

Abstract
Pharmacologic blockade of monoamine oxidase A (MAOA) or serotonin transporter (5-HTT) has antidepressant and anxiolytic efficacy in adulthood. Yet, genetically conferred MAOA or 5-HTT hypo-activity is associated with altered aggression and increased anxiety/depression. Here we test the hypothesis that increased monoamine signaling during development causes these paradoxical aggressive and affective phenotypes. We find that pharmacologic MAOA blockade during early postnatal development (P2-P21) but not during peri-adolescence (P22-41) increases anxiety- and depression-like behavior in adult (> P90) mice, mimicking the effect of P2-21 5-HTT inhibition. Moreover, MAOA blockade during peri-adolescence, but not P2-21 or P182-201, increases adult aggressive behavior, and 5-HTT blockade from P22-P41 reduced adult aggression. Blockade of the dopamine transporter, but not the norepinephrine transporter, during P22-41 also increases adult aggressive behavior. Thus, P2-21 is a sensitive period during which 5-HT modulates adult anxiety/depression-like behavior, and P22-41 is a sensitive period during which DA and 5-HT bi-directionally modulate adult aggression. Permanently altered DAergic function as a consequence of increased P22-P41 monoamine signaling might underlie altered aggression. In support of this hypothesis, we find altered aggression correlating positively with locomotor response to amphetamine challenge in adulthood. Proving that altered DA function and aggression are causally linked, we demonstrate that optogenetic activation of VTA DAergic neurons increases aggression. It therefore appears that genetic and pharmacologic factors impacting dopamine and serotonin signaling during sensitive developmental periods can modulate adult monoaminergic function and thereby alter risk for aggressive and emotional dysfunction.
Keywords: Serotonin, Dopamine, Development, Aggression, Anxiety, Depression, Mouse
Introduction
Most neuropsychiatric disorders have developmental origins, and an emerging model postulates that such developmental vulnerability is often restricted to sensitive periods 1–3. The concept of sensitive developmental periods for the indelible modulation of complex behaviors is similar to that described for sensory systems (e.g. visual cortex, ocular dominance plasticity) 4, 5, but affected behaviors, modulating factors, and underlying mechanisms are much less well understood. Furthering our knowledge of sensitive periods that determine the developmental trajectory of complex behaviors is a necessary step towards improving prevention and treatment approaches for neuropsychiatric disorders. Therefore, we here investigate how genetic and environmental risk factors act during sensitive periods of brain development to alter adult behavior and thereby potentially confer vulnerability to neuropsychiatric disorders.
Monoamine oxidase A (MAOA) inactivates bioamines, including serotonin (5-HT), norepinephrine (NE), dopamine (DA), and trace amines through oxidative deamination. Pharmacologic MAOA blockade is an effective treatment for depression and anxiety disorders in adults 6. Unlike the salutary effects of pharmacologic MAOA inhibition, constitutive mutations of maoa function result in a syndrome characterized by antisocial/aggressive behavior in human males 7. Moreover, low expressing maoa variants are also associated with aggression and anxiety traits in rhesus macaques and humans 8–10. Consistent with higher primates, mice with genetic inactivation of maoa exhibit heightened levels of aggression and neophobia 11–13. The divergent effects of genetic (lifelong) mutations versus pharmacologic inhibition (during adulthood) lead us to hypothesize that lower MAOA function during sensitive periods of brain maturation increases the likelihood of aggressive and anxiety traits later in life.
The role of monoamine signaling in brain development has been firmly established. During the embryonic and early postnatal period, monoamines act as trophic factors, modulating neurodevelopmental processes, such as cell division, migration, and differentiation 14–16. In mice, genetic maoa ablation increases brain tissue concentrations of 5-HT, NE, and DA during development and early adulthood 11. The most prominent teratogenic consequence of murine maoa ablation, the disruption of barrel fields in the somatosensory cortex, has been linked to increased 5-HT signaling during perinatal development 17–19. After closure of this sensitive window relating to the establishment of somatosensory cortex organization, increased 5-HT signaling during early postnatal development increases anxiety/depression-like behavior including neophobia in adult mice 20, 21. Thus, we hypothesized first that increased 5-HT signaling during early postnatal development causes neophobia in maoa deficient mice. Secondarily, we predicted that the origins of aggressive behaviors are also developmental and postnatal, because re-expressing maoa from P1 onwards restores normal aggressive behavior in conditional maoa knock-out mice 22. To test both hypotheses, we compared the effects of developmental MAOA inhibition and monoamine transporter blockade on adult behavior and monoamine signaling in mice.
Materials and methods
Subjects
Mice (129SvEv/Tac) were bread at Columbia Psychiatry, New York State Psychiatric Institute. Mice used for experiments were born to litters containing 4-6 pups. Mice were separated by sex and weaned into groups of 5 mice per cage at P22. Mice were treated during postnatal development (P2-21, as previously described 20, 21) or periadolescence (P22-41). One additional control group was treated during adulthood (P182-201). Treatment consisted of the following intraperitoneal (i.p.) drug injections: vehicle (VEH, 0.9% NaCl, 5 ml/kg), fluoxetine (FLX, 10 mg/kg, 5 ml/kg, 5-HTT blocker), clorgyline (CLO, 20 mg/kg, MAOA inhibitor), desipramine (DMI, 20 mg/kg, NET blocker), or GBR12909 (GBR, 20 mg/kg, DAT blocker). The dose for each treatment was chosen based on previous experiments: FLX at 10 mg/kg/day and DMI at 20 mg/kg/day produce serum and brain drug and metabolite levels that are comparable to those with therapeutic efficacy in humans 20, 21; GBR at 20 mg/kg is a standard dose to achieve functional DAT blockade in mice 23; and a CLO dose of 20 mg/kg/day produces partial disruption of MAOA-dependent barrel field formation (to achieve full disruption of barrel field formation, CLO has to be administered every 8h) 19. Treatment assignment was randomized in each cage. Injections were administered daily between 3 pm and 5 pm. Animals were maintained on a 12-hour light-dark cycle (lights on at 8 am) and provided with food and water ad libitum. Animal testing was conducted in accordance with the Principles of Laboratory Animal Care National Institute of Health (NIH) guidelines and the institutional animal committee guidelines.
Optogenetics
In order to selectively express channelrhodopsin2 (ChR2) in dopaminergic cells we crossed a Dopamine-transporter Cre-driver-line (DATIRESCre 24,) with a ROSA26-floxedSTOP ChR2-eYFP line (ai32 line 25). Under isofluorane surgery, optical fiber ferrules (Ceramic ferrules (OD 1.2 mm): Precision fiber products; Optical fiber (OD 0.2mm): Thor labs), were implanted over the VTA (AP:-3.5; ML:0.5; DV:-4.2). After recovery, mice were subjected to the isolation-induced aggression test as described bellow, but pairing DATIRESCre:ai32 mice with single mutant controls. During each encounter only one mouse of the pair was stimulated using blue light pulses (473 nm, 20 Hz, 10 ms pulse duration, 8-10mW) originating from a laser source. Each mouse of the pair was stimulated in consecutive days, counterbalancing the stimulation-order by genotype. After behavioral tests were completed, mice were euthanized, and genotypes and fiber placements were confirmed.
Behavioral testing
For neophobia/anxiety-related behavioral testing, male and female mice were investigated and all animals were exposed to the same series of behavioral paradigms starting at 3 months of age. The tests were administered in the following order: open field, novelty suppressed feeding, and shock escape with a minimum of 7 days between each test. Aggression and amphetamine response was tested in behavioral naïve independent male cohorts starting at 5 months of age for the P2-21 and P22-41 groups and at 10 months of age for the P182-201 group. All behavioral testing took place during the light cycle between 12:00 pm and 7:00 pm. To eliminate odor cues, each apparatus was thoroughly cleaned after each animal.
Open field test
Exploration and reactivity to a novel environment was assessed using the open field test as previously described 26. In brief, square Plexiglas activity chambers equipped with infrared detectors to track animal movement in the horizontal and vertical planes were used as the novel environment. The conflicting innate tendencies to avoid bright light and open spaces but explore novel environments influence locomotor behavior. Mice were placed into the center of the open field and activity was recorded for 30 minutes. Testing took place under bright ambient light conditions. Total distance, total ambulatory time, and vertical activity were measured.
Novelty-suppressed feeding test
The novelty-suppressed feeding test is a behavior conflict paradigm that is sensitive to chronic but not acute antidepressant treatment as well as acute benzodiazepine treatment. The test was performed as described previously 20: the testing apparatus consisted of a plastic box (50 x 50 x 20 cm). The floor was covered with 2 cm of wooden bedding. Twenty-four hours before behavioral testing, animals were deprived of food in the home cage. At the time of testing, two food pellets were placed on a round filter paper (12 cm diameter) positioned in the center of the box. Animals were placed in a corner of the box and the latency to approach the pellet and begin feeding was recorded (maximum time, 10 min). Each mouse was weighed before food deprivation and before testing to determine the percentage of body weight lost.
Shock escape test
Shock escape latency is the primary dependent measure affected by uncontrollable stress and chronic antidepressant treatment. The shock escape test was performed in a 2-chambered Plexiglas shuttle box with the two chambers separated by an automated guillotine door as previously described 26. Each apparatus is equipped with infrared detectors to track animal movement and located within a sound-attenuated chamber. At the beginning of each trial, the door was raised and a mild scrambled foot shock (0.2 mA; 10 s duration) was delivered to the subject through the floor bars. The end of a trial was signaled by the closing of the guillotine door and was triggered either by a transition to the opposite chamber or after 10 s. Transition latencies were recorded. If the subject failed to make a transition during the 10s duration of the foot shock, a maximum latency of 10 s was recorded. A session consisted of 30 trials separated by a 30 s inter-trial interval. Locomotor activity was assessed during the pre-shock habituation phase with the guillotine door open.
Isolation-induced aggression test
We used a rectangular cage that was divided in half by a perforated partition made of clear plastic. A pair of mice with the same treatment was placed in the cage, one in each compartment. The mouse in one compartment is able to see, hear and smell the other mouse through the holes in the plastic divider, but physical interaction is blocked. Mice were housed for at least 10 days before experiment was performed. On the test day, dividers were taken out and mice were allowed to freely interact for 10 min. All behaviors were videotaped. The time spent mounting, tail rattling, and biting was scored.
Amphetamine response
Adult mice were injected with amphetamine (3 – 5 mg/kg, i.p.) or VEH and locomotor activity was assessed in open field apparati (see above).
High Pressure Liquid Chromatography (HPLC)
HPLC was carried out on several brain regions as previously described 27. Concentrations of biogenic amines and metabolites were measured using reversed-phase high-performance liquid chromatography (HPLC) with electrochemical detection. The dissected brain samples in a 1.5-ml microfuge tube were homogenized in 0.5-1.0 ml of 0.4 M perchloric acid with an Ultra Cell Disruptor (Microson, NY). The homogenate was centrifuged for 5 minutes at 14,000 g and 4°C. 50 μl of typically four-fold diluted aliquot of the supernatant was injected over the HPLC system. The HPLC system, equipped with Waters Millennium software, consists of a Waters 515 HPLC pump, a Waters 717 Autosampler, a Varian Microsorb 100-5 C18 reverse-phase column (DYNAMAX 150x4.6 mm) attached with a Guard column (4.6 mm) and an ESA Coulochem electrochemical detector (Model 5100A) with a guard cell (Model 5020) and a dual analytical cell (Model 5011A). The electrochemical detector was set at potential of +0.05 V for the first cell and +0.5 V for the second cell. The mobile phase contained 0.75 mM sodium phosphate (pH 3.1), 1.4 mM 1-Octanesulfonic acid, 10 μM sodium EDTA and 8% acetonitrile. The mobile phase was filtered through a Millipore 0.22 μm filter (Type GV) and degassed in vaccuo. The flow rate was maintained at 0.8 ml/min. A chromatography software package (Waters Millennium) was used for data acquisition and analysis. Values are calculated based on peak area and compared to standard solutions. The inter- and intra-assay coefficients of variation of the assay were each less than 5%. The sensitivity of the assay was less than 0.5 pmol/injection.
Immunohistochemistry
For immunohistochemistry, animals were deeply anesthetized with ketamine/xylazine and perfused with PBS followed by 4 % PFA in PBS. Brains were then removed and post-fixed over-night (4 % PFA in PBS), and subsequently cryoprotected in 30 % sucrose and frozen. Coronal sections (50 μm thick) were cut with a cryostat and fluorescence immunohistochemistry was performed on free-floating sections. Immunohistochemistry was performed using a rabbit primary antibody against GFP (rabbit anti-GFP; 1:1000; Life technologies). As secondary antibody, we used cy3 donkey anti-rabbit (1:350; Jackson Immunoresearch). Antibodies were diluted in blocking solution containing 2 % bovine serum albumin and 0.2 % Triton X-100 dissolved in PBS. Sections were incubated in primary antibody for 24 h at 4° C, washed with PBS, and then incubated with secondary antibody for 2 h at room temperature. After washing, sections were mounted with Vectashield mounting medium (Vector Labs).
Statistic analysis
Statistical analysis was performed using StatView 5.0 software (SAS Institute, Cary, NC). Data were analyzed using Student’s t test, one-way, or two-way ANOVA with Student–Newman–Keuls posthoc testing; survival analysis, Kaplan-Meier, Logrank Mantel-Cox test; or non-parametric Mann-Whitney test as indicated. The criterion for significance for all analyses was p < 0.05. No statistically significant interactions of independent variables and gender were detected and therefore data were collapsed for sex. Results from data analyses are expressed as mean ± SEM.
Results
5-HTT and MAOA blockade during early postnatal development increase adult anxiety/depression-like behavior
Early postnatal 5-HTT blockade increases anxiety/depression-like behavior in adult mice 20, 21. Because 5-HTT and MAOA blockade both increase 5-HT signaling, we hypothesized that early postnatal MAOA blockade would also increase adult anxiety/depression-like behavior. To test this hypothesis, we administered the 5-HTT blocker fluoxetine (FLX, 10 mg/kg/day, i.p.), the MAOA inhibitor clorgyline (CLO, 20 mg/kg/day, i.p.), or vehicle (VEH, 0.9% NaCl, i.p.) to mice in the early postnatal period (PN), from postnatal day 2 to 21 (P2-P21) and assessed emotional behavior in adulthood (P90). To investigate developmental specificity, we also tested mice that received FLX, CLO, and VEH in the peri-adolescent period (ADO), from postnatal day 22 to 41 (P22-P41). To control for the effects of the injection procedure, we included naïve littermates in our behavioral tests.
Using the novel open field, we assessed neophobic behaviors by measuring exploratory activity. We observed that FLX and CLO treatment from P2-P21 decreased total ambulatory time (treatment effect: F(2,127) = 6.225, p = 0.0026) and total time spent rearing (treatment effect: F(2,127) = 5.42, p = 0.0055; developmental timing effect: F(1,127) = 13.56, p = 0.0003) when compared to VEH-treated mice (Fig. 1a and b). Neither FLX nor CLO treatment from P22-P41 altered exploratory behavior compared with VEH treated mice. Behaviors in VEH-treated mice and naïve littermates did not differ in either measure.
Fig. 1. Increased adult anxiety/depression-like behavior after early postnatal 5-HTT or MAOA blockade.

Anxiety/depression-like behavior was assessed in mice using the open field test (a, b), the novelty suppressed feeding test (c, d), and the shock escape test (e, f). FLX or CLO treatment from P2-P21 reduced the time ambulating (a) and the time rearing (b) when compared to control mice treated with VEH from P2-P21. FLX or CLO treatment from P2-P21 increased the latency to feed when compared to control mice treated with VEH from P2-P21 (c). No effect of treatment, period, or treatment x period interaction was detected for the weight loss after 24 hours of food deprivation (d). FLX or CLO treatment from P2-P21 increased the latency to escape when compared to control mice treated with VEH from P2-P21 (e). No effect of treatment, period, or treatment x period interaction was detected for activity in the shuttle box before the onset of shock (f). (n = 17–32 mice per group). (*p < 0.05; **p < 0.01, ***p < 0.001).
To further probe neophobia phenotypes, we used the novelty suppressed feeding paradigm, which assesses approach-avoidance behavior when a food-deprived animal is presented with a familiar food pellet placed in the center of a brightly-lit novel arena. Mice exposed to FLX or CLO from P2-P21 exhibited longer latencies to approach and feed when compared to PN-VEH controls (treatment effect: p = 0.0001, survival analysis, Kaplan-Meier, Logrank Mantel-Cox) (Fig. 1c). However, neither FLX treatment nor CLO treatment from P22-P41 produced such behavioral effect. No effect for treatment, developmental timing or treatment x developmental timing was detected for weight loss during food deprivation (Fig. 1d). Behavior between VEH-treated mice and naïve littermates did not differ in either measure.
To assess behavioral response to stress, we examined escape latency in the shock escape paradigm. Consistent with our findings in tests of neophobia, mice exposed to FLX or CLO from P2-P21 but not P22-P41 exhibited significantly increased escape latencies when compared to PN-VEH controls (treatment effect: F(2,133) = 21.106, p < 0.0001; developmental timing effect: F(1,133) = 46.749, p < 0.0001; treatment x developmental timing interaction: F(2,133) = 24.613, p < 0.0001) (Fig. 1e). No effect of treatment, developmental timing or treatment x developmental timing was detected for pre-shock activity in the dark chambers with the door open (Fig. 1f), indicating that the effect on escape latencies was not caused by reduced overall activity. Behavior between VEH-treated mice and naïve littermates did not differ in either measure.
Peri-adolescent MAOA blockade increases and 5-HTT blockade reduces adult aggressive behavior
Next we investigated aggressive behaviors in adult mice that had received FLX, CLO, or VEH during early postnatal or peri-adolescent development using the isolation-induced aggression paradigm. We measured the time mouse dyads spent engaged in aggressive behaviors consisting of mounting, tail rattling, or biting.
We found that CLO treatment from P22-P41 but not from P2-P21 increased aggressive behavior, when compared to VEH controls (effect of treatment: F(2,69) = 7.258, p = 0.0014; effect of developmental timing: F(1,69) = 4.737, p = 0.033; treatment x developmental timing interaction: F(2,69) = 3.828, p = 0.0265) (Fig. 2a). FLX treatment reduced aggressive behavior when compared to VEH treatment, with no indication of developmental timing specificity. Aggressive behavior between VEH-treated mice and naïve littermates did not differ. Next we investigated period specificity for the effect of transient CLO-treatment on adult aggression, using mice that had received VEH or CLO treatment during adulthood (P182-P201) and were tested 4 months later (P300). We detected no effect of treatment on aggression (Fig. 2b).
Fig. 2. Altered aggression after developmental 5-HTT or MAOA blockade.

Isolation induced aggressive behavior was assessed in mice by scoring the time spent mounting, tail rattling, or biting during a 10-minute encounter. (a) Aggressive behavior was increased in mice treated with CLO from P22-P41 when compared to control mice treated with VEH from P22-P41. Mice treated with FLX displayed reduced aggression when compared to VEH-treated control mice. Of note, FLX treated mice did not display any tail rattling or biting behavior. (n = 7–16 pairs per group). (b) Aggressive behavior was unaffected by transient CLO-treatment from P182-P201 (n = 7 pairs per group). (@ indicates main effect of treatment: p < 0.05; **p < 0.01).
Peri-adolescent MAOA blockade reduces the metabolism of 5-HT, NE and DA
The differential consequences of P22-P41 5-HTT and MAOA blockade on adult aggressive behavior suggest that non-5-HT effects of peri-adolescent MAOA blockade increase adult aggression. In peri-adolescence, maoa is expressed in 5-HTergic, DAergic and NEergic neurons 28. To determine the consequences of P22-P41 CLO treatment on brain monoamine signaling, we assessed tissue monoamine and metabolite levels at P23 (24h after treatment initiation) and at P42 (24 h after treatment cessation). Specifically, we quantified 5-HT and its main metabolite 5-hydroxyindoleacetic acid (5-HIAA), DA and its two main metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), and NE. These measures serve as a proxy for enzyme activity.
CLO treatment significantly increased 5-HT and reduced 5-HIAA levels in the brainstem at P23 (F(1,12) = 51.972, p < 0.0001 and F(1,12) = 31.92, p = 0.0001, respectively) and P42 (F(1,11) = 62.475, p < 0.0001 and F(1,11) = 18.8, p = 0.0012, respectively), when compared to VEH treated controls (Fig. 3a–d). Norepinephrine (NE) levels in the brainstem were also increased by CLO treatment at P23 and P42 (F(1,12) = 68.717, p < 0.0001 and F(1,11) = 40.45, p < 0.0001, respectively) (Fig. 3e,f). Striatal DA levels were not affected at P23, but significantly increased at P42 after chronic CLO treatment when compared to VEH treated controls (F(1,12) = 0.033, p = 0.8587 and F(1,11) = 61.745, p < 0.0001, respectively) (Fig. 3g,h). Striatal DOPAC levels were lower at P23 and P42 (F(1,12) = 30.292, p = 0.0001 and F(1,11) = 13.444, p = 0.0037, respectively), while HVA levels were unchanged (F(1,12) = 0.003, p = 0.9586 and F(1,11) = 2.41, p = 0.1489, respectively) (Fig. 3i–l). These data demonstrate that MAOA blockade using CLO treatment during peri-adolescence inhibits 5-HT, NE, and DA metabolism and raises levels of 5-HT, DA and NE.
Fig. 3. Altered monoamine and –metabolite levels during and after peri-adolescent MAOA blockade.

Mice were injected daily with CLO from P22-P41 and tissue monoamine and monoamine-metabolite levels were assessed on P23 (a, c, e, g, i, k) and P42 (b, d, f, h, j, l) by high performance liquid chromatography. 5-HT (a, b), 5-HIAA (c, d), and NE (e, f) were measured in the brainstem (a–f). DA (g, h), DOPAC (i, j), and HVA (k, l) levels were measured in the striatum (g–l). Monoamine levels were increased and monoamine-metabolite levels were decreased during CLO treatment when compared to VEH treatment as indicated. (n = 6–8 mice per group;). (**p < 0.01; ***p < 0.001).
Peri-adolescent DAT but not NET blockade increases adult aggressive behavior
Next we sought to identify the monoamine component in P22-P41 MAOA blockade, which leads to increased adult aggressive behavior. Because peri-adolescent MAOA blockade robustly increases central 5-HT, NE and DA levels, we examined whether increased peri-adolescent NE or DA signaling could increase adult aggressive behavior. To selectively and transiently increase NE or DA signaling we administered drugs blocking the NE transporter (NET) or the DA transporter (DAT). We used desipramine (DMI, 20 mg/kg/day) to block the NET and GBR12909 (GBR, 20 mg/kg/day) to block the DAT. DMI treatment from P22-P41, either alone or in combination with FLX (10 mg/kg/day) treatment, did not result in increased aggressive behavior in adulthood (Fig. 4a). GBR treatment from P22-P41 however lead to increased time spent in active aggressive behaviors (Mann Whitney, p = 0.0104) (Fig. 4b).
Fig. 4. Increased adult aggressive behavior after peri-adolescent DAT blockade.
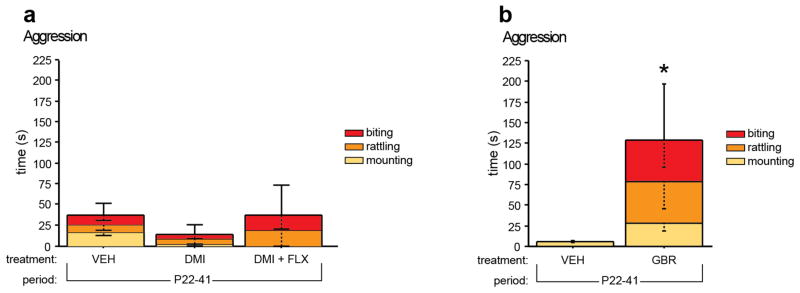
Isolation induced aggressive behavior was assessed in mice by scoring the time spent mounting, tail rattling, or biting during a 10 minute encounter (a, b). Aggressive behavior was not altered in mice treated with DMI or DMI+FLX from P22-P41 when compared to VEH treated control mice (a). Aggressive behavior was increased in mice treated with GBR from P22-P41 when compared to VEH treated control mice (b). (n = 5-15 per group) (*p < 0.05).
Peri-adolescent MAOA but not 5-HTT blockade reduces adult brainstem 5-HIAA levels and increases striatal DA and DOPAC levels
To start investigating the underlying neural substrate which gives rise to altered adult aggressive behavior after peri-adolescent MAOA, 5-HTT, and DAT blockade, we next examined if these developmental interventions produce long-lasting changes in brain monoamine or metabolite levels. P22-P41 CLO-treatment significantly reduced 5-HIAA levels in the brain stem at P180 when compared to either VEH- or FLX-treatment (F(2,15) = 8.505, p = 0.0034) (Table 1a). P22-P41 CLO treatment also significantly increased striatal DA (F(2,15) = 4.085, p = 0.0384) and DOPAC levels (F(2,15) = 5.847, p < 0.0133) at P180 when compared to VEH-treatment and VEH- or FLX-treatment, respectively (Table 1a). However, peri-adolescent FLX or GBR treatment did not produce significant changes in monoamines or their metabolites in the brainstem or striatum at P180, when compared to VEH treated controls (Table 1a,b). Taken together, this dissociation indicates that while altered tissue monoamine levels might be sufficient to alter adult aggressive behavior, they are not necessary.
Table 1. Adult Monoamine and –metabolite levels after peri-adolescent MAOA, 5-HTT and DAT blockade.
Tissue 5-HT, 5-HIAA, NE, DA, DOPAC, and HVA levels were measured by high performance liquid chromatography. (a) Consequences of ADO-CLO and ADO-FLX on adult monoamine and –metabolite levels. (b) Consequences of ADO-GBR on adult monoamine and –metabolite levels. (n = 5-7 mice per group; colored fields indicate differences compared to VEH with p at least < 0.05; red indicates a decrease and green indicates an increase when compared to VEH treatment).
| a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p180 | ||||||||||||
| VEH (N=5) | CLO (N=7) | FLX (N=6) | treatment | VEH vs. CLO | VEH vs. FLX | |||||||
| mean | SE | mean | SE | mean | SE | p % | change | p % | change | p | ||
| Brainstem | 5-HT (pmol/g) | 8699 | 283 | 8250 | 355 | 9547 | 323 | 0.037 | −5.2 | 0.362 | 9.7 | 0.107 |
| 5-HIAA (pmol/g) | 9471 | 191 | 7553 | 414 | 8555 | 238 | 0.003 | −20.3 | 0.001 | −9.7 | 0.078 | |
| NE (pmol/g) | 6754 | 1122 | 5355 | 868 | 6451 | 1122 | 0.492 | |||||
| Striatum | DA (pmol/g) | 16931 | 605 | 19217 | 460 | 17903 | 651 | 0.038 | 13.5 | 0.013 | 5.7 | 0.266 |
| DOPAC (pmol/g) | 2493 | 190 | 3206 | 229 | 2299 | 172 | 0.013 | 28.6 | 0.029 | −7.8 | 0.534 | |
| HVA (pmol/g) | 2957 | 174 | 3605 | 321 | 3026 | 199 | 0.174 | |||||
| b | ||||||
|---|---|---|---|---|---|---|
| P180 | ||||||
| VEH (N=7) | GBR (N=6) | treatment | ||||
| mean | SE | mean | SE | p | ||
| Brainstem | 5-HT (pmol/g) | 9476 | 994 | 9150 | 1327 | 0.845 |
| 5-HIAA (pmol/g) | 7223 | 286 | 9124 | 1075 | 0.094 | |
| NE (pmol/g) | 3753 | 511 | 3671 | 508 | 0.913 | |
| Striatum | DA (pmol/g) | 15626 | 373 | 15576 | 373 | 0.946 |
| DOPAC (pmol/g) | 4051 | 327 | 4513 | 391 | 0.380 | |
| HVA (pmol/g) | 1555 | 31 | 1614 | 103 | 0.569 | |
Peri-adolescent MAOA or DAT blockade increases while 5-HTT blockade reduces the behavioral response to amphetamine in adulthood
Dopaminergic hyper-activity has been implicated in aggression 29. We find a positive association between aggression and striatal DA and DOPAC levels after peri-adolescent MAOA, but not 5-HTT or DAT blockade. Here we further investigate the hypothesis that dysfunction of the DAergic system might underlie altered aggression, using behavioral response to amphetamine challenge (as opposed to total monoamine tissue levels) as a proxy for DAergic function.
First, we analyzed the behavioral response to amphetamine (3 mg/kg, i.p.) and VEH challenge in adult mice (P180), which had received VEH or CLO during P22-P41. Adult VEH injection did not alter behavior in P22-P41 VEH or CLO-treated mice (Fig. 5a); adult amphetamine injection increased ambulatory activity in P22-P41 VEH and CLO-treated mice (effect of time in the amphetamine treated group: F(16,416) = 18.812, p < 0.0001, Fig. 5a); In comparison with P22-P41 VEH treated mice, P22-P41 CLO treated mice showed a significantly increased response to amphetamine (effect of peri-adolescent treatment: F(1,26) = 6.576, p = 0.0165; effect of peri-adolescent x adult treatment interaction: F(16,416) = 3.810, p < 0.0001; Fig. 5a). Analysis of only post-challenge activity confirmed these results: We detected an effect of peri-adolescent treatment, an effect of challenge, and an interaction between both (F(1,26) = 6.768, p = 0.0165, F(1,26) = 45.932, p < 0.0001, and F(1,26) = 5.549, p = 0.0263, respectively; Fig. 5b). To test for period specificity, we next investigated the consequences of transient CLO-treatment during adulthood on behavioral response to amphetamine later in life. Specifically, mice, which had received VEH or CLO during P182-P201, were challenged with amphetamine (3 mg/kg, i.p.) or VEH 4 months later. We detected an effect of amphetamine but no effect of peri-adolescent treatment or peri-adolescent x adult treatment interaction (F(1,44) = 34.464, p < 0.0001; Fig. 5c). Next, we investigated the behavioral response to amphetamine challenge in adult mice, which had received VEH or FLX during P22-P41. We detected an effect of peri-adolescent treatment, an effect of amphetamine and a peri-adolescent x adult treatment interaction (F(1,18) = 4.719, p = 0.0434, F(1,18) = 4.480, p = 0.0485, and F(1,18) = 4.484, p = 0.0484, respectively; Fig. 5d). To our surprise, P22-P41 FLX treated mice displayed no behavioral response to amphetamine, while P22-P41 VEH treated littermates did respond as expected (Fig. 5d). We hypothesized that peri-adolescent FLX treatment shifts the amphetamine dose-response curve as opposed to making mice generally insensitive to amphetamine. Hence we tested the effect of amphetamine challenge at 5 mg/kg in adult mice, which had received VEH, CLO or FLX during P22-P41. Analyzing behavior as a function of amphetamine-dose, we detected an effect of peri-adolescent treatment and an effect of amphetamine-dose (F(2,30) = 6.364, p = 0.005, F(2,60) = 58.639, p < 0.0001, respectively; Fig. S1). Posthoc analysis revealed that FLX treated mice do behaviorally respond to amphetamine (F(2,8) = 6.259, p = 0.0231), but that their response is diminished (Fig. S1). Finally, we tested if peri-adolescent GBR-treatment impacts adult amphetamine response. Strengthening the positive correlation between aggressive behavior and a sensitized/hyper-active DA system, we found that P22-P41 GBR treated mice display an exaggerated response to adult amphetamine challenge when compared to adult VEH injection or P22-P41 VEH treated mice (effect of peri-adolescent treatment: F(1,21) = 9.273, p = 0.0062; effect of amphetamine challenge: F(1,21) = 40.730, p < 0.0001; peri-adolescent treatment x adult amphetamine challenge interaction: F(1,21) = 6.847, p = 0.0161, Fig. 5e). These data demonstrate that P22-P41 CLO, GBR, and FLX treatment differentially impact the sensitivity of the adult DA system. Furthermore, the behavioral sensitivity to amphetamine correlates positively with levels of adult aggression.
Fig. 5. Altered behavioral response to amphetamine in adulthood after peri-adolescent MAOA, 5-HTT, or DAT blockade.

Behavioral amphetamine response was assessed using the open field. (a) Amphetamine injection (AMP, 3 mg/kg, i.p.) induced locomotor hyperactivity in ADO-VEH and ADO-CLO treated mice. Peri-adolescent CLO-treatment altered the response to AMP (indicated significance refers to peri-adolescent treatment x time interaction). (b) Post-injection activity demonstrates increased behavioral response to AMP in ADO-CLO when compared to ADO-VEH. (c) Transient adult CLO treatment did not impact behavioral response to AMP challenge. (d) ADO-FLX treatment abolished the behavioral response to AMP challenge. (e) ADO-GBR treatment increased the behavioral response to AMP challenge. (n = 5-15 per group) (*p < 0.05, ***p < 0.001).
Optogenetic stimulation of VTA DAergic neurons increases aggressive behavior
Peri-adolescent monoamine levels impact adult amphetamine-response and aggressive behavior, suggesting that altered DAergic sensitivity is determining aggression levels. Alternatively, DAergic activity may be correlated with but not causally related to aggressive phenotypes. To investigate these alternative hypotheses we studied the direct casual relationship between DAergic neuronal activity and aggressive behavior using optogenetics.
To selectively target the dopaminergic system we crossed mice expressing Cre-recombinase under the control of the dopamine transporter promoter (DATIRESCre 24) with a ROSA26-reporter line conditionally expressing ChR2-eYFP in a Cre-dependent fashion (ai32 line 25) (Fig. 6a). Optical fibers were implanted over the VTA region in double mutant mice (DATIRESCre:ai32) and in single mutant controls (Fig. 6b, c). In order to assess aggressive behavior we used the isolation-induced aggression paradigm, pairing DATIRESCre:ai32 mice with single mutant controls. Blue light pulses (473 nm, 20 Hz, 10 ms pulse duration) were delivered to the VTA for the duration of the test. Only one mouse of the pair was stimulated during each 10 min encounter. Under these conditions, we detected an effect of genotype on aggressive behavior (F(1,27) = 5.146, p = 0.0315), with stimulation of DatIRESCre:ai32 mice leading to significantly longer time engaging in aggressive contacts, when compared to stimulation of single mutant controls (Fig. 6d).
Fig. 6. Optogenetic stimulation of DAergic VTA neurons increases aggression.
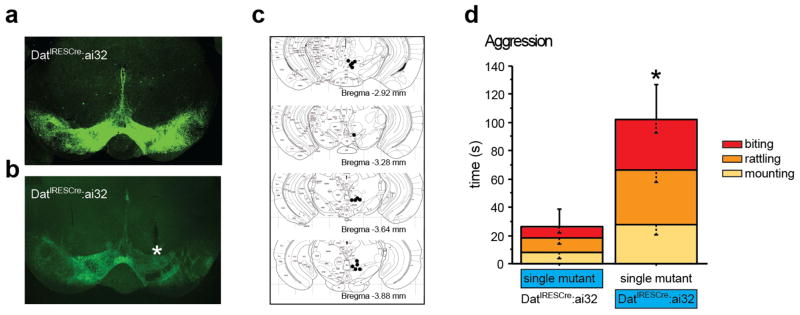
DATIRESCre:ai32 double mutant mice expressing ChR2-eYFP in VTA neurons as visualized by immunohistochemistry against eYFP (a) and autofluorescence (b). A fiberoptic cable track demarks the position of an implant with its tip just dorsal of the VTA (indicated by a star, b). Tip locations were assessed histologically after behavioral experiments were concluded (c). DATIRESCre:ai32 double transgenic mice and single mutant controls were co-housed in mixed pairs and isolation induced aggressive behavior was assessed by scoring the time spent mounting, tail rattling, or biting during a 10 minute encounter (d). Only one mouse in a pair was stimulated (blue). Aggressive behavior was increased in pairs when DATIRESCre:ai32 double mutant mice were stimulated when compared to pairs where the single mutant controls were stimulated (d). (n = 11-18 per group) (*p < 0.05).
These data demonstrate that VTA DAergic neuronal activity is causally related to aggressive behaviors.
Discussion
Our experiments demonstrate the existence of two developmental periods during which early-life perturbation of monoamine signaling alters adult behavior: an early postnatal (P2-P21) 5-HT-sensitive period that affects anxiety and depression-related behaviors and a later peri-adolescent (P22-P41) 5-HT and DA-sensitive period altering aggression. Serotonergic and dopaminergic signaling perturbations during peri-adolescence also exert profound and opposite effects on the behavioral response to psychostimulant challenge in adulthood, suggesting that altered DA-function underlies the changes in aggressive behaviors. In support of this hypothesis we demonstrate that direct optogenetic stimulation of VTA DAergic neurons increases aggressive behavior.
Our findings can explain the paradoxical high neophobia/anxiety phenotypes of 5-htt−/− mice 26, 30, maoa−/− mice 11–13, and pre-synaptic htr1a deficient mice 31, because these mouse lines lack their respective gene products throughout life, including the sensitive P2-P21 period. Likewise, the model can explain the increased aggression seen in constitutive maoa, dat, and comt loss-of-function mouse lines 11, 12, 32, 33. The moderating effect of developmental 5-HTT blockade on adult aggressive behavior does not follow the strict timing specificity (5-HTT blockade during P2-P21 or P22-P41 both decreased adult aggression), but nonetheless supports a developmental mechanism for the low-aggression phenotype of 5-htt−/− mice 34. Because we performed developmental manipulations in wild-type mice but not in the respective mutant mouse lines, we can only compare phenotypes qualitatively but not quantitatively. Hence we cannot draw conclusions as to whether the phenotypes we have observed are fully or only partially recapitulating the magnitude of behavioral consequences elicited by the life-long genetic mutations. Furthermore, we cannot exclude the involvement of off-target effects or downstream developmental effects in contributing to the phenotypes examined here. For example, even though clorgyline preferentially inhibits MAOA, it might also inhibit MAOB in our experiments. To overcome this drawback, we base our conclusions not on single drug effects but on the convergence of multiple pharmacological interventions that resonate with a large literature on genetic effects in both mice and humans. For example, our conclusion that increased DAergic activity during P22-41 increases adult aggression and the behavioral response to amphetamine, is supported by our finding that we can mimic the effect of CLO through DAT but not 5-HTT or NET blockade.
Our findings comport with human vulnerabilities to anxiety, depression and aggression conferred by functional genetic polymorphisms. For example, endophenotypes of affective disorders have been associated with low expressing alleles for the 5-htt 35–39, maoa 9, 40 and presynaptic htr1a 41. Likewise, aggressive behavior has been associated with loss-of-function and low-expressing maoa alleles 7, 8, 42, 43, the 10R variant of dat1 44, 45, and the low activity met allele of the comt 46. Our model predicts that these risk alleles act primarily during sensitive developmental periods to alter brain maturation and circuit formation leading to altered behaviors. The temporal dynamics of transcription in the human brain and their genetic moderation support this hypothesis 47. An important aspect to consider when comparing our interventions or gene ablation studies in mice to genetic risk factors in humans is that human polymorphisms rarely lead to complete loss of function whereas the murine interventions often do. Thus, it is possible that in humans multiple risk factors that impact monoamine signaling during adolescence must converge to elicit a phenotype.
Based on our data, pharmacologic interventions during sensitive periods of human development would be predicted to impact affective and aggressive behavior. The murine P2-P21 period roughly corresponds to the third trimester of human gestation and early childhood, while P22-P41 corresponds to peri-adolescent development 48. Thus, SSRIs taken by pregnant mothers might impact fetal brain maturation and adult mood and anxiety, while stimulant exposure during peri-adolescence could alter adult aggressive behavior. Indeed, chronic stimulant exposure increases aggressive behavior in rodents, non-human primates and humans 49–51, even in abstinent stimulant users 52. The long-term effects of gestational SSRI exposure on affect, aggression, and cognition are unknown 15, 53, but prenatal SSRI exposure has been associated with a 2-fold increased risk of autism spectrum disorder 54.
Our findings support the fundamental notion that neural circuitry and consequently behavior are most vulnerable to long lasting modulation during periods of maturation and high plasticity 4, 55. The 5-HT sensitive period described here coincides with the emergence of anxiety- and fear-related behaviors in rodents and the maturation of the underlying circuitry. For example, at around P10 the consequence of odor shock conditioning switches from preference to aversion, with amygdala engagement and hypothalamic-pituitary-adrenal axis integration 56, 57. Limbic structures including amygdala, prefrontal cortex and hippocampus are maturing during postnatal development and human s allele carriers as well as 5-htt−/− mice exhibit disrupted mPFC-amygdala functional connectivity that likely arises during early circuit maturation 58, 59. The DA-sensitive, peri-adolescent period coincides with the developmental onset of play fighting in rodents 60, 61 and non-human primates 62, and human adolescence is characterized by dynamic changes in aggressive behavior 63.
Insight into how developmental monoamine signaling establishes long-lasting alterations in brain function and behavior remains scarce. The best-studied example is the establishment of the cyto- and circuit-architecture in the somatosensory cortex of mice. MAOA and 5-HTT deficient mice lack topographically defined barrel fields 17–19, 64. This deficit is caused by increased 5-HT signaling through 5-HT1B receptors located on thalamocortical axon terminals, which may inhibit the glutamate release 17 necessary for the formation of barrel columns 65. The sensitive period for this developmental process starts at P0 and ends at P4 19, and therefore partially dissociates from the 5-HT and fully dissociates from the DA-sensitive period under investigation here. Thus, thalamocortical wiring abnormalities are unlikely to contribute to the emotional and aggressive phenotypes. Focusing on the peri-adolescent period we identified changes in monoamine levels and more importantly, changes in behavioral reactivity to amphetamine challenge that correlate with aggressive behavior. Specifically, we report that increased adult aggressive behavior elicited by peri-adolescent MAOA or DAT blockade correlates with increased locomotor-stimulating effects of amphetamine challenge in adulthood. Conversely, reduced adult aggressive behavior elicited by peri-adolescent 5-HTT blockade correlates with decreased locomotor-stimulating effects of amphetamine challenge in adulthood. Supporting phylogenetic conservation, human individuals with antisocial traits also show mesolimbic dopamine hypersensitivity to amphetamine, as impulsivity is positively correlated with the magnitude of amphetamine-induced DA release in the striatum 66. Highlighting the notion that altered DAergic signaling impacts adult behavior as a function of developmental timing, transient pre-adolescent methylphenidate exposure decreases responsiveness to cocaine’s locomotor-activating effects in adult rats 67. In contrast, transient adult methylphenidate exposure does not alter cocaine’s locomotor-activating effects in later adulthood 67.
The positive correlation between levels of aggression and behavioral sensitivity to amphetamine across our peri-adolescent treatment regimes comports with the DA-hypothesis of aggression, which states that DAergic hyperfunction increases aggression 29, 68. However, the DA hypothesis of aggression is largely based on correlative data and has thus far not been tested using direct and specific activation of DAergic neuronal activity. We here therefore performed such an experiment using optogenetics and demonstrate the existence of a direct causal relationship between DAergic neuronal activity and aggressive behavior. Hence, our data support a model in which monoamine signaling during peri-adolescence permanently alters the sensitivity/activity of DAergic neurons/pathways to impact adult aggression. However, many questions remain unanswered: are DAergic changes located pre-synaptically or post-synaptically? Do they occur at the cell-intrinsic level or do they involve altered micro- or macro-circuitry? How do DA and 5-HT exert their opposing effects on behavior and how is DA-dominance established (aggressive MAOA deficient mice have both, elevated 5-HT and DA levels)? Future studies examining these questions will be critical to understand both the mechanism for the establishment of DAergic sensitivity levels during development as well as the nature of alterations in adult DA-related circuit function.
Taken together, our findings support the existence of sensitive periods that influence lifelong vulnerability to anxiety, depression, aggression, and substance abuse. Such sensitive periods have been most extensively characterized for sensory systems (e.g. visual cortex), but conceptually similar principles may apply to the development and organization of brain circuitry that mediate the more complex behaviors described here. With increased knowledge about sensitive periods during which genetic and environmental factors act to confer risk for neuropsychiatric disorders, we will not only be able to improve classification and diagnosis, but also treatment and prevention.
Supplementary Material
Behavioral amphetamine response was assessed using the open field. (a) Amphetamine injections (AMP, 3 and 5 mg/kg, i.p.) induced locomotor hyperactivity in ADO-VEH, ADO-FLX and ADO-CLO treated mice. The amphetamine response was diminished in ADO-FLX mice and exacerbated in ADO-CLO mice when compared to ADO-VEH controls as revealed by posthoc comparison (P = 0.054 and P = 0.0451, respectively) (n = 5-15 per group) (*p < 0.05, ***p < 0.001).
Acknowledgments
We thank D. Lin, A. Teissier, and C. Kellendonk for their critical review of the manuscript. This work was supported by the Sackler Institute for Developmental Psychobiology and the National Institute of Mental Health (R01 MH080116; R00 MH083044; MH062185).
Footnotes
Supplementary information is available at Molecular Psychiatry’s website.
Figure S1 (jpg file format) contains data on the behavioral response to amphetamine after peri-adolescent MAOA and 5-HTT blockade.
Conflict of interest. All authors declare that there are NO competing financial interests in relation to the work described.
References and Notes
- 1.Ansorge MS, Hen R, Gingrich JA. Neurodevelopmental origins of depressive disorders. Curr Opin Pharmacol. 2007;7(1):8–17. doi: 10.1016/j.coph.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Leonardo ED, Hen R. Anxiety as a developmental disorder. Neuropsychopharmacology. 2008;33(1):134–140. doi: 10.1038/sj.npp.1301569. [DOI] [PubMed] [Google Scholar]
- 3.Beneyto M, Lewis DA. Insights into the neurodevelopmental origin of schizophrenia from postmortem studies of prefrontal cortical circuitry. Int J Dev Neurosci. 2011;29(3):295–304. doi: 10.1016/j.ijdevneu.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- 5.Knudsen EI. Sensitive periods in the development of the brain and behavior. J Cogn Neurosci. 2004;16(8):1412–1425. doi: 10.1162/0898929042304796. [DOI] [PubMed] [Google Scholar]
- 6.Krishnan KR. Revisiting monoamine oxidase inhibitors. J Clin Psychiatry. 2007;68(Suppl 8):35–41. [PubMed] [Google Scholar]
- 7.Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science. 1993;262(5133):578–580. doi: 10.1126/science.8211186. [DOI] [PubMed] [Google Scholar]
- 8.Buckholtz JW, Meyer-Lindenberg A. MAOA and the neurogenetic architecture of human aggression. Trends Neurosci. 2008;31(3):120–129. doi: 10.1016/j.tins.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Tadic A, Rujescu D, Szegedi A, Giegling I, Singer P, Moller HJ, et al. Association of a MAOA gene variant with generalized anxiety disorder, but not with panic disorder or major depression. Am J Med Genet B Neuropsychiatr Genet. 2003;117B(1):1–6. doi: 10.1002/ajmg.b.10013. [DOI] [PubMed] [Google Scholar]
- 10.Karere GM, Kinnally EL, Sanchez JN, Famula TR, Lyons LA, Capitanio JP. What is an “adverse” environment? Interactions of rearing experiences and MAOA genotype in rhesus monkeys. Biol Psychiatry. 2009;65(9):770–777. doi: 10.1016/j.biopsych.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, et al. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science. 1995;268(5218):1763–1766. doi: 10.1126/science.7792602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott AL, Bortolato M, Chen K, Shih JC. Novel monoamine oxidase A knock out mice with human-like spontaneous mutation. Neuroreport. 2008;19(7):739–743. doi: 10.1097/WNR.0b013e3282fd6e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Godar SC, Bortolato M, Frau R, Dousti M, Chen K, Shih JC. Maladaptive defensive behaviours in monoamine oxidase A-deficient mice. Int J Neuropsychopharmacol. 2010:1–13. doi: 10.1017/S1461145710001483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaspar P, Cases O, Maroteaux L. The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci. 2003;4(12):1002–1012. doi: 10.1038/nrn1256. [DOI] [PubMed] [Google Scholar]
- 15.Homberg JR, Schubert D, Gaspar P. New perspectives on the neurodevelopmental effects of SSRIs. Trends Pharmacol Sci. 2010;31(2):60–65. doi: 10.1016/j.tips.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Souza BR, Tropepe V. The role of dopaminergic signalling during larval zebrafish brain development: a tool for investigating the developmental basis of neuropsychiatric disorders. Rev Neurosci. 2011;22(1):107–119. doi: 10.1515/RNS.2011.012. [DOI] [PubMed] [Google Scholar]
- 17.Rebsam A, Seif I, Gaspar P. Refinement of thalamocortical arbors and emergence of barrel domains in the primary somatosensory cortex: a study of normal and monoamine oxidase a knock-out mice. J Neurosci. 2002;22(19):8541–8552. doi: 10.1523/JNEUROSCI.22-19-08541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P. Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron. 1996;16(2):297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 19.Vitalis T, Cases O, Callebert J, Launay JM, Price DJ, Seif I, et al. Effects of monoamine oxidase A inhibition on barrel formation in the mouse somatosensory cortex: determination of a sensitive developmental period. J Comp Neurol. 1998;393(2):169–184. doi: 10.1002/(sici)1096-9861(19980406)393:2<169::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 20.Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science. 2004;306(5697):879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- 21.Ansorge MS, Morelli E, Gingrich JA. Inhibition of serotonin but not norepinephrine transport during development produces delayed, persistent perturbations of emotional behaviors in mice. J Neurosci. 2008;28(1):199–207. doi: 10.1523/JNEUROSCI.3973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen K, Cases O, Rebrin I, Wu W, Gallaher TK, Seif I, et al. Forebrain-specific expression of monoamine oxidase A reduces neurotransmitter levels, restores the brain structure, and rescues aggressive behavior in monoamine oxidase A-deficient mice. J Biol Chem. 2007;282(1):115–123. doi: 10.1074/jbc.M609830200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirate K, Kuribara H. Characteristics of the ambulation-increasing effect of GBR-12909, a selective dopamine uptake inhibitor, in mice. Jpn J Pharmacol. 1991;55(4):501–511. doi: 10.1254/jjp.55.501. [DOI] [PubMed] [Google Scholar]
- 24.Backman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, et al. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis. 2006;44(8):383–390. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- 25.Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, et al. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci. 2012;15(5):793–802. doi: 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lira A, Zhou M, Castanon N, Ansorge MS, Gordon JA, Francis JH, et al. Altered depression-related behaviors and functional changes in the dorsal raphe nucleus of serotonin transporter-deficient mice. Biol Psychiatry. 2003;54(10):960–971. doi: 10.1016/s0006-3223(03)00696-6. [DOI] [PubMed] [Google Scholar]
- 27.Underwood MD, Arango V, Bakalian MJ, Ruggiero DA, Mann JJ. Dorsal raphe nucleus serotonergic neurons innervate the rostral ventrolateral medulla in rat. Brain Res. 1999;824(1):45–55. doi: 10.1016/s0006-8993(99)01181-6. [DOI] [PubMed] [Google Scholar]
- 28.Vitalis T, Fouquet C, Alvarez C, Seif I, Price D, Gaspar P, et al. Developmental expression of monoamine oxidases A and B in the central and peripheral nervous systems of the mouse. J Comp Neurol. 2002;442(4):331–347. doi: 10.1002/cne.10093. [DOI] [PubMed] [Google Scholar]
- 29.de Almeida RM, Ferrari PF, Parmigiani S, Miczek KA. Escalated aggressive behavior: dopamine, serotonin and GABA. Eur J Pharmacol. 2005;526(1–3):51–64. doi: 10.1016/j.ejphar.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Holmes A, Murphy DL, Crawley JN. Abnormal behavioral phenotypes of serotonin transporter knockout mice: parallels with human anxiety and depression. Biol Psychiatry. 2003;54(10):953–959. doi: 10.1016/j.biopsych.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Richardson-Jones JW, Craige CP, Nguyen TH, Kung HF, Gardier AM, Dranovsky A, et al. Serotonin-1A autoreceptors are necessary and sufficient for the normal formation of circuits underlying innate anxiety. J Neurosci. 2011;31(16):6008–6018. doi: 10.1523/JNEUROSCI.5836-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguiz RM, Chu R, Caron MG, Wetsel WC. Aberrant responses in social interaction of dopamine transporter knockout mice. Behav Brain Res. 2004;148(1–2):185–198. doi: 10.1016/s0166-4328(03)00187-6. [DOI] [PubMed] [Google Scholar]
- 33.Gogos JA, Morgan M, Luine V, Santha M, Ogawa S, Pfaff D, et al. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc Natl Acad Sci U S A. 1998;95(17):9991–9996. doi: 10.1073/pnas.95.17.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmes A, Murphy DL, Crawley JN. Reduced aggression in mice lacking the serotonin transporter. Psychopharmacology (Berl) 2002;161(2):160–167. doi: 10.1007/s00213-002-1024-3. [DOI] [PubMed] [Google Scholar]
- 35.Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274(5292):1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- 36.Canli T, Lesch KP. Long story short: the serotonin transporter in emotion regulation and social cognition. Nat Neurosci. 2007;10(9):1103–1109. doi: 10.1038/nn1964. [DOI] [PubMed] [Google Scholar]
- 37.Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009;301(23):2462–2471. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry. 2010;167(5):509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry. 2011;68(5):444–454. doi: 10.1001/archgenpsychiatry.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt LG, Sander T, Kuhn S, Smolka M, Rommelspacher H, Samochowiec J, et al. Different allele distribution of a regulatory MAOA gene promoter polymorphism in antisocial and anxious-depressive alcoholics. J Neural Transm. 2000;107(6):681–689. doi: 10.1007/s007020070069. [DOI] [PubMed] [Google Scholar]
- 41.Le Francois B, Czesak M, Steubl D, Albert PR. Transcriptional regulation at a HTR1A polymorphism associated with mental illness. Neuropharmacology. 2008;55(6):977–985. doi: 10.1016/j.neuropharm.2008.06.046. [DOI] [PubMed] [Google Scholar]
- 42.Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297(5582):851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- 43.Zalsman G, Huang YY, Harkavy-Friedman JM, Oquendo MA, Ellis SP, Mann JJ. Relationship of MAO-A promoter (u-VNTR) and COMT (V158M) gene polymorphisms to CSF monoamine metabolites levels in a psychiatric sample of caucasians: A preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;132(1):100–103. doi: 10.1002/ajmg.b.30094. [DOI] [PubMed] [Google Scholar]
- 44.Bedard AC, Schulz KP, Cook EH, Jr, Fan J, Clerkin SM, Ivanov I, et al. Dopamine transporter gene variation modulates activation of striatum in youth with ADHD. Neuroimage. 2010;53(3):935–942. doi: 10.1016/j.neuroimage.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo G, Roettger ME, Shih JC. Contributions of the DAT1 and DRD2 genes to serious and violent delinquency among adolescents and young adults. Hum Genet. 2007;121(1):125–136. doi: 10.1007/s00439-006-0244-8. [DOI] [PubMed] [Google Scholar]
- 46.Volavka J, Bilder R, Nolan K. Catecholamines and aggression: the role of COMT and MAO polymorphisms. Ann N Y Acad Sci. 2004;1036:393–398. doi: 10.1196/annals.1330.023. [DOI] [PubMed] [Google Scholar]
- 47.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478(7370):519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24(4):417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- 49.Dawe S, Davis P, Lapworth K, McKetin R. Mechanisms underlying aggressive and hostile behavior in amphetamine users. Curr Opin Psychiatry. 2009;22(3):269–273. doi: 10.1097/YCO.0b013e32832a1dd4. [DOI] [PubMed] [Google Scholar]
- 50.Sokolov BP, Schindler CW, Cadet JL. Chronic methamphetamine increases fighting in mice. Pharmacol Biochem Behav. 2004;77(2):319–326. doi: 10.1016/j.pbb.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 51.Martin SP, Smith EO, Byrd LD. Effects of dominance rank on d-amphetamine-induced increases in aggression. Pharmacol Biochem Behav. 1990;37(3):493–496. doi: 10.1016/0091-3057(90)90018-d. [DOI] [PubMed] [Google Scholar]
- 52.Sekine Y, Ouchi Y, Takei N, Yoshikawa E, Nakamura K, Futatsubashi M, et al. Brain serotonin transporter density and aggression in abstinent methamphetamine abusers. Arch Gen Psychiatry. 2006;63(1):90–100. doi: 10.1001/archpsyc.63.1.90. [DOI] [PubMed] [Google Scholar]
- 53.Oberlander TF, Gingrich JA, Ansorge MS. Sustained neurobehavioral effects of exposure to SSRI antidepressants during development: molecular to clinical evidence. Clin Pharmacol Ther. 2009;86(6):672–677. doi: 10.1038/clpt.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Croen LA, Grether JK, Yoshida CK, Odouli R, Hendrick V. Antidepressant Use During Pregnancy and Childhood Autism Spectrum Disorders. Arch Gen Psychiatry. 2011 doi: 10.1001/archgenpsychiatry.2011.73. [DOI] [PubMed] [Google Scholar]
- 55.Crews F, He J, Hodge C. Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol Biochem Behav. 2007;86(2):189–199. doi: 10.1016/j.pbb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sullivan RM, Landers M, Yeaman B, Wilson DA. Good memories of bad events in infancy. Nature. 2000;407(6800):38–39. doi: 10.1038/35024156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moriceau S, Sullivan RM. Maternal presence serves as a switch between learning fear and attraction in infancy. Nat Neurosci. 2006;9(8):1004–1006. doi: 10.1038/nn1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8(6):828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- 59.Heinz A, Braus DF, Smolka MN, Wrase J, Puls I, Hermann D, et al. Amygdala-prefrontal coupling depends on a genetic variation of the serotonin transporter. Nat Neurosci. 2005;8(1):20–21. doi: 10.1038/nn1366. [DOI] [PubMed] [Google Scholar]
- 60.Pellis SM, Pellis VC. The prejuvenile onset of play fighting in laboratory rats (Rattus norvegicus) Dev Psychobiol. 1997;31(3):193–205. [PubMed] [Google Scholar]
- 61.Pellis SM, Pasztor TJ. The developmental onset of a rudimentary form of play fighting in C57 mice. Dev Psychobiol. 1999;34(3):175–182. [PubMed] [Google Scholar]
- 62.Suomi SJ. Risk, resilience, and gene x environment interactions in rhesus monkeys. Ann N Y Acad Sci. 2006;1094:52–62. doi: 10.1196/annals.1376.006. [DOI] [PubMed] [Google Scholar]
- 63.Moffitt TE. Adolescence-limited and life-course-persistent antisocial behavior: a developmental taxonomy. Psychol Rev. 1993;100(4):674–701. [PubMed] [Google Scholar]
- 64.Persico AM, Mengual E, Moessner R, Hall FS, Revay RS, Sora I, et al. Barrel pattern formation requires serotonin uptake by thalamocortical afferents, and not vesicular monoamine release. J Neurosci. 2001;21(17):6862–6873. doi: 10.1523/JNEUROSCI.21-17-06862.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H, Fertuzinhos S, Mohns E, Hnasko TS, Verhage M, Edwards R, et al. Laminar and columnar development of barrel cortex relies on thalamocortical neurotransmission. Neuron. 2013;79(5):970–986. doi: 10.1016/j.neuron.2013.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buckholtz JW, Treadway MT, Cowan RL, Woodward ND, Benning SD, Li R, et al. Mesolimbic dopamine reward system hypersensitivity in individuals with psychopathic traits. Nat Neurosci. 2010;13(4):419–421. doi: 10.1038/nn.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Andersen SL, Arvanitogiannis A, Pliakas AM, LeBlanc C, Carlezon WA., Jr Altered responsiveness to cocaine in rats exposed to methylphenidate during development. Nat Neurosci. 2002;5(1):13–14. doi: 10.1038/nn777. [DOI] [PubMed] [Google Scholar]
- 68.Seo D, Patrick CJ, Kennealy PJ. Role of Serotonin and Dopamine System Interactions in the Neurobiology of Impulsive Aggression and its Comorbidity with other Clinical Disorders. Aggress Violent Behav. 2008;13(5):383–395. doi: 10.1016/j.avb.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Behavioral amphetamine response was assessed using the open field. (a) Amphetamine injections (AMP, 3 and 5 mg/kg, i.p.) induced locomotor hyperactivity in ADO-VEH, ADO-FLX and ADO-CLO treated mice. The amphetamine response was diminished in ADO-FLX mice and exacerbated in ADO-CLO mice when compared to ADO-VEH controls as revealed by posthoc comparison (P = 0.054 and P = 0.0451, respectively) (n = 5-15 per group) (*p < 0.05, ***p < 0.001).