

Significance
Rhinovirus is the most frequent cause of the common cold, as well as one of the most important causes of asthma exacerbations. Most rhinovirus strains replicate better at the cooler temperatures found in the nasal cavity than at lung temperature, but the underlying mechanisms are not known. Using a mouse-adapted virus, we found that airway epithelial cells supporting rhinovirus replication initiate a more robust antiviral defense response through RIG-I–like receptor (RLR)–dependent interferon secretion and enhanced interferon responsiveness at lung temperature vs. nasal cavity temperature. Airway cells with genetic deficiencies in RLR or type I interferon receptor signaling supported much higher levels of viral replication at 37 °C. Thus, cooler temperatures can enable replication of the common cold virus, at least in part, by diminishing antiviral immune responses.
Keywords: rhinovirus, common cold, airway, RIG-I, innate immunity
Abstract
Most isolates of human rhinovirus, the common cold virus, replicate more robustly at the cool temperatures found in the nasal cavity (33–35 °C) than at core body temperature (37 °C). To gain insight into the mechanism of temperature-dependent growth, we compared the transcriptional response of primary mouse airway epithelial cells infected with rhinovirus at 33 °C vs. 37 °C. Mouse airway cells infected with mouse-adapted rhinovirus 1B exhibited a striking enrichment in expression of antiviral defense response genes at 37 °C relative to 33 °C, which correlated with significantly higher expression levels of type I and type III IFN genes and IFN-stimulated genes (ISGs) at 37 °C. Temperature-dependent IFN induction in response to rhinovirus was dependent on the MAVS protein, a key signaling adaptor of the RIG-I–like receptors (RLRs). Stimulation of primary airway cells with the synthetic RLR ligand poly I:C led to greater IFN induction at 37 °C relative to 33 °C at early time points poststimulation and to a sustained increase in the induction of ISGs at 37 °C relative to 33 °C. Recombinant type I IFN also stimulated more robust induction of ISGs at 37 °C than at 33 °C. Genetic deficiency of MAVS or the type I IFN receptor in infected airway cells permitted higher levels of viral replication, particularly at 37 °C, and partially rescued the temperature-dependent growth phenotype. These findings demonstrate that in mouse airway cells, rhinovirus replicates preferentially at nasal cavity temperature due, in part, to a less efficient antiviral defense response of infected cells at cool temperature.
Rhinovirus (RV) is the most frequent cause of the common cold and has recently been recognized as the most frequent cause of exacerbations of asthma, a disease affecting ∼10% of the US population (1, 2). RV is also increasingly recognized to be a major cause of lung symptoms in patients with other chronic respiratory diseases and in young children (3). Previously, RV was thought to cause disease primarily in the nasal cavity, consistent with the observation that most RV strains replicate more robustly at the cooler temperatures found in the nasal cavity (33–35 °C) than at lung temperature (37 °C) (4, 5). However, the recent recognition that RV is an important cause of disease in the lung (2, 3) compels further investigation of the mechanisms that control the optimal replication temperature of this virus, which are unknown.
Previous studies of the replication machinery of RV have not identified a virus-intrinsic reason for temperature-dependent growth, including studies of cell entry, uncoating, and polymerase activity (6, 7). Therefore, we considered the possibility that other factors, such as temperature-dependent host antiviral responses, might contribute to this phenotype. To investigate this possibility, we examined the effect of incubation temperature on the response to RV infection by the infected host cell. Using a mouse primary airway cell infection model, we observed that incubating cells at the lower temperature of the nasal cavity (33 °C) greatly diminishes the antiviral defense response elicited by RV infection in airway epithelial cells, and that host cells genetically deficient in the innate immune signaling molecules that mediate this response support robust RV replication at 37 °C.
Results
Mouse-Adapted RV-1B Exhibits Robust, Temperature-Dependent Growth in Mouse Epithelial Cells.
To study viral infection using genetic knockouts and diverse types of primary cells, we chose to investigate temperature-dependent replication of RV using a mouse model system. To do this, we created a mouse-adapted variant of the minor-group rhinovirus RV 1B (RV-1B). Minor-group rhinoviruses, which use the LDL receptor and related receptors for cellular entry, have been shown to enter mouse cells and undergo limited replication, which can be improved by serial passage in mouse cells (8–10). Consistently, we found that RV-1B replicated to a limited extent in the mouse airway epithelial cell line (LA-4) but that replication efficiency was dramatically improved following serial passage of the virus 27 times in LA-4 cells (Fig. 1 A and B). During serial adaptation to mouse cells, RV-1B acquired mutations corresponding to 14 amino acid changes, as shown in Fig. S1A. Mutations affected 7 out of 11 RV proteins, including regions implicated in species specificity in previous studies within the genes encoding 2BC and 3A (8, 10–12), suggesting that changes in these proteins are important for species adaptation. The mouse-adapted strain, RV-1BM, displayed temperature-dependent growth in LA-4 cells with robust replication at 33 °C but limited replication at 37 °C (Fig. 1B). Both the parent virus, RV-1B, and RV-1BM displayed similar temperature-dependent growth patterns in the permissive human epithelial cell line HeLa (Fig. 1 C and D). In addition, like replication of RV-1B in human epithelial cells (13), replication of RV-1BM in mouse LA-4 epithelial cells and in mouse embryonic fibroblasts was potently inhibited by addition of exogenous interferon β (IFN-β; Fig. S1 B–D).
Fig. 1.

Temperature-dependent replication of rhinovirus and host response. (A–D) Cells were inoculated with a multiplicity of infection (MOI) of 0.001 of the indicated virus, and then incubated at 33 °C (blue line) or at 37 °C (red line). (A) At 33 °C, RV-1B exhibited ∼105-fold amplification in the human cell line, HeLa (upper line) but <50-fold increase in titer in mouse LA-4 cells (lower line). (B) Mouse-adapted RV-1B, RV-1BM, displayed ∼104-fold amplification in LA-4 cells at 33 °C (circles, solid line) compared to the minimal replication of RV-1B (squares, dashed line). At 37 °C, RV-1BM replicated less than 50-fold (solid red line) and RV-1B replication was not observed (not shown). (C and D) Growth curve of RV-1BM and RV-1B in HeLa cells at 33 °C (blue line) or at 37 °C (red line). (E–L) Primary mouse AECs were inoculated with RV-1BM, MOI 20, and incubated at 33 °C or 37 °C following the initial 1-h inoculation at 33 °C. (E) Fold change in viral RNA. (F) RNA-Seq results showing representation of the RNA viral genome at 7 h postinfection in cells incubated at 37 °C vs. 33 °C. The y axis shows the coverage at each position: (number of reads at each position/total number of mapped reads in the sample) × 106; the x axis represents the position in the viral genome. (G and H) DAVID analysis of host mRNAs differentially enriched during RV-1BM infection at 37 °C compared with 33 °C. (G) Transcripts that differed in expression by at least twofold were included in the analysis (364 transcripts). Gene Ontology (GO) database clusters up-regulated at 37 °C (P < 0.01) are shown with fold enrichment (Left) and Bonferroni-corrected P values (Right). (H) For the immune response cluster, genes differentially expressed by greater than twofold are shown, arranged from greatest (left) to least (right) differential expression. (I) Fold change in host cell IFN-β mRNA during the single-step RV-1BM infection shown in E, normalized to Hprt. (J) IFN-β protein detected by ELISA in cell lysates prepared from replicate cultures of the single-step growth curve experiment shown in E. (K) Fold change in Oas1a mRNA during the single-step RV-1BM infection shown in E. (L) Viral titers of cell lysates prepared from replicate cultures of the single-step growth curve experiment shown in E were determined by plaque assay. Points and error bars represent the mean and SEM of two or three replicates per condition. Data are representative of at least three independent experiments.
The Airway Epithelial Cell Antiviral Response to RV Infection Is Temperature-Dependent.
Next, we used RV-1BM to examine host–virus interactions during a single replication cycle in primary airway epithelial cells (AECs), the major target cells of RV infection in the airway. To do this, we isolated primary AECs from mouse tracheas as described previously (14) and compared a single viral growth cycle in AECs incubated ex vivo at 33 °C or 37 °C. Initially, viral replication proceeded rapidly at both temperatures, but the replication rate declined earlier at the nonpermissive temperature (Fig. 1E), similar to kinetics previously observed with RV2 (15). At early time points postinfection, more viral RNA was present at 37 °C than at 33 °C, as assessed by quantitative (q)PCR at 5 h (Fig. 1E) and by the percentage of reads mapping to the viral genome by RNA sequencing (RNA-Seq) at 7 h (3.84% of total reads vs. 2.55% of total reads, at 37 °C vs. 33 °C, respectively; P < 2.2 × 10−16; Fig. 1F and Table S1). No significant differences were observed in the regions of the viral genome represented at 37 °C vs. 33 °C at 7 h postinfection (Fig. 1F and Fig. S2). To better understand the cellular events occurring during the restriction of viral replication at 37 °C, we performed differential RNA-Seq analysis of host cell gene expression at 7 h postinfection by comparing the abundance of host mRNAs present in infected cells incubated at 33 °C vs. 37 °C. Analysis of differentially expressed cellular transcripts revealed a striking enrichment at 37 °C of genes related to the antiviral IFN response, including Gene Ontology clusters for “immune response,” “response to virus,” and “defense response” (P < 10−15; Fig. 1 G and H). These observations suggested that RV induces a more robust IFN-dependent innate immune response at 37 °C than at 33 °C. To further investigate this possibility, we performed qRT-PCR and ELISA to assess the production of IFN during the single-step replication cycle. Consistent with the RNA-Seq results, mouse AECs displayed greatly increased production of IFN-β mRNA and protein and increased induction of the IFN-stimulated gene (ISG) 2′,5′-oligoadenylate synthase (Oas1a) at 37 °C compared with 33 °C during the RV replication cycle (Fig. 1 I–K). Further analysis revealed that increased expression of IFN and ISGs at 37 °C relative to 33 °C also correlated with an earlier plateau in viral titer at 37 °C during single-step replication (Fig. 1L). IFN induction by RV-1BM infection was consistently significantly higher when infected cells were incubated at 37 °C during viral replication, even when cells were preincubated at 33 °C or inoculated at 37 °C instead of 33 °C (Fig. S3). Additional analysis revealed that both type I (IFN-α, IFN-β) and type III (IFN-λ) IFNs were induced at much higher levels at 37 °C than at 33 °C during the RV replication cycle (Fig. S4 A–C). Both of these IFN subtypes have been reported to limit RV replication (16–18). Thus, RV infection of AECs results in more rapid accumulation of viral genome, higher levels of IFN gene and ISG expression, and lower levels of infectious virus production at core body temperature relative to nasal cavity temperature.
The RIG-I–Like Receptor Pathway Mediates the Temperature-Dependent IFN Response to RV Infection.
To gain further insight into the mechanism of enhanced IFN production at 37 °C vs. 33 °C during RV replication, we next sought to identify the innate immune signaling pathway(s) responsible for IFN expression in response to RV infection in AECs. Upon viral infection, host cells can detect pathogen-associated molecular patterns (PAMPs) via several distinct innate sensors. RV, a picornavirus, has a positive-sense single-stranded (ss)RNA genome and generates double-stranded (ds)RNA replication intermediates in infected cells. These viral nucleic acids could serve as PAMPs for endosomal innate immune receptors Toll-like receptor 7 (TLR7) and TLR3 and cytoplasmic RIG-I–like receptors (RLRs) MDA5 and RIG-I. Previous studies have indicated roles for TLR3 and RLRs in the induction of type I and type III IFNs following RV infection (19–21). To investigate pathways used in RV recognition, we measured type I IFN induction following RV infection at 33 °C and 37 °C in primary cells derived from wild-type (WT) or knockout (KO) mice lacking innate immune signaling molecules. In primary mouse AECs, RV triggered a greater than 103-fold up-regulation of IFN-β mRNA in WT, Tlr7−/−, and Tlr3−/− AECs, but this response was greatly diminished in MAVS−/− AECs (Fig. S4D), indicating that RLRs were primarily responsible for the temperature-dependent IFN response to RV replication (Fig. 1). To probe whether temperature-dependent IFN induction was specific to the RLR pathway, we next examined IFN secretion in response to RV in two other primary cell types, dendritic cells (DCs) and plasmacytoid dendritic cells (pDCs). These cell types are important in vivo in the antiviral IFN response and in linking innate to adaptive immunity (22). We observed that the IFN response to RV infection in DCs, as in airway epithelial cells, was MAVS-dependent (Fig. S4E). In contrast, in pDCs, IFN induction did not require MAVS but was dependent on TLR7 in this cell type (Fig. S4F). Notably, as in AECs, IFN secretion was higher at 37 °C compared with 33 °C in DCs, but in pDCs, IFN secretion was similarly robust at 33 °C and 37 °C (Fig. S4 G–I). The RLR pathway is thought to recognize picornaviruses via recognition of dsRNA structures that are generated in the cytosol during viral replication, whereas TLR7 recognizes endocytosed ssRNA genomes, which are present even in the absence of viral replication. Because RLRs recognize viral replication intermediates, these results suggested that replicating viral RNA was the PAMP eliciting temperature-dependent IFN secretion. To further probe this possibility, we compared IFN induction in each cell type in response to infection with RV-1BM, which replicates robustly in mouse cells, with RV-1B, which undergoes limited replication (Fig. 1B). Supporting our hypothesis, we observed that in cell types using the RLR pathway for viral recognition, RV-1BM elicited a more robust IFN response than RV-1B. In contrast, in pDCs, RV-1BM and RV-1B elicited similar levels of IFN secretion (Fig. S4 A–C and G–I). Therefore, these data indicated that the temperature-dependent IFN response elicited by RV infection is a feature of RLR- but not TLR-dependent recognition.
RLR-Mediated Induction of IFN and IFN-Stimulated Genes Is Temperature-Dependent.
Next, we sought to determine the mechanism whereby an incubation temperature of 37 °C results in more robust IFN responses to RV infection than observed at 33 °C. We reasoned that enhanced IFN and ISG expression at 37 °C could result from (i) increased stimulation of RLRs at 37 °C due to more rapid RV genome replication at early time points, (ii) enhanced RLR function at 37 °C, and/or (iii) enhanced IFN-αβR signaling at 37 °C. To probe whether the increase in RLR ligand accumulation accounts for enhanced IFN and ISG induction in response to RV, we used a synthetic RLR ligand, poly I:C (PIC), which stimulates the RLR receptor MDA-5 upon transfection (23, 24). This approach enabled us to ask whether temperature-dependent IFN and ISG induction occurs when the RLR pathway is stimulated with an equivalent level of ligand at each temperature. To do this, we transfected PIC into the cytoplasm of airway epithelial cells and then removed the stimulus and incubated the cells at either 33 °C or 37 °C and measured IFN secretion into the supernatant (Fig. 2A) and induction of mRNA for ISGs (Fig. 2 B and C). We found that at early time points poststimulation (3–5 h), higher levels of IFN-β were found in supernatants from cells incubated at 37 °C than those incubated at 33 °C. However, by 7–9 h poststimulation, IFN-β levels in the supernatants of cells incubated at each temperature were similar; this was also observed at higher and lower PIC concentrations (Fig. S5). Next, we asked whether incubation temperature had an impact on ISG induction following PIC stimulation. We observed that, to varying degrees, PIC stimulation of mouse AECs induced more robust induction of ISGs, including Oas1 and Stat-1, in cells incubated at the warmer temperature (Fig. 2 B and C). RLR expression itself is known to be enhanced by type I IFNs, and therefore positive feedback via IFN-αβR signaling might contribute to the increase in IFN induction observed at core body temperature. To probe whether enhanced IFN induction observed at 37 °C during RV infection requires positive feedback from IFN-αβR signaling, we examined the RLR-dependent IFN response to RV infection in IFN-αβR knockout AECs (Fig. S6). Both IFN-β and IFN-α4 were induced to higher levels at 37 °C compared with 33 °C. These results indicated that RLR signaling is more robust at 37 °C than at 33 °C even in the absence of positive feedback from IFN-αβR signaling. We next examined induction of IFN-β mRNA at early time points following a brief PIC stimulation (15 min at 33 °C) using LA-4 respiratory epithelial cells (Fig. 2D). Induction of IFN-β mRNA was significantly more robust at 37 °C relative to 33 °C at early time points poststimulation, with mRNA levels equalizing by 3 h postexposure to PIC. To directly examine RLR activity at these temperatures, we conducted an enzymatic assay for both RIG-I and MDA5 to assess their ability to catalyze ATP hydrolysis, a hallmark of the duplex RNA-activated ATPase superfamily to which the RLRs belong. Analysis of kcat (the maximal rate constant for ATP hydrolysis at both ATP and RNA saturation) revealed that the ATPase activity of RLRs is significantly higher at 37 °C compared with 33 °C for both RLRs (Fig. 2E). The magnitude of the changes in ATP hydrolysis from 33 °C to 37 °C (∼65% increase for RIG-I and ∼20% increase for MDA-5) are within a range that would be expected to impact receptor function, based on previous studies correlating ATP hydrolysis with RLR-mediated IFN induction (25, 26).
Fig. 2.

RLR signaling and activity in response to poly I:C is enhanced at 37 °C. (A–C) Primary mouse airway epithelial cells were stimulated with Lipofectamine + poly I:C, 0.5 μg/mL, at 33 °C (0–1 h), and then stimulus was replaced with medium and cells were incubated at 33 °C (blue bars) or 37 °C (red bars) until 3, 5, 7, or 9 h poststimulation. At the indicated time points, supernatants were collected for IFN-β ELISA (A) and cells were collected for RNA isolation. RT-qPCR was performed to assess induction of ISGs at each time point (B and C). In addition, mock-treated cells (Lipofectamine only) were incubated at 33 °C or 37 °C for 9 h (9M). Bars represent the mean and SEM of two or three replicates per condition. Asterisks indicate significant a difference (P < 0.05) between 33 °C and 37 °C IFN-β protein or ISG mRNA levels as determined by an unpaired t test. (D) LA-4 respiratory epithelial cells were exposed to Lipofectamine + poly I:C, 0.5 μg/mL, at 33 °C for 15 min, and then the stimulus was removed and replaced with medium. Cells were incubated at 33 °C (blue bars) or 37 °C (red bars) until 1, 2, or 3 h poststimulation, at which time cells were collected for RNA isolation. RT-qPCR was performed to assess induction of IFN-β mRNA at each time point relative to untreated cells. (E) The maximal ATP hydrolysis rate constants for recombinant RIG-I and MDA5 were measured in vitro using saturating RNA and ATP concentrations (50 nM RIG-I or MDA5, 15 ng/μL PIC, and 2 mM ATP). The reactions were performed in triplicate and resulted in the following kcat values and SDs at the indicated temperatures for RIG-I: 1.04 ± 0.12 s−1 (30 °C), 2.78 ± 0.15 s−1 (33 °C), 4.59 ± 0.22 s−1 (37 °C), and 1.08 ± 0.07 s−1 (42 °C); and for MDA5: 5.39 ± 0.14 s−1 (30 °C), 9.55 ± 0.67 s−1 (33 °C), 11.35 ± 0.55 s−1 (37 °C), and 12.14 ± 0.34 s−1 (42 °C). Student t test on ATP hydrolysis rates for MDA5 and RIG-I at 33 °C vs. 37 °C determined P = 0.02 and P = 0.0003, respectively.
IFN-αβR–Dependent Signaling Is Enhanced at 37 °C Compared with 33 °C.
Next, to probe whether IFN responsiveness is enhanced at 37 °C, we measured ISG expression in LA-4 airway epithelial cells in response to recombinant IFN-β (Fig. 3). We observed a higher level of ISG induction in cells incubated at 37 °C relative to 33 °C. Among the ISGs tested, the degree of temperature dependence of ISG induction was most pronounced for the ISGs with the greatest fold change from unstimulated levels, and no significant differences were noted in baseline ISG mRNA levels at the two temperatures, consistent with the idea that the observed differences in ISG mRNA levels result from temperature-dependent differences in IFN receptor signaling rather than other factors that might influence mRNA levels at different incubation temperatures. These results indicate that, in addition to the impact that differences in accumulation of RLR ligands may have on IFN induction (Fig. 1 E and F), RLR signaling and IFN-αβR signaling are enhanced at 37 °C compared with 33 °C (Figs. 2 and 3), and suggest that the combination of these differences contributes to the greater restriction of RV replication at core body temperature compared with nasal cavity temperature.
Fig. 3.

IFN responsiveness is enhanced at 37 °C compared with 33 °C. (A–F) LA-4 respiratory epithelial cells were exposed to medium containing 0, 10, or 50 U/mL recombinant mouse IFN-β and incubated at 33 °C or 37 °C for 6 h, at which time cells were collected for RNA isolation. Expression levels of ISGs were determined with RT-qPCR. Expression levels of each mRNA are plotted as the fold change from the expression level in mock-treated cells incubated at 33 °C for 6 h. The mean and SD for two or three replicates per condition are shown. Asterisks indicate a significant difference (P < 0.05) in expression level at 33 °C vs. 37 °C as determined by an unpaired t test. Panels showing ISG induction are arranged from highest (A) to lowest (F) level of induction.
Host Cells Deficient in RLR or Type I IFN Signaling Support Robust Replication of RV at 37 °C.
Next, we sought to investigate the impact of the temperature-dependent IFN and ISG induction observed during RV replication on limiting viral replication at 37 °C. To do this, we examined RV-1BM replication in AECs from mice deficient in MAVS or the IFN-αβ receptor (Ifnar1−/−) (Fig. 4). RV growth was greatly enhanced at both temperatures in the absence of MAVS or IFN-αβR. Notably, the effects of MAVS or IFN-αβR deficiency on viral titer were significantly more pronounced at 37 °C than at 33 °C, partially rescuing the temperature-dependent growth phenotype. For example, in WT AECs, a 100-fold difference in the final viral titer was observed at 37 °C vs. 33 °C, in contrast to a 5-fold difference in final titer observed in MAVS-deficient host cells and a 9-fold difference in IFN-αβR–deficient host cells (Fig. 4). The observation that a difference in viral titer at 37 °C vs. 33 °C was observed even in the absence of MAVS or IFN-αβR indicates that additional factors contribute to the temperature dependence of viral replication. Nonetheless, replication levels in knockout AECs incubated at 37 °C were equal to or higher than replication levels observed in wild-type cells incubated at the permissive temperature for viral growth (33 °C). Therefore, the MAVS- and type I IFN receptor-dependent innate immune response to RV contributes significantly to restricting viral growth at 37 °C in mouse airway cells and to the temperature dependence of RV replication (see the model in Fig. S7).
Fig. 4.
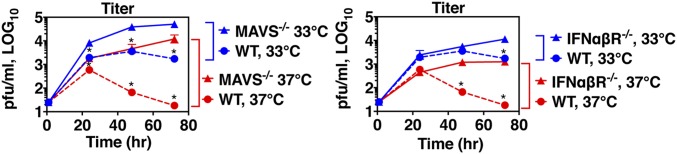
Replication of RV-1BM at 37 °C is partially restored in airway cells deficient in RLR or type I IFN receptor signaling. Primary airway cells derived from WT, Mavs−/−, or Ifnar1−/− mice were inoculated with RV-1BM, MOI 0.01, at 33 °C for 1 h, and then incubated for 24, 48, or 72 h at 33 °C or 37 °C. Cell lysates were prepared at the indicated time points and titers were determined by plaque assay on HeLa cells. Asterisks indicate a significant difference in the titer observed in WT vs. KO airway cells incubated at the same temperature (P < 0.05).
Discussion
When rhinoviruses were first cultured in the 1960s, isolates were observed to replicate more robustly at temperatures slightly below core body temperature (33–35 °C) than at 37 °C (5). This growth pattern fit with the idea that RV caused disease in the nasal cavity (the common cold), which is cooled by inhalation of environmental air, but not in the lungs. In this study, we examined the host response to a well-studied temperature-dependent RV serotype, RV-1B. Using a mouse model system and mouse-adapted RV-1B, we observed that RV replication elicits more robust IFN secretion and ISG induction by primary airway epithelial cells when infected cells are incubated at 37 °C vs. 33 °C. IFN secretion in response to RV is mediated by the RLR pathway, which senses cytoplasmic double-stranded RNA generated during picornavirus replication. Host cells that cannot mount this response or cannot respond via the type I IFN receptor support viral replication to levels equal to or greater than those observed in wild-type cells incubated at the permissive 33-°C temperature. Collectively, our results indicate that incubating airway epithelial cells at the higher temperature during RV replication leads to higher levels of RLR ligand accumulation at early time points, enhanced RLR function, and increased IFN responsiveness, leading to more robust antiviral gene expression and restriction of the virus. Although many temperature-sensitive viruses are limited by mechanisms intrinsic to the viral replication machinery, these findings clearly demonstrate that a temperature-dependent host response to infection contributes greatly to the temperature-dependent replication phenotype of RV. Although our findings do not rule out additional effects of temperature on other aspects of RV biology, they do indicate that effects of temperature on host innate immune defense play a significant role in the temperature dependence of RV replication.
To determine whether there is an intrinsic increase in RLR function at core body temperature that contributes to the temperature-dependent IFN induction triggered by RV infection, we examined the temperature dependence of IFN induction stimulated by the synthetic RLR ligand poly I:C. We found that in response to fixed concentrations of this RLR ligand, airway cells induced higher levels of IFN secretion at 37 °C than at 33 °C. Moreover, we demonstrate that the enzymatic activity of both RIG-I and MDA5 in the presence of ligand is temperature-dependent, in that both receptors demonstrate increased ATPase activity at 37 °C compared with 33 °C.
There are many reasons why activation of RLR signaling by RV infection might result in more sustained temperature dependence of IFN induction than results from PIC stimulation. One contributing factor may be that viral replication produces a continuous increase in RLR ligands during infection, whereas our PIC stimulation experiments provided a single bolus of RLR ligand at the start of the experiment. In addition, the effect of incubation temperature on the rate of accumulation of viral replication intermediates might contribute to greater IFN induction at 37 °C, particularly at early points postinfection (Fig. 1 E and F). Other biological effects of RV infection could also contribute to enhancing the temperature dependence of the IFN response. For example, RV, like other picornaviruses, is known to inhibit host protein synthesis. Initiation of such a blockade during the course of viral replication might prevent IFN secretion at 33 °C from “catching up” to 37-°C levels during RV infection, as it does following PIC stimulation. The exaggeration of the temperature dependence of IFN induction in the setting of RV infection has many possible biological implications for host and virus. For example, developing mechanisms that exaggerate the temperature dependence of the IFN response elicited by infection might have allowed RV to evolve means to undergo robust replication only in the cooler areas of the host such as the nasal cavity, thereby limiting pathogenicity to the host and also facilitating more efficient transmission.
IFN exerts its antiviral effects, in large part, through induction of >300 IFN-stimulated genes with diverse antiviral functions (27). Testing the temperature dependence of ISG induction revealed that many ISGs exhibit a higher level of induction when epithelial cells are stimulated with IFN-β at 37 °C relative to 33 °C. Intriguingly, one of the ISGs exhibiting this pattern was Stat-1, a key component of the signaling cascade downstream of IFN receptors that results in ISG induction. It will be important to investigate whether temperature-dependent STAT-1 induction results in more efficient IFN receptor signaling in general, thereby influencing the temperature dependence of induction of other ISGs. In addition, many of the other molecules involved in both IFN receptor signaling and RLR signaling are themselves ISGs (28). We observe that RV infection leads to increased IFN induction at 37 °C relative to 33 °C even in the absence of type I IFN receptor signaling (Fig. S6); however, we hypothesize that in wild-type hosts, amplification of RLR-dependent innate immune responses via increased type I IFN receptor signaling at 37 °C likely further contributes to restricting the virus at core body temperature.
Based on our findings, it is intriguing to consider possible implications of the relatively less robust IFN and ISG response observed in respiratory epithelial cells at cool temperature. Many respiratory viruses initiate infection in the nasal cavity or infect the nasal cavity to cause “colds” without infecting the lung. In addition to the greater accessibility of the nasal cavity to pathogens in the environment, diminished innate immune induction at cooler temperatures could also contribute to a more permissive environment for respiratory infections in the nasal cavity compared with the warmer airways of the lung. Along these lines, it is also intriguing to consider the possibility that inhaling cool air might diminish resistance to respiratory virus infections by lowering the temperature of potential host cells lining the nasal cavity. Our observations therefore provide a possible mechanism for the popular but controversial idea that exposure to cool weather conditions can increase susceptibility to common colds (29).
Although the relevance of our findings to other respiratory viruses remains to be determined, our results clearly show that temperature-dependent innate immune responses have a great impact on determining the permissive temperature range for rhinovirus infection. In the last decade, it has become more important to re-examine the temperature dependence of RV replication, largely due to better viral detection techniques that have demonstrated that RV is a major causative agent of symptoms in the lung, most notably as the most frequent cause of asthma exacerbations but also as a cause of respiratory symptoms in patients with other chronic respiratory diseases and in young children. Mounting evidence supports the view that RV can replicate in the airways of the lung in some cases, including demonstration of RV in lung biopsies of experimentally infected subjects and in patients with asthma (30–32). Explanations to support the idea that rhinoviruses can replicate in lung airways have included the observation that temperatures in the large airways of the lung may reach temperatures in the permissive range for RV infection and the finding that RV strains vary in the extent to which they exhibit temperature dependence (33–36). An additional possibility is that restriction factors responsible for diminished viral replication at body temperature are not intrinsic to the biology of the virus but also reflect the biology of the host and therefore may vary from host to host. In such a case, both host genetics and host conditions at the time of infection may impact the degree to which RV can productively replicate at core body temperature. Intriguingly, there is some experimental evidence that asthma patients mount a less robust IFN response to rhinovirus infection than control subjects, although this is not always observed (16, 37, 38). Reports on innate immunity to RV in human cells indicate a role for RLR signaling in mediating the IFN response to infection (19, 20). However, how temperature regulates innate immune responses to RV in human cells is unknown. Therefore, in future studies it will be important to examine the contribution of IFN- and ISG-mediated innate immunity to temperature-dependent growth of RV in human cells.
In conclusion, using a mouse model system, we have demonstrated that a temperature-dependent host–virus interaction contributes significantly to the temperature-dependent growth phenotype of rhinovirus, an important respiratory pathogen and the most frequent cause of the common cold. We show that lowering the temperature changes the virus–host interaction, leading to a reduced innate immune response by infected airway cells. We also show that the innate immune competency of host cells is a critical determinant of the temperature range permissive for RV replication. These findings compel further investigation of how host responses to infection affect the temperature range permissive for RV replication and thereby impact the pathogenesis of RV-associated diseases.
Materials and Methods
Growth Curves and Plaque Assay.
H1-HeLa cells and LA-4 cells were plated the day before infection at 80% confluence and infected at a multiplicity of infection (MOI) of 0.001. Cell lysates were prepared by freeze/thaw, and titer was determined by plaque assay on H1-HeLa cells using a procedure modified from Fiala and Kenny (39).
Primary Cell Culture and Infection.
Primary cells were isolated from WT or KO B6 mice as indicated below.
Primary Mouse Airway Epithelial Cells.
Primary mouse AECs were cultured from mouse tracheas based on a previously described protocol (14). Cells were maintained in Millipore airway medium plus supplements (SCML, SCML002-S) in collagen-coated flasks and plated on collagen-coated 12-well plates 2–8 d before infection or stimulation.
DCs.
WT and KO bone marrow (BM) cells were plated at 106 per well in a 24-well plate and cultured in complete RPMI medium containing GM-CSF for 5 d. Cells were inoculated with 100 μL virus in PBS + 0.1% BSA (1 h, 33 °C, with rocking), after which medium was added and cells were incubated at 33 °C and/or 37 °C for 20–24 h.
pDCs.
BM cells were cultured in complete RPMI containing 0.1 μg/mL FLT3L (Gemini Bio-Products; 300-118P) for 7 d and then plated in 48-well plates and infected by addition of virus to the medium.
ELISA.
To assess mouse IFN-β levels in AEC supernatants, ELISA was performed using a PBL kit according to the manufacturer’s instructions. To assess mouse IFN-α levels in dendritic cell supernatants, ELISA was performed using coating antibody (Novus Biologicals; NB100-64387) and detection antibody (PBL InterferonSource; 32100-1).
Quantitative RT-PCR.
To assess viral RNA levels and cellular IFN mRNA levels, cellular RNA was extracted from cultured cells using an RNeasy Kit (Qiagen) and reverse-transcribed using an iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR was performed using SYBR Green (Qiagen; QuantiTect SYBR Green Kit 204145) or iTaq Universal SYBR (Bio-Rad). Primer sequences are shown in SI Materials and Methods.
RNA-Seq.
RNA was isolated from RV-1BM–infected mouse primary airway epithelial cells as above and used to generate libraries that were sequenced on the Illumina HiSeq 2000 using paired-end sequencing at the Yale Center for Genomic Analysis. Raw reads were mapped to the mouse reference genome (mm10) with the TopHat (version 2.0.6) algorithm (40) and mapped to the virus RNA sequence (HRV-1BM) with the bwa (version 0.6.2) algorithm (41). RNA abundance of both was calculated by Cufflinks (version 2.0.2) (42). Gene set enrichment of differentially expressed genes in mouse in the Gene Ontology database was performed by the online tool DAVID (43, 44).
Poly I:C Treatment.
Cells were incubated with poly I:C (Sigma; P9582) complexed with Lipofectamine 2000 (Invitrogen; 1168-019) for 1 h at 33 °C, and then the mixture was removed and complete medium was added.
IFN-β.
Recombinant mouse IFN-β was obtained from PBL InterferonSource (12400-1). For the IFN-β stock used in these experiments, 10 U/mL is equivalent to 1 ng/mL. All procedures used in this study complied with federal guidelines and institutional policies of the Yale Animal Care and Use Committee.
Supplementary Material
Acknowledgments
We thank B. Yordy, M. Tokuyama, and R. Medzhitov for critical reading of the manuscript, G. Shadel and N. Ijima for helpful discussions, and H. Dong, R. Bian, and A. Hoang for technical support. E.F.F. is supported by NIH Awards AI054359S1 and T32 HL007974. J.A.S. is funded by NIH Training Grant 2T32AI007640. This work is supported in part by the NIH/National Institute of Allergy and Infectious Diseases under Award U54AI057160 to the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research, and funding from the NIH (AI054359 and AI064705; to A.I.), the National Science Foundation (DEB-1021243; to P.E.T.), and NIH Award UL1 TR000142 (to L.H. and H.Z.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequence of mouse-adapted rhinovirus 1B reported in this paper has been deposited in the GenBank database (accession no. KC881035), and the sequence of the parent rhinovirus 1B used in this study has also been deposited (accession no. KC881032).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411030112/-/DCSupplemental.
References
- 1.World Health Organization . Global Surveillance, Prevention and Control of Chronic Respiratory Diseases: A Comprehensive Approach. WHO; Geneva: 2007. [Google Scholar]
- 2.Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol. 2010;84(15):7418–7426. doi: 10.1128/JVI.02290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobs SE, Lamson DM, St George K, Walsh TJ. Human rhinoviruses. Clin Microbiol Rev. 2013;26(1):135–162. doi: 10.1128/CMR.00077-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tyrrell DA, Parsons R. Some virus isolations from common colds. III. Cytopathic effects in tissue cultures. Lancet. 1960;1(7118):239–242. doi: 10.1016/s0140-6736(60)90168-9. [DOI] [PubMed] [Google Scholar]
- 5.Turner RB, Couch RB. 2007. in Fields Virology, eds Knipe DM, Howley PM (Lippincott Williams & Wilkins, Philadelphia), Vol 1, pp 895–909. [Google Scholar]
- 6.Stott EJ, Heath GF. Factors affecting the growth of rhinovirus 2 in suspension cultures of L132 cells. J Gen Virol. 1970;6(1):15–24. doi: 10.1099/0022-1317-6-1-15. [DOI] [PubMed] [Google Scholar]
- 7.Yin FH, Knight E., Jr In vivo and in vitro synthesis of human rhinovirus type 2 ribonucleic acid. J Virol. 1972;10(1):93–98. doi: 10.1128/jvi.10.1.93-98.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin FH, Lomax NB. Host range mutants of human rhinovirus in which nonstructural proteins are altered. J Virol. 1983;48(2):410–418. doi: 10.1128/jvi.48.2.410-418.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuthill TJ, et al. Mouse respiratory epithelial cells support efficient replication of human rhinovirus. J Gen Virol. 2003;84(Pt 10):2829–2836. doi: 10.1099/vir.0.19109-0. [DOI] [PubMed] [Google Scholar]
- 10.Rasmussen AL, Racaniello VR. Selection of rhinovirus 1A variants adapted for growth in mouse lung epithelial cells. Virology. 2011;420(2):82–88. doi: 10.1016/j.virol.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris JR, Racaniello VR. Amino acid changes in proteins 2B and 3A mediate rhinovirus type 39 growth in mouse cells. J Virol. 2005;79(9):5363–5373. doi: 10.1128/JVI.79.9.5363-5373.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris JR, Racaniello VR. Changes in rhinovirus protein 2C allow efficient replication in mouse cells. J Virol. 2003;77(8):4773–4780. doi: 10.1128/JVI.77.8.4773-4780.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cakebread JA, et al. Exogenous IFN-beta has antiviral and anti-inflammatory properties in primary bronchial epithelial cells from asthmatic subjects exposed to rhinovirus. J Allergy Clin Immunol. 2011;127(5):1148–1154. doi: 10.1016/j.jaci.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 14.Brockman-Schneider RA, Amineva SP, Bulat MV, Gern JE. Serial culture of murine primary airway epithelial cells and ex vivo replication of human rhinoviruses. J Immunol Methods. 2008;339(2):264–269. doi: 10.1016/j.jim.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Killington RA, Stott EJ, Lee D. The effect of temperature on the synthesis of rhinovirus type 2 RNA. J Gen Virol. 1977;36(3):403–411. doi: 10.1099/0022-1317-36-3-403. [DOI] [PubMed] [Google Scholar]
- 16.Contoli M, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12(9):1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 17.Rotbart HA. Antiviral therapy for enteroviruses and rhinoviruses. Antivir Chem Chemother. 2000;11(4):261–271. doi: 10.1177/095632020001100402. [DOI] [PubMed] [Google Scholar]
- 18.Becker TM, et al. Exogenous interferons reduce rhinovirus replication and alter airway inflammatory responses. Ann Allergy Asthma Immunol. 2013;111(5):397–401. doi: 10.1016/j.anai.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, et al. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J Immunol. 2009;183(11):6989–6997. doi: 10.4049/jimmunol.0901386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slater L, et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010;6(11):e1001178. doi: 10.1371/journal.ppat.1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Q, et al. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 2011;7(5):e1002070. doi: 10.1371/journal.ppat.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25(3):373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 23.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 24.Gitlin L, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103(22):8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohlway A, Luo D, Rawling DC, Ding SC, Pyle AM. Defining the functional determinants for RNA surveillance by RIG-I. EMBO Rep. 2013;14(9):772–779. doi: 10.1038/embor.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rawling DC, Kohlway AS, Luo D, Ding SC, Pyle AM. The RIG-I ATPase core has evolved a functional requirement for allosteric stabilization by the Pincer domain. Nucleic Acids Res. 2015;42(18):11601–11611. doi: 10.1093/nar/gku817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: A complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mourtzoukou EG, Falagas ME. Exposure to cold and respiratory tract infections. Int J Tuberc Lung Dis. 2007;11(9):938–943. [PubMed] [Google Scholar]
- 30.Papadopoulos NG, et al. Rhinoviruses infect the lower airways. J Infect Dis. 2000;181(6):1875–1884. doi: 10.1086/315513. [DOI] [PubMed] [Google Scholar]
- 31.Gern JE, Galagan DM, Jarjour NN, Dick EC, Busse WW. Detection of rhinovirus RNA in lower airway cells during experimentally induced infection. Am J Respir Crit Care Med. 1997;155(3):1159–1161. doi: 10.1164/ajrccm.155.3.9117003. [DOI] [PubMed] [Google Scholar]
- 32.Wos M, et al. The presence of rhinovirus in lower airways of patients with bronchial asthma. Am J Respir Crit Care Med. 2008;177(10):1082–1089. doi: 10.1164/rccm.200607-973OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McFadden ER, Jr, et al. Thermal mapping of the airways in humans. J Appl Physiol (1985) 1985;58(2):564–570. doi: 10.1152/jappl.1985.58.2.564. [DOI] [PubMed] [Google Scholar]
- 34.Papadopoulos NG, Sanderson G, Hunter J, Johnston SL. Rhinoviruses replicate effectively at lower airway temperatures. J Med Virol. 1999;58(1):100–104. doi: 10.1002/(sici)1096-9071(199905)58:1<100::aid-jmv16>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 35.Ashraf S, Brockman-Schneider R, Bochkov YA, Pasic TR, Gern JE. Biological characteristics and propagation of human rhinovirus-C in differentiated sinus epithelial cells. Virology. 2013;436(1):143–149. doi: 10.1016/j.virol.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tapparel C, et al. Growth and characterization of different human rhinovirus C types in three-dimensional human airway epithelia reconstituted in vitro. Virology. 2013;446(1-2):1–8. doi: 10.1016/j.virol.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 37.Wark PA, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201(6):937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sykes A, et al. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax. 2014;69(3):240–246. doi: 10.1136/thoraxjnl-2012-202909. [DOI] [PubMed] [Google Scholar]
- 39.Fiala M, Kenny GE. Enhancement of rhinovirus plaque formation in human heteroploid cell cultures by magnesium and calcium. J Bacteriol. 1966;92(6):1710–1715. doi: 10.1128/jb.92.6.1710-1715.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 44.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.