

Significance
Glioblastoma is an incurable brain tumor. It is characterized by intratumoral phenotypic and genetic heterogeneity, but the functional significance of this heterogeneity is unclear. We devised an integrated functional and genomic strategy to obtain single cell-derived tumor clones directly from patient tumors to identify mechanisms of aggressive clone behavior and drug resistance. Genomic analysis of single clones identified genes associated with clonal phenotypes. We predict that integration of functional and genomic analysis at a clonal level will be essential for understanding evolution and therapeutic resistance of human cancer, and will lead to the discovery of novel driver mechanisms and clone-specific cancer treatment.
Keywords: cancer, glioblastoma, clonal heterogeneity, genomic analysis, functional analysis
Abstract
Glioblastoma (GBM) is a cancer comprised of morphologically, genetically, and phenotypically diverse cells. However, an understanding of the functional significance of intratumoral heterogeneity is lacking. We devised a method to isolate and functionally profile tumorigenic clones from patient glioblastoma samples. Individual clones demonstrated unique proliferation and differentiation abilities. Importantly, naïve patient tumors included clones that were temozolomide resistant, indicating that resistance to conventional GBM therapy can preexist in untreated tumors at a clonal level. Further, candidate therapies for resistant clones were detected with clone-specific drug screening. Genomic analyses revealed genes and pathways that associate with specific functional behavior of single clones. Our results suggest that functional clonal profiling used to identify tumorigenic and drug-resistant tumor clones will lead to the discovery of new GBM clone-specific treatment strategies.
Glioblastoma (GBM) is the most common and most aggressive primary malignant brain tumor in adults. Despite major efforts to improve GBM survival, radiation therapy with concurrent temozolomide (TMZ) chemotherapy achieves only a median survival of 15 months with few long-term survivors. Many patients fail to respond to TMZ, and treatment of all patients at disease progression uniformly fails.
Glioblastoma is one of the first cancer types systematically studied at a genomic and transcriptomic level (1–3). Transcriptional profiling of GBM samples has revealed a landscape of intertumoral heterogeneity with distinct molecular tumor subtypes, although only slight prognostic differences are apparent in patients except for a clearly better prognosis in the CIMP+/IDH1 mutant subgroup (1, 4–6). Increasing evidence suggests that cancer tissues are more complex than previously thought, as tumors comprise considerable intratumoral heterogeneity with mixtures of genetically distinct subclones that likely escape therapy and cause disease progression (4, 7–10). In particular, GBM is a cancer type comprised of morphologically and phenotypically diverse cells (11). Recent studies have also uncovered genetic diversity apparent in subsamples of individual patient GBMs (12–15), and more recently in single GBM cells (9). Disease recurrence is associated with mutational events that are not shared with the primary tumor suggesting evolution from minority populations present at time of initial diagnosis (10).
Understanding the links between genetic and functional behavior of individual GBM clones, derived from single patient samples, will be essential to decipher patient-specific molecular mechanisms of GBM progression and therapeutic resistance. The study of bulk tumors provides a mixture of different signals from a heterogeneous set of clones and current single cell approaches are amenable to genomics but not functional studies. Here, we provide a detailed analysis of single cell derived clones from patient GBM samples using a method that directly links clonal molecular changes with functional properties. Parallel phenotypic and genomic analysis of these clones reveals a diverse landscape of functional and genetic heterogeneity, as well as insights into drug sensitivity pathways in glioblastoma.
Results
Single Cell-Derived Clones of Human Glioblastoma Exhibit Functional Heterogeneity.
To enable functional characterization of heterogeneous clones in glioblastoma, we devised a method to isolate and grow individual clones from patient tumors using flow cytometry sorting (FACS) and single cell plating in stem cell culture conditions (Fig. 1A). We generated 44 individual single cell derived clones from four patient tumors. All tested clones established typical GBM tumors after orthotopic injection in mice, indicating their tumorigenic potential (SI Appendix, Table S1 and Fig. S1 A–D and Fig. 1B).
Fig. 1.

Single cell-derived clonal cultures from primary GBMs show a neural precursor phenotype and are tumorigenic. (A) Functional characterization of single cell-derived clones. Glioblastoma samples were dissociated into single cell suspensions. Cells were then sorted using stem cell markers to enrich for clonogenic activity. Single live cells were then expanded in EGF/FGF media and analyzed functionally and genomically. (B) Immunostaining of a representative xenograft shows tumor formation with an infiltrative phenotype reminiscent of human GBM cell behavior in situ (green, human nestinl; red, human GFAP; blue, DAPI).
We next functionally characterized individual clones. We identified heterogeneous expression of key GBM proteins phosphatase and tensin homolog (PTEN), epidermal growth factor receptor (EGFR), and the constitutively active EGFR deletion mutant, EGFRvIII (2, 3, 10, 16), in three tumors, indicating that distinct known molecular GBM drivers vary at a clonal level (Fig. 2A and SI Appendix, Fig. S2) and suggesting limitations to targeted therapies based solely on bulk GBM analysis. In identical culture conditions and with each result consistent in at least two independent experiments with clones of different passages, the clones exhibited wide and independent variation in proliferation and differentiation abilities (Fig. 2B and SI Appendix, Figs. S3 and S4 A–C), demonstrating intrinsic distinct functional properties.
Fig. 2.
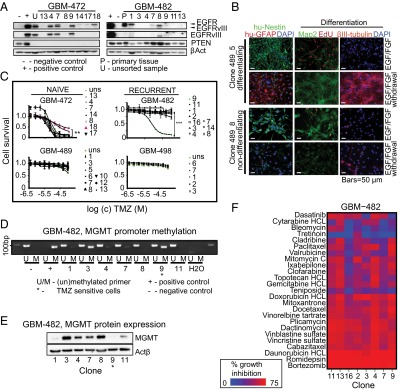
Individual clones within the same tumor show phenotypic heterogeneity. (A) Differential expression of EGFR, EGFRvIII, and PTEN in individual clones from two patient tumors. EGFRvIII-transfected human fetal brain cell line HF7450NS was used as control. (B) Individual clones of tumor GBM-489 vary in their ability to express markers of differentiation. Staining for human Nestin (green), human GFAP (red), Map2 (green), βIII-tubulin (red), and EdU (red). (C) Variable temozolomide (TMZ) drug responses of individual clones from four different tumors. Clones were treated once with TMZ (0.39–100 µM) and measured for viability. Significant differences in TMZ response were observed among clones from two tumors, GBM-472 (P = 0.0011), and GBM-482 (P < 10−4). (D) Heterogeneous promoter methylation of MGMT in GBM-482. Asterisks indicate sensitive cell populations. (E) Heterogeneous protein expression of MGMT in GBM-482. (F) NCI drug library screen reveals clones with variable drug sensitivity in GBM-482. Drugs with at least 50% growth inhibition on individual samples are shown and are ranked by response variability.
We then assessed clonal population response to the conventional GBM chemotherapeutic agent TMZ (17), in at least three independent experiments with clones of different passages. Two tumors contained clones with heterogeneous TMZ responses: 2 of 8 tested populations in the naïve tumor GBM-472 showed significantly increased resistance to TMZ (P = 0.0011, t test with Welch correction; clones 472_8, 472_18), whereas 1 clone of 10 in the recurrent tumor GBM-482 was relatively sensitive (P < 10−4; 482_9; Fig. 2C). We observed only TMZ-resistant clones from the two additional naïve and recurrent tumors. TMZ response and proliferation ability was not significantly correlated (SI Appendix, Figs. S5 and S6). Clonal analysis of O6-methylguanin-DNA methyltransferase (MGMT), a common biomarker of TMZ resistance in GBM (18), showed variable promoter methylation and protein expression levels that were inconsistent with TMZ response (Fig. 2 D and E and SI Appendix, Fig. S7). These data show that TMZ-resistant clones are present ab initio and stably in untreated patient tumors and are not necessarily acquired under therapeutic selection pressure.
Given that many GBM clones were TMZ resistant, we sought to identify potential alternative chemotherapeutic agents. We screened six to eight individual clones from each of three tumors with the National Cancer Institute (NCI) oncology library of 98 drugs and found that every tested tumor showed clone by clone variability in responses to multiple drugs (Fig. 2F and SI Appendix, Table S2 and Figs. S8 and S9). These data readily suggest reasons why this disease is so refractory to chemotherapies and suggest that clone-specific therapies could be developed for GBM treatment.
Integrated Analysis of Functional and Genomic Clonal Heterogeneity Predicts Pathways of Drug Resistance.
Copy number (CN) profiling of all samples with Affymetrix SNP6.0 microarrays reveals a landscape of clonal genetic heterogeneity, consistent with published observations (2, 3, 9) (Fig. 3A). All tumors carried hallmark GBM alterations such as chr7 gains and chr10 losses, validating our genetic data. Phylogenetic analysis of CN profiles confirms the heterogeneous genetic composition of GBMs and shows that clones of common origin are genetically closer to each other than to those from other tumor samples (Fig. 3B and SI Appendix, Fig. S10). We did not find single clones from one patient crossing into the genomic space of another. These results suggest that the impact of our stem cell culturing on genetic stability is negligible compared with clonal genetic heterogeneity. They also suggest that unsorted populations have one or more dominant clones: two unsorted populations (GBM-489, GBM-498) most resemble single clones (489_13, 498_5) and thus likely have single dominant clones, whereas the unsorted population of tumor GBM-472 is similar to multiple clones or the primary tumor sample and thus is likely polyclonal.
Fig. 3.

Heterogeneous clonal genetics as mapped by copy number (CN) analysis. (A) Probe-level CN heat map shows genetic variation in clonal populations. Samples are grouped vertically by patient tumor (left vertical color bar; right label: asterisk, primary sample; diamond, unsorted population). (B) Phylogenetic analysis confirms the common origin of derived clones (colors) and shows that primary samples (asterisks) are relatively distant from derived clones. (C) Circos plot of segmented CN alterations in GBM-482. Inner circle represents baseline (no CN change), and lines expanding toward the center or the periphery of the diagram represent CN losses or gains, respectively (primary, black; clonal, colored). (D) FISH confirms chr3 gain in primary tumor tissue of GBM-482. (E) Heat map of frequently altered genes with clonal heterogeneity (at least five alterations).
CN analysis reveals clonal chromosomal and focal genomic alterations (Fig. 3C and SI Appendix, Fig. S11 A–C). For example, gain of chr7 appears in a subset of clones from tumor GBM-482, whereas chr3 gain is only observed in clone 482_3 (Fig. 3D and SI Appendix, Fig. S12). Fluorescence in situ hybridization (FISH) validates these amplifications and highlights gain of chr3 in clone 482_3 as a minor cell population of the primary tissue, as well as the genetic homogeneity of derived clones (Fig. 3D and SI Appendix, Fig. S13). These data confirm genetic polyclonality in sample GBM-482 and provide evidence that our derived clones are homogeneous and originate from the primary tumor sample.
Clonal genetic heterogeneity is also apparent at the gene level (Fig. 3E and SI Appendix, Table S3 and Fig. S14). The most frequent clonal variation comprises hallmark GBM events such as EGFR amplification (2). In total, we found copy number alterations in 456 genes, including 31 of 100 published frequently CN altered genes in GBM (2) (P = 1.2 × 10−25; OR, 19; Fisher’s exact test; SI Appendix, Fig. S15). Clonally altered genes are enriched in functions such as regulation of fibroblast proliferation [6 genes; false discovery rate (FDR), P = 0.022 from g:Profiler (19)], and signaling pathways such as PI3K/AKT (2) (13 genes; FDR, P = 0.014), TGFβ (20) (3 genes; FDR, P = 0.0080), and mTOR (21) (5 genes; FDR, P = 0.020), suggesting that genetic alterations contribute to functional clonal heterogeneity (SI Appendix, Table S4 and Fig. S16). However, we did not observe statistically significant correlations between genetic pathway alterations and clonal chemoresistance.
Clonal heterogeneity is also evident at the level of gene expression (Fig. 4). We performed microarray analysis of 32 GBM clones from all four tumors and two unsorted populations (GBM-472 and GBM-489). Principal component analysis of clonal expression profiles shows extensive heterogeneity within each tumor (Fig. 4A). Most clones from the same tumor were more similar to each other than to clones from a different tumor at the level of transcription, generally confirming our observed pattern of genetic heterogeneity. However, a few clones were more similar to clones from another tumor, in agreement with previous observations from single cell RNA sequencing (9). In particular, the TMZ-sensitive clone 482_9 is clearly distinct from other clones of GBM-482.
Fig. 4.
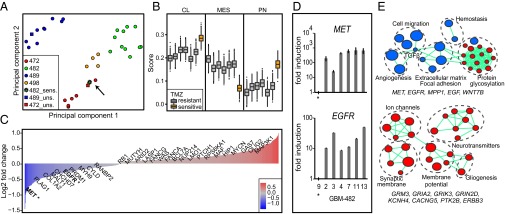
Clonal function is correlated with gene expression and pathway function. (A) Principal component analysis of gene expression profiles of 32 GBM clones and unsorted populations highlights divergence of the TMZ-sensitive clone 482_9. (B) Transcriptional analysis of GBM-482 predicts that the TMZ-sensitive clone (orange, 482_9) belongs to a different GBM subtype than the six TMZ-resistant clones (gray). Three transcriptional subtypes of GBM (classical, mesenchymal, proneural) were analyzed with bootstrapping and permutation tests. (C) Barplot shows differentially expressed genes in the TMZ-sensitive clone 482_9 compared with mean expression in six TMZ-resistant clones of tumor GBM-482 (absolute z-score > 2). Genes are ranked according to fold change. (D) qRT-PCR analysis confirms significantly lower gene expression of MET and EGFR in the TMZ-sensitive clone 482_9 compared with six TMZ-resistant clones of tumor GBM-482. (E) Pathway analysis illustrates differentially expressed pathways in the TMZ-sensitive clone with increased (red) and decreased (blue) gene expression. Selected genes associated with pathways are shown below network diagrams.
Transcriptional profiling of bulk tumors shows that GBM comprises several distinct subtypes, generally agreed to be classical, proneural, and mesenchymal (1, 4, 6, 22). We sought to analyze the relationship of clonal subtypes and TMZ sensitivity. Using the previously defined subtype signatures, we calculated subtype scores for 29 GBM clones from all four tumors and two unsorted populations (GBM-472 and GBM-489) to distinguish between the three subtypes. Consistent with recent RNA-seq based subtyping of single GBM cells (9), these clones show a hybrid cellular state with high scores in more than one subtype (Fig. 4B and SI Appendix, Fig. S17). The TMZ-sensitive clone 482_9 scores lower in mesenchymal and higher in proneural and classical subtypes compared with resistant clones of tumor GBM-482, suggesting that functional heterogeneity of drug response is driven by transcriptional heterogeneity and that a mixture of clones of different subtypes is present in the bulk tumor.
To further explore variable TMZ sensitivity within single patient tumors, we compared gene expression profiles of GBM-482 clones. We ranked 777 genes with the most pronounced differential expression in the TMZ-sensitive clone relative to mean expression in six TMZ-resistant clones (absolute z-score > 2; Fig. 4C and SI Appendix, Table S5). This list includes a significant enrichment of 28 known cancer genes (P = 2.5 × 10−4; OR, 2.2), suggesting that known cancer pathways are involved in differential drug response of clones of GBM-482. For example, the well-defined GBM oncogenes hepatocyte growth factor receptor (MET) and EGFR show reduced expression in the TMZ-sensitive clone of GBM-482. qRT-PCR assays confirm the results of microarray analysis and validate dramatic up-regulation of EGFR and MET in TMZ-resistant clones (>10-fold; Fig. 4D).
Pathway enrichment analysis (19) of differentially expressed genes in the TMZ-sensitive clone highlights multiple significant pathways and processes (FDR, P < 0.05; Fig. 4E and SI Appendix, Table S6). These include ion channels, neurotransmitter signaling, and synaptic membrane genes with increased expression (e.g., KCNH4, GRM3, and CACNG5), whereas genes involved in cell migration (MET) and angiogenesis (WNT7B, EGF) are down-regulated compared with resistant clones. GRM3 encodes a glutamate receptor involved in growth, proliferation, and survival of glioma and melanoma cells (23). Activation of MET enhances GBM cell migration (24) and tumor cell resistance in response to DNA damage (25). WNT7B and EGF have been shown to induce vasculature in the central nervous system (26, 27). Interestingly, the TMZ-sensitive clone shows increased expression of several genes involved in neurotransmitter signaling such as glutamate receptors (GRIA2, GRIK3, and GRIN2D) (28). Neurotransmitters are involved in brain development and proliferation of progenitor cells (29); however, their role in tumor biology is less established. This protein family includes targets of approved drugs and therefore is an interesting candidate for drug repurposing studies. Our data suggest that clonal chemo-response in GBM may be altered by modulation of neurotransmitter signaling, and this deserves future exploration. We demonstrate an example where transcriptional clonal heterogeneity correlates with variable drug response and points to potential therapeutic mechanisms that could render resistant clones as sensitive.
Discussion
The very name of this tumor type, glioblastoma multiforme, has long defined its morphologic heterogeneous nature. There is increasing appreciation that intratumoral genetic heterogeneity is central to GBM biology (9, 14) and therapeutic resistance (10).
A number of recent studies demonstrated intratumoral heterogeneity in GBM by showing regional variation and cell-to-cell differences in expression of receptor tyrosine kinases (12, 13, 15). Also, recent studies of GBM geography demonstrate maps of regional copy number and gene expression differences that importantly shed light on mechanisms of intratumoral clonal evolution in space and time and show that individual tumors can contain cells corresponding to different GBM subgroups (14). These observations have been extended by a single cell genomic analysis of a large number of GBM cells from five patient tumors, which strikingly reveals the genomic complexity of this tumor (9).
To date, however, these features of genetic heterogeneity have not been directly linked to functional readouts such as drug response, growth, or differentiation potential. Unless we consider functional analysis from single cell derived populations, it will be difficult to find the precise driver genes and molecular pathways responsible for aggressive behavior of individual clones in a heterogeneous tumor, particularly if those clones are rare. Direct functional-genomic correlations at a clonal level will be essential to developing new GBM therapies.
In this study, we developed a strategy to analyze prospectively isolated single GBM clones from fresh tumors that links genotypes of individual subclones with tumorigenic, proliferative, differentiation, and, most importantly, drug responsiveness properties. Our proof-of-principle drug screen shows that single tumors contain clones that respond differently to known cancer drugs, thus highlighting the opportunity to identify resistant clones, define prognostic biomarkers, and develop clone-specific combination therapies. Importantly, a clonal analysis of the standard GBM biomarker MGMT did not correlate with TMZ responsiveness, suggesting that new biomarkers of drug responsiveness are sorely needed, consistent with more recent bulk GBM genomic analyses which highlight the subgroup limitations of this marker (1). We predict that further studies of larger groups of patient tumors and derived clones are likely to yield additional clonal vulnerabilities that will have clinical relevance.
Understanding the significance of cancer genetic heterogeneity and the impact on cancer relapse is enormously challenging and will require multiple approaches. The integration of genomics techniques with sophisticated bioinformatic analysis and, most importantly, clonal functional assays, provide a direct starting point, as it will identify tumor subpopulations that drive growth and therapeutic resistance. Future developments of this strategy would consider deep sequencing of bulk tumors and clones combined with computational inference of intratumoral clonal structure (30). In addition, combining single cell approaches (9) with single clone derived functional analysis are likely to give a clearer picture of GBM heterogeneity and the significance of genomic diversity. Although our approach may not capture all relevant clones in the primary patient sample, our study focuses on the critical tumorigenic fraction, as functional assays for the bulk population have not been developed. We predict that clone-specific functional profiling of GBMs will help identify aggressive clones, new cancer driver mechanisms, molecular signatures, and therapeutic vulnerabilities emphasizing the potential of cancer treatment at a clone-specific level. We envisage a similar clonal functional analysis strategy will be applicable to deciphering heterogeneity in other types of cancer. One potential application of this approach will be the development of anticipatory therapy, directed at the most aggressive relapse-initiating clones identified at the time of patient diagnosis.
Materials and Methods
Two naïve and two recurrent tumors originated from four individual patients. Single cell-derived clonal populations were retrieved by FACS live sorting and expanded in stem cell conditions. Intracranial cell transplantation involved injection of 100,000 cells into immuno-compromised (NSG) mice. Immunohistochemistry was performed on paraffin-embedded tissue. Clonal protein expression of EGFRvIII was analyzed with Western blots using EGFRvIII-transfected human fetal brain cells (HF7450NS) as a control. Differentiation assays were carried out in growth factor withdrawal conditions. AlamarBlue assay was performed for cell survival analysis after drug treatment (temozolomide; National Cancer Institute oncology drug library). MGMT promoter methylation was determined by nested two-stage methylation-specific PCR (18), using CpGenome Universal Methylated DNA (Millipore) and patient blood DNA as controls. Genetic profiling of GBM samples was carried out with Affymetrix Human SNP Array 6.0 microarrays, using CRMAv2 (31) for preprocessing, the CBS algorithm (32) for genome segmentation, and custom R scripts for filtering and global analyses. Broad genomic segments were filtered. Matched blood reference was used for three tumors, whereas a median diploid genome of 226 patients (33) combined with filtering of polymorphic copy number variants (34) was used on the unmatched tumor GBM-489. Phylogenetic analysis of copy number variation was conducted with a Markov Chain Monte Carlo procedure using the MrBayes software (35). To enumerate copy number changes of chromosomes 7, 3, and 17, the Urovision probe kit (Abbott Laboratories) was used. Transcriptional profiling was performed with Affymetrix Human Gene 1.0 ST microarrays, using the RMA algorithm for preprocessing and z-scores for differential expression estimation. Pathway enrichment analysis was performed with g:Profiler (19) and visualized as Enrichment Maps (36) with Cytoscape. All experiments were conducted on cell cultures below 20 passages. The identity of all samples was confirmed by genotyping analysis with Genotyping Console (Partek), and by short tantem repeats (STR) analysis. Extended description of methods is available in SI Appendix, SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank P. A. Penttilä, A. Khandani, S. Zhao, T. Velauthapillai, and L. Jamieson for preparative flow cytometry, Dr. A. Guha for kindly providing the EGFRvIII plasmid, and the support of the Toronto Western Hospital Brain Tumor Bank led by Dr. Gelareh Zadeh. Flow cytometry was performed in The SickKids-University Health Network Flow Cytometry Facility. P.B.D. is supported by grants from Ontario Institute for Cancer Research with funds from the Government of Ontario, Canadian Institutes for Health Research (CIHR), Genome Canada, Canadian Cancer Society, the Hospital for Sick Children Foundation, Jessica’s Footprint Foundation, and the Hopeful Minds Foundation. G.D.B. is supported by National Resource for Network Biology (US National Institutes of Health Grant P41 GM103504), and M.W. is supported by a CIHR grant.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Gene expression profiles and copy number data are available at ArrayExpress, www.ebi.ac.uk/arrayexpress/ (accession nos. E-MTAB-2693 and E-MTAB-2694).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320611111/-/DCSupplemental.
References
- 1.Brennan CW, et al. TCGA Research Network The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verhaak RG, et al. Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noushmehr H, et al. Cancer Genome Atlas Research Network Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sturm D, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 7.Shah SP, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson BE, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 12.Little SE, et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012;72(7):1614–1620. doi: 10.1158/0008-5472.CAN-11-4069. [DOI] [PubMed] [Google Scholar]
- 13.Snuderl M, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Sottoriva A, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110(10):4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szerlip NJ, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA. 2012;109(8):3041–3046. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikawa R, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA. 1994;91(16):7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stupp R, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 18.Esteller M, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 19.Reimand J, Arak T, Vilo J. g:Profiler—a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39(web server issue):W307-15. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikushima H, et al. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5(5):504–514. doi: 10.1016/j.stem.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 21.Akhavan D, Cloughesy TF, Mischel PS. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro-oncol. 2010;12(8):882–889. doi: 10.1093/neuonc/noq052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sturm D, et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14(2):92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prickett TD, Samuels Y. Molecular pathways: Dysregulated glutamatergic signaling pathways in cancer. Clin Cancer Res. 2012;18(16):4240–4246. doi: 10.1158/1078-0432.CCR-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70(2):217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci USA. 2011;108(24):9951–9956. doi: 10.1073/pnas.1016912108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldman CK, et al. Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: A model of glioblastoma multiforme pathophysiology. Mol Biol Cell. 1993;4(1):121–133. doi: 10.1091/mbc.4.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stenman JM, et al. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322(5905):1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 28.Chen RS, Deng TC, Garcia T, Sellers ZM, Best PM. Calcium channel gamma subunits: A functionally diverse protein family. Cell Biochem Biophys. 2007;47(2):178–186. doi: 10.1007/s12013-007-0002-0. [DOI] [PubMed] [Google Scholar]
- 29.Diamandis P, et al. Chemical genetics reveals a complex functional ground state of neural stem cells. Nat Chem Biol. 2007;3(5):268–273. doi: 10.1038/nchembio873. [DOI] [PubMed] [Google Scholar]
- 30.Roth A, et al. PyClone: Statistical inference of clonal population structure in cancer. Nat Methods. 2014;11(4):396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bengtsson H, Wirapati P, Speed TP. A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6. Bioinformatics. 2009;25(17):2149–2156. doi: 10.1093/bioinformatics/btp371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5(4):557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 33.Silversides CK, et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012;8(8):e1002843. doi: 10.1371/journal.pgen.1002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 35.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 36.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE. 2010;5(11):e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.