrin-1. Sections of hearts were stained with 2,3,5-triphenyl tetrazolium chloride (TTC) and infarct area calculated as percentage of risk area. (B) Representative TTC staining of I/R-injured hearts isolated from mice transfected in vivo siRNAs for SIAH1/2 with or without netrin-1 perfusion. (C) Quantitative grouped data of B, mean ± SEM (n = 3–4). **P < 0.01, ***P < 0.001.
SIAH Inhibition Attenuates I/R-Induced Myocardial Infarction and Improves Cardiac Function in Vivo.
To explore whether lowered SIAH levels have any effect on I/R damage in vivo, RNAi-treated mice were subjected to a 30 min of ischemia by coronary occlusion, followed by a 24-h reperfusion (Fig. 7A) (9). As shown in Fig. 7 B–E, in agreement with our ex vivo data, SIAH1/2 siRNA alone significantly decreased infarct size compared with the scrambled siRNA group. Mice receiving scrambled siRNA displayed a 35.7 ± 4.5% infarct size per area at risk (Inf/AAR) and a 19.6 ± 3.2% infarct size per left ventricle (LV) size (Inf/LV). The SIAH1/2 siRNA-treated group, however, developed infarct of 17.4 ± 2.3% for Inf/AAR and 9.5 ± 1.1% for Inf/LV. The percentage AAR per LV (AAR/LV) was similar among different groups, indicating similar severities of myocardial ischemia.
Fig. 7.

SIAH inhibition attenuates I/R-induced myocardial infarction and improves cardiac function in vivo. (A) SIAH1/2 siRNAs were delivered by tail vain injection. I/R injury of the heart was induced by 30 min of left coronary artery (LCA) ligation, followed by a 24 h of reperfusion in WT C57BL6 mice in vivo. Evans blue was used to visualize the nonischemic area. Sections of hearts were stained with 2,3,5-TTC. (B) Representative TTC staining of I/R-injured hearts. The white area indicates infarct zone, whereas the blue area indicates noninfarcted area. The red and white areas represent area at risk. (C–E) Infarct size analyzed by (C) AAR/LV, (D) Inf/LV, and (E) Inf/AAR. The results are represented as means ± SEM (n = 4). *P < 0.05. (F–H) Echocardiography was performed on in vivo RNAi-ed WT mice reperfused for 24 h after 30 min of LCA ligation. Ejection fraction and fractional shortening were measured. (F) Representative echocardiography data of scrambled siRNA group and SIAH1/2 siRNA group. (G) Grouped ejection fraction (EF) and (H) grouped fractional shortening (FS) data (n = 6). *P < 0.05.
To further validate cardioprotective properties of SIAH inhibition in vivo, we measured cardiac function via echocardiography on animals that underwent I/R injury. The results, shown in Fig. 7 F–H, illustrated that both fractional shortening and ejection fraction were significantly increased in SIAH1/2 siRNA-treated hearts at 1 d after I/R, compared with scrambled siRNA treated hearts, suggesting improvement in cardiac function.
Inhibition of SIAH Augments Netrin-1 Induced Cardioprotection in Vivo.
To further examine whether SIAH inhibition is effective in potentiating cardioprotection induced by netrin-1 in vivo, the same coronary artery occlusion assay was performed. Netrin-1 was injected into the LV lumen at the onset of reperfusion at a dosage as small as 5 μg/kg. Our recent findings have established netrin-1’s cardioprotective function in this model (9). Data in Fig. 8 demonstrated that inhibition of SIAH markedly augmented the cardioprotective effect of netrin-1 (Fig. 8 A–D). The scrambled siRNA plus netrin-1 group displayed an 18.5 ± 2.3% Inf/AAR and a 9.8 ± 0.9% Inf/LV. The SIAH1/2 siRNA plus netrin-1 group showed a 12.1 ± 1.0% Inf/AAR and a 6.7 ± 0.9% Inf/LV. These data indicate robust additive effects of cardioprotection induced by in vivo RNAi of SIAH and netrin-1 perfusion. Of note, AAR/LV was similar for both groups, indicating similar degree of ischemic injury.
Fig. 8.

Inhibition of SIAH augments netrin-1–induced cardioprotection in vivo. SIAH1/2 siRNAs were delivered by tail vain injection. I/R injury of the heart was induced by 30 min of LCA ligation followed by 24 h of reperfusion in WT C57BL6 mice in vivo. Netrin-1 was injected into the LV lumen at the onset of reperfusion at doses of 5 μg/kg. Evans blue was used to visualize the nonischemic area. Sections of hearts were stained with 2,3,5-TTC. (A) Representative TTC staining of I/R-injured hearts. The white area indicates infarct zone, whereas the blue area indicates noninfarcted area. The red and white areas represent area at risk. (B–D) Infarct size analyzed by (B) AAR/LV, (C) Inf/LV, and (D) Inf/AAR. The results are represented as Means ± SEM, n = 5. *P < 0.05. (E–G) Echocardiography was performed on in vivo RNAi-ed WT mice reperfused for 72 h after 30 min of LCA ligation. Ejection fraction and fractional shortening were measured. (E) Representative echocardiography data of scrambled siRNA with netrin-1 group and SIAH1/2 siRNA with netrin-1 group. (F) Grouped EF and (G) grouped FS data (n = 4). **P < 0.01.
Moreover, cardiac function was measured by echocardiography. There was no obvious difference between the two groups at 1 d after I/R. However, fractional shortening and ejection fraction were significantly increased in SIAH1/2 siRNA plus netrin-1-treated hearts 3 d after I/R, compared with scrambled siRNA plus netrin-1-treated hearts (Fig. 8 E–G).
SIAH Inhibition-Induced Cardioprotection Is Absent in DCC-Deficient Mice.
To further confirm whether SIAH inhibition-induced cardioprotection is mediated by DCC, hearts isolated from DCC+/− mice and WT littermates were subjected to I/R injury using a Langendroff system. DCC deficiency on its own increased infarct size after I/R injury (52.4 ± 3.2% vs. 39.2 ± 2.5% for DCC+/− mice vs. WT mice, P < 0.05; Fig. 9 A and B), implicating an important role of endogenous DCC in cardioprotection. Moreover, as shown in Fig. 9 C and D, in vivo RNAi inhibition of SIAH1/2 had no additional effect on infarct size in DCC+/− mice (SIAH1/2 siRNA group, 55.4 ± 5.3% vs. scrambled siRNA group, 54.4 ± 7.5%). These data indicate that cardioprotection afforded by SIAH inhibition is mediated by DCC.
Fig. 9.

SIAH inhibition-induced cardioprotection is absent in DCC-deficient mice. (A and B) Hearts harvested from DCC+/− mice and WT littermates were subjected to I/R injury using a Langendorff perfusion system with protocol described in Fig. 6. Sections of hearts were stained with 2,3,5-TTC and infarct area calculated as percentage of risk area. (A) Representative TTC staining of I/R-injured hearts isolated from WT and DCC+/− mice. (B) Quantitative grouped data of A, mean ± SEM (n = 4). *P < 0.05. (C and D) SIAH1/2 siRNAs and scrambled siRNA were delivered into DCC+/− mice before subjecting the animals to identical I/R protocols. Sections of hearts were stained with 2,3,5-TTC and infarct area calculated as percentage of risk area. (C) Representative TTC staining of I/R-injured hearts isolated from DCC+/− mice with scrambled siRNA and SIAH1/2 siRNA. (D) Quantitative grouped data of C, mean ± SEM (n = 3).
Discussion
The most significant finding of the study is the innovative identification of an NO/SIAH/DCC pathway in DCC-dependent cardioprotection, and its role in potentiating netrin-1/NO–provoked cardioprotection. NO down-regulates total SIAH protein abundance, leading to decreased degradation of DCC via a ubiquitin-proteasome pathway. This feed-forward loop is believed to augment cardioprotective signaling of netrin-1/DCC/ERK1/2/eNOS/NO by accumulation of DCC receptor. SIAH inhibition not only effectively reduced infarct size after I/R injury, but also markedly improved cardiac function. More importantly, it also markedly potentiated netrin-1’s cardioprotective effects. These findings define a central mediator role of SIAH in DCC-dependent cardioprotection provoked by netrin-1/NO.
As we described earlier, DCC protein level is critical for netrin-1 signaling. Several previous studies have provided evidence for mechanisms of regulation for DCC. For example, Kuo et al. (22) have shown that a zinc finger transcription factor, Bcl11A/CTIP1, regulates expression of DCC to control axon branching and dendrite outgrowth. In addition, Hu and colleagues identified that Drosophila Sina and its mammalian homologs SIAH can regulate DCC protein level via proteasome-dependent mechanisms (15, 16). In the present study, endothelial DCC protein accumulated in response to NO donor within a very short time. Because DCC is a large protein, we hypothesized that this quick response is at the posttranslational level, and likely through a proteasome signaling pathway. Indeed, reduced ubiquitination of endogenous DCC after NO donor treatment was observed. A proteasome signaling pathway has been considered as an important mechanism in regulating protein degradation. Three enzymes, known as E1, E2, and E3, act in series to catalyze ubiquitination (23). Among them, E3 ligase is thought to play a role in recognizing the specific substrate protein (24). We therefore next examined a role of SIAH E3 ubiquitin ligase in NO modulation of DCC protein stability.
Our findings showed that both SIAH family members SIAH1 and SIAH2 regulate DCC protein abundance through direct interaction, resulting in increased ubiquitination of DCC in HEK293T cells, which suggests functional redundancy among the members of the family. It is not surprising that the same substrate can be degraded by one or more E3 ubiquitin ligase family members, because they share a similar motif for substrate binding. For example, TGF-β receptor can be degraded by four members of the Nedd4-like family, including Smurf1, Smurf2, WWP1, and Nedd4-2 (25). House et al. (26) identified a peptide motif (RPVAxVxPxxR) that mediates the interaction of SIAH protein with an array of protein partners. The relative contribution of E3 members might be dependent on the cellular context and distribution of each member in different cell types or tissues. More importantly, we have further identified the involvement of SIAH in NO up-regulation of DCC, to our knowledge for the first time. In our study, NO donor diminished SIAH protein abundance, whereas overexpression of SIAH blocked NO up-regulation of DCC. These data establish a novel rapidly responsive NO/SIAH/DCC signaling pathway in endothelial cells that is important for induction of cardioprotection.
Another interesting finding is that SIAH1 and SIAH2 compensate for each other in both BAECs and C2C12 cells. The inhibition of one isoform increases protein abundance of the other, in which the organism ensures enough functional proteins at work. This seems to share some similarities with other E3 systems. For example, loss of Pellino 1 E3 ubiquitin ligase in peripheral innate immune cells results in functional compensation by Pellino 2 and/or Pellino 3 (27). Therefore, to regulate DCC protein abundance, two SIAH isoforms must be properly and coordinately controlled. Indeed, SIAH1/2 RNAi together effectively increased DCC protein abundance in both endothelial cells and mouse heart. These data indicate that SIAH isoforms compensate for each other to synergistically regulate DCC. Both SIAH1 and SIAH2 ought to be considered as targets to potentiate netrin-1–induced cardioprotective signaling via modulation of DCC.
Of note, combined silencing of SIAH1/2 in vivo significantly reduced infarct size and improved cardiac function. The cardioprotective role of SIAH1/2 inhibition alone was mediated by endogenous SIAH/DCC pathway, because DCC+/− mice displayed no cardioprotection in response to SIAH inhibition. In addition, in vivo SIAH1/2 RNAi, combined with netrin-1 perfusion, markedly augmented cardioprotective effects of netrin-1. The effects and mechanisms of netrin-1 alone have been established by our previous work (8–10, 28). Taken together, reduced SIAH1/2 can work very well either alone or in combination with netrin-1 to induce robust cardioprotection.
In summary, the present study characterized a novel signaling mechanism whereby NO up-regulates DCC by inhibition of its E3 ubiquitin ligase SIAH, leading to sustained netrin-1/NO signaling to provoke cardioprotection. SIAH can quickly regulate DCC protein abundance through a ubiquitin-proteasome pathway. RNAi inhibition of SIAH proved to be beneficial in reducing infarct size and improving cardiac function by accumulation of DCC. In addition to our previously established netrin-1/DCC/ERK1/2/eNOS/NO signaling, the feed-forward NO/SIAH/DCC signaling also augments cardioprotection induced by netrin-1/NO. This study may facilitate development of novel therapeutics targeting SIAH, for the treatment of myocardial infarction, particularly in patients suffering from cardiac I/R injury.
Materials and Methods
Detailed information of materials, cell culture and transfection, immunoprecipitation and Western blotting, in vivo RNAi and Langendorff perfusion, infarct size analysis, in vivo murine model of myocardial I/R injury, echocardiography, immunofluorescence microscopy, and statistical analysis are provided in SI Materials and Methods. The use of animals and experimental procedures were approved by the Institutional Animal Care and Usage Committee at the University of California, Los Angeles (UCLA).
Supplementary Material
Acknowledgments
This study was supported by NIH National Heart, Lung and Blood Institute Grants HL077440 (to H.C.), HL088975 (to H.C.), HL108701 (to H.C. and David G. Harrison), and HL119968 (to H.C.); American Heart Association Established Investigator Award 12EIA8990025 (to H.C.); and American Heart Association Postdoctoral Fellowship Award 14POST20380966 (to Q.L.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1420695112/-/DCSupplemental.
References
- 1.Fearon ER, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990;247(4938):49–56. doi: 10.1126/science.2294591. [DOI] [PubMed] [Google Scholar]
- 2.Keino-Masu K, et al. Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell. 1996;87(2):175–185. doi: 10.1016/s0092-8674(00)81336-7. [DOI] [PubMed] [Google Scholar]
- 3.Liu G, et al. Netrin requires focal adhesion kinase and Src family kinases for axon outgrowth and attraction. Nat Neurosci. 2004;7(11):1222–1232. doi: 10.1038/nn1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehlen P, et al. The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature. 1998;395(6704):801–804. doi: 10.1038/27441. [DOI] [PubMed] [Google Scholar]
- 5.Mazelin L, et al. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature. 2004;431(7004):80–84. doi: 10.1038/nature02788. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen A, Cai H. Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed-forward mechanism. Proc Natl Acad Sci USA. 2006;103(17):6530–6535. doi: 10.1073/pnas.0511011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park KW, et al. The axonal attractant Netrin-1 is an angiogenic factor. Proc Natl Acad Sci USA. 2004;101(46):16210–16215. doi: 10.1073/pnas.0405984101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Cai H. Netrin-1 prevents ischemia/reperfusion-induced myocardial infarction via a DCC/ERK1/2/eNOS s1177/NO/DCC feed-forward mechanism. J Mol Cell Cardiol. 2010;48(6):1060–1070. doi: 10.1016/j.yjmcc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouhidel JO, et al. Netrin-1 improves post-injury cardiac function in vivo via DCC/NO-dependent preservation of mitochondrial integrity, while attenuating autophagy. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbadis.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bouhidel JO, Wang P, Li Q, Cai H. Pharmacological postconditioning treatment of myocardial infarction with netrin-1. Front Biosci (Landmark Ed) 2014;19(3):566–570. doi: 10.2741/4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 12.Powell SR, Herrmann J, Lerman A, Patterson C, Wang X. The ubiquitin-proteasome system and cardiovascular disease. Prog Mol Biol Transl Sci. 2012;109(chap 9):295–346. doi: 10.1016/B978-0-12-397863-9.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narmoneva DA, Vukmirovic R, Davis ME, Kamm RD, Lee RT. Endothelial cells promote cardiac myocyte survival and spatial reorganization: implications for cardiac regeneration. Circulation. 2004;110(8):962–968. doi: 10.1161/01.CIR.0000140667.37070.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuramochi Y, et al. Cardiac endothelial cells regulate reactive oxygen species-induced cardiomyocyte apoptosis through neuregulin-1beta/erbB4 signaling. J Biol Chem. 2004;279(49):51141–51147. doi: 10.1074/jbc.M408662200. [DOI] [PubMed] [Google Scholar]
- 15.Hu G, et al. Mammalian homologs of seven in absentia regulate DCC via the ubiquitin-proteasome pathway. Genes Dev. 1997;11(20):2701–2714. doi: 10.1101/gad.11.20.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu G, Fearon ER. Siah-1 N-terminal RING domain is required for proteolysis function, and C-terminal sequences regulate oligomerization and binding to target proteins. Mol Cell Biol. 1999;19(1):724–732. doi: 10.1128/mcb.19.1.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu G, et al. Characterization of human homologs of the Drosophila seven in absentia (sina) gene. Genomics. 1997;46(1):103–111. doi: 10.1006/geno.1997.4997. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt RL, et al. Inhibition of RAS-mediated transformation and tumorigenesis by targeting the downstream E3 ubiquitin ligase seven in absentia homologue. Cancer Res. 2007;67(24):11798–11810. doi: 10.1158/0008-5472.CAN-06-4471. [DOI] [PubMed] [Google Scholar]
- 19.Lee SH, et al. Novel tumor suppressive function of Smad4 in serum starvation-induced cell death through PAK1-PUMA pathway. Cell Death Dis. 2011;2(12):e235. doi: 10.1038/cddis.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhanot M, Smith S. TIN2 stability is regulated by the E3 ligase Siah2. Mol Cell Biol. 2012;32(2):376–384. doi: 10.1128/MCB.06227-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Youn JY, Gao L, Cai H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia. 2012;55(7):2069–2079. doi: 10.1007/s00125-012-2557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo TY, Hong CJ, Hsueh YP. Bcl11A/CTIP1 regulates expression of DCC and MAP1b in control of axon branching and dendrite outgrowth. Mol Cell Neurosci. 2009;42(3):195–207. doi: 10.1016/j.mcn.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 23.Welchman RL, Gordon C, Mayer RJ. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol. 2005;6(8):599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- 24.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol Rev. 2002;82(2):373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 25.Kuratomi G, et al. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem J. 2005;386(Pt 3):461–470. doi: 10.1042/BJ20040738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.House CM, et al. A binding motif for Siah ubiquitin ligase. Proc Natl Acad Sci USA. 2003;100(6):3101–3106. doi: 10.1073/pnas.0534783100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moynagh PN. The roles of Pellino E3 ubiquitin ligases in immunity. Nat Rev Immunol. 2014;14(2):122–131. doi: 10.1038/nri3599. [DOI] [PubMed] [Google Scholar]
- 28.Siu KL, Lotz C, Ping P, Cai H. Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J Mol Cell Cardiol. 2015;78C:174–185. doi: 10.1016/j.yjmcc.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.