

Abstract
Hydrogen peroxide (H2O2) is produced endogenously in a number of cellular compartments, including the mitochondria, the endoplasmic reticulum, peroxisomes, and at the plasma membrane, and can play divergent roles as a second messenger or a pathological toxin. It is assumed that the tuned production of H2O2 within neuronal and non-neuronal cells regulates a discreet balance between survival and death. However, a major challenge in understanding the physiological versus pathological role of H2O2 in cells has been the lack of validated methods that can spatially, temporally, and quantitatively modulate H2O2 production. A promising means of regulating endogenous H2O2 is through the expression of peroxide-producing enzyme D-amino acid oxidase (DAAO from Rhodotorula gracilis lacking a peroxisomal targeting sequence). Using viral vectors to express DAAO in distinct cell types and using targeting sequences to target DAAO to distinct subcellular sites, we can manipulate H2O2 production by applying the substrate D-alanine or permeable analogs of D-alanine. In this chapter, we describe the use of DAAO to produce H2O2 in culture models and the real-time visual validation of this technique using two-photon microscopy and chemoselective fluorescent probes.
1. INTRODUCTION
Neuronal death in stroke, Alzheimer’s disease, Parkinson’s disease, and other neurological conditions is known to share common cellular signaling pathways. One group of highly studied, yet not fully understood signals come from toxic metabolites generated from oxygen, also known as reactive oxygen species (ROS; Barnham, Masters, & Bush, 2004; Lin & Beal, 2006). Endogenous ROS are produced in the mitochondria as a normal by-product of cellular metabolism as superoxide (O2−), which is then converted into more reactive and lipid soluble ROS and reactive nitrogen species (RNS) such as hydrogen peroxide (H2O2) and peroxynitrate (ONOO−), respectively (Perez-Pinzon, Dave, & Raval, 2005; Thompson, Narayanan, & Perez-Pinzon, 2012). Following neuronal injury, the overproduction of ROS and the destruction or consumption of antioxidant defenses lead to an imbalance between oxidants and antioxidants, otherwise known as oxidative stress. Oxidative stress has the potential to damage proteins, lipids, or DNA, but whether this damage is a mediator of neuronal death or a consequence of oxidative death is unclear. Despite the widely held belief that oxidants induce damage to cells in disease, therapeutic strategies aimed at reducing ROS levels using antioxidants have thus far been unsuccessful in clinical trials for diabetes and related neuropathies (Cowell & Russell, 2004; Johansen, Harris, Rychly, & Ergul, 2005).
The failure to translate antioxidant treatments to the clinic is likely multifactorial, but may be due to the possibility that ROS do not serve primarily as direct toxins. There is growing recognition that ROS, specifically peroxide, may act as a second messenger molecule by activating kinases (i.e., MAP kinases), inhibiting protein tyrosine phosphatases (PTPs) and inducing transcription factor activation (i.e., NFkB, FOXO, and p53) (Essers et al., 2004; Lange et al., 2008; Rhee, 2006; Ryu et al., 2003; Schreck, Rieber, & Baeuerle, 1991; Sundaresan, Yu, Ferrans, Irani, & Finkel, 1995; Yin et al., 1998). In addition to being a second messenger molecule, H2O2 is also involved in cellular mechanisms such as neurogenesis, chemotaxis, apoptosis, and peripheral neuroregeneration following injury (Bao et al., 2009; Le Belle et al., 2011; Rieger & Sagasti, 2011; Sundaresan et al., 1995; Terman, Mao, Pasterkamp, Yu, & Kolodkin, 2002; Yin et al., 1998). Some of these peroxide-related injury responses suggest that H2O2 is not only involved in cell damage following injury, but also acts as a messenger signal within cells that promote survival. These observations suggest that the inhibition of all ROS within a cell would not only inhibit the damaging effects on free radicals but may also inadvertently suppress beneficial physiological signaling.
Physiological ROS involved with signaling appear to be generated in a number of subcellular compartments, including the mitochondria, and the cellular membrane. For example, in response to ligand activation of receptor tyrosine kinases (RTK), phosphatidylinositol 3,4,5-trisphosphate (PIP3) is produced by phosphatidylinositol 3-kinase activation. PIP3 activates the nicotinamide adenine dinucleotide phosphate oxidase complex at the cellular membrane, which produces localized H2O2. Under physiological conditions, localized H2O2 accumulation can inactivate PTPs, such as the tumor suppressor protein phosphate and tensin homolog (PTEN), by oxidizing a catalytic cysteine residue (Kwon et al., 2004; Rhee, 2006; Rhee, Chang, Bae, Lee, & Kang, 2003). Similarly, localized H2O2 can also activate tyrosine kinases, such as SRC, by oxidizing two cysteine residues (Giannoni, Buricchi, Raugei, Ramponi, & Chiarugi, 2005). Cysteine has a low pKa (where Ka is a disassociation constant), and under normal pH form a thiolate ion, which can be easily oxidized by peroxide to form a disulfide bond (Rhee, 2006). Cell membrane production of H2O2 can also inactivate anti-oxidant enzymes, such as peroxiredoxins, to further accumulate H2O2 (Kwon et al., 2004). This combination of RTK-dependent localized peroxide production and H2O2-dependent inactivation of PTPs demonstrates that H2O2 can modulate phosphorylation states of second messenger pathways.
The challenge of understanding the differences between the physiological versus pathological roles of H2O2 in various cell types in the nervous system is the lack of validated methods that can spatially, quantitatively, and temporally manipulate peroxide both in vivo and in vitro. The most common method used to manipulate H2O2 uses exogenous changes in oxygen or its metabolites such as application of nonphysiological concentrations of H2O2 and O2− or normobaric hyperoxia (elevated ambient oxygen) (Gille & Joenje, 1992). Another method currently used in our lab to produce endogenous oxidative stress involves nonreceptor mediated glutamate toxicity (oxidative glutamate toxicity; Ratan, Murphy, & Baraban, 1994). This model involves treating immature neurons with glutamate or its analogs (i.e., homocysteate acid, HCA), where glutamate competitively inhibits the activity of the glutamate-cystine (Xc(−)) antiporter, which reduces uptake of cystine. Within the cell, cystine is rapidly reduced to cysteine which is required for glutathione synthesis (Ratan & Baraban, 1995). This results in a depletion of glutathione, which is required for removing naturally produced ROS, and leads to a buildup of ROS in the cell and oxidative damage. Some of the limitations of these models in studying oxidative stress include (a) the use of immature neurons; (b) the lack of synaptic activity (glutathione depletion model only); (c) the inability to manipulate spatially and temporally specific ROS production both in a single cell type and heterologous cell cultures; and (d) the inability to distinguish physiological and pathological ROS production. Since current models of oxidative stress are limited, our lab has been exploring enzymatic methods to endogenously manipulate specific ROS.
In this chapter, we will first discuss techniques developed in our lab using the H2O2 producing enzyme DAAO from Rhodotorula gracilis (R. gracilis DAAO) with its C-terminal peroxisomal targeting sequence deleted to endogenously manipulate peroxide concentrations. This model used viral vectors to selectively express R. gracilis DAAO in astrocytes, followed by the subsequent application of exogenously applied D-alanine (D-ala) and flavin adenine dinucleotide (FAD) to produce H2O2 (Haskew-Layton et al., 2010). The latter half of the chapter will discuss our validation method using two-photon microscopy (TPM) to detect site-specific fluorescent boronate probes sensitive to H2O2 (Guo et al., 2013). The advantage of this method over other ROS detection methods is that it detects only intracellular H2O2 concentrations (as opposed to other ROS). As well, the addition of a TPM allows for real-time visualization that prevents photobleaching and can be used both in vivo and in vitro (Chen et al., 2013).
2. PRODUCTION OF H2O2 USING DAAO
DAAO is a peroxisomal flavoenzyme that exists in a wide array of species from yeast to humans; the enzyme oxidatively deaminates D-amino acids into their corresponding imino acids and uses the cofactor FAD to produce H2O2 as a by-product (Pollegioni, Piubelli, Sacchi, Pilone, & Molla, 2007). Unlike other enzymatic models of peroxide production, such as glucose oxidase, galactose oxidase, monoamine oxidase, and superoxide dismutase, DAAO allows for precise H2O2 production by manipulating the concentration of its substrate D-ala (Stegman et al., 1998). The D-ala substrate was also appealing to use in rodent models since the D enantiomer is generally scarce in mammalian cells. Our model uses adenoviruses containing flag-tagged cDNA for R. gracilis (red yeast) DAAO lacking its peroxisomal targeting sequence. The absence of targeting sequence allows for R. gracilis DAAO expression only in the cytoplasm and thus prevents scavenging of H2O2 in peroxisomes by catalase (Stegman et al., 1998). R. gracilis DAAO also has a higher catalytic activity and is less prone to auto-oxidation-induced inactivation in comparison to endogenous mammalian DAAO.
We have used this model to manipulate H2O2 production in astrocytes (Guo et al., 2013; Haskew-Layton et al., 2010). When R. gracilis DAAO expressing astrocytes are cocultured with neurons, we found that low concentrations of peroxide production (3.7 nmol/min/mg protein) protected neighboring neurons from glutamate-induced oxidative stress, thus showing that this model can be used to distinguish protective and nonprotective concentrations of peroxide (Haskew-Layton et al., 2010). In this section, we describe the methods developed in our lab using R. gracilis DAAO to endogenously produce peroxide in cells.
2.1. Cell cultures
2.1.1 Astrocyte cultures
Primary astrocyte cultures are prepared from the cerebral cortices of Sprague–Dawley rat pups (P1–3) as described in Haskew-Layton et al. (2010). In brief, the brain tissue is dissociated using the protease Papain (Cat.: LS003127 Worthington Biochemical Corp., Lakewood, NJ). Astrocyte cultures are seeded at a low density (15,000/mL) on Primaria plates (BD Falcon, San Jose, CA) in minimal essential medium (MEM; Cat.: 41090-101; Life Technologies, Grand Island, NY) supplemented with 10% horse serum (Cat.: 26050-088) and 25 U/mL penicillin plus 25 g/mL streptomycin (Cat.: 15140122; Life Technologies). Cultures are grown for ~2 weeks in a 37 °C incubation chamber with 5% CO2 with regular media changes until astrocytes reach confluency. Once confluent, astrocytes are treated with 8 μM cytosine-D-arabinofuranoside (Ara-C; Cat.: C1768; Sigma-Aldrich, St Louis, MO), a mitotic inhibitor, for ~3 days to kill O2A progenitor and microglia contaminating cells. Astrocytes are maintained for 2–3 weeks in culture prior to experimental use. To confirm astrocyte culture purity (~95% purity) use glial fibrillary acidic protein staining, which stains primarily astrocytes.
2.1.2 HT22 cultures
HT22 is an immortalized neuroblast line originating from hippocampal neurons. Culture HT22 cells on clear bottom untreated plastic plates in a Dulbecco’s modified Eagle medium (DMEM; Cat.: 11965-118; Life Technologies) supplemented with 10% fetal bovine serum (Cat.: 16140-071; Life Technologies) and 25 U/mL penicillin plus 25 g/mL streptomycin. Cultures are incubated at 37 °C with 5% CO2 until they are ready to use. HT22 cultures can be used for experiments when grown to 60–80% confluence. At 100% confluence, HT22 cells must be split using 0.05% trypsin–EDTA (Cat.: 25300-120; Life Technologies) and plated in new culture dishes with DMEM culture media.
2.1.3 Culture plates
Culture plate sizes can be modified for experimental requirements. It is recommended that cells are cultured on glass bottom culture dishes when using the two-photon system (described in Section 3) to measure H2O2 and on opaque 96-well plates when using the horseradish peroxidase (HRP)/Amplex Red assay (described in Section 2.3.2). When using opaque plates, also plate cells at the same density in a clear bottom plate to determine confluence.
2.2. DAAO transduction into cells
2.2.1 DAAO viral construct generation
The R. gracilis DAAO viral construct was generated prior to experimentation. cDNA encoding flag-tagged R. gracilis DAAO (gift of B. D. Ross and A. Rehemtulla, University of Michigan, Ann Arbor, MI) was subcloned into a pVQAd5CMVK-NpA shuttle plasmid containing a CMV promoter. In addition to the R. gracilis DAAO plasmid, we also generated an empty pVQAd5CMVK-NpA plasmid (for negative control) and a GFP cDNA pVQAd5CMVK-NpA plasmid to calculate multiplicity of infection (MOI). Shuttle plasmids were incorporated into adenoviral backbones and replication-deficient adeno viruses were generated by Viraquest Inc. Were commend using viral vector-based transduction of the DAAO construct due to its high efficiency. Liposomal transfection of nonviral plasmids can be used, but may cause complications due to inconsistent and low transfection rates, particularly in primary cells. Here, we will describe the viral transduction method only.
2.2.2 R. gracilis DAAO transduction
Prior to transduction, remove media and wash with the serum-free Opti-MEM media (Cat.: 31985-088; Life Technologies). Adenovirus containing the R. gracilis DAAO construct are diluted in Opti-MEM at an MOI of 15. Then cells are treated with R. gracilis DAAO+Opti-MEM media for 4 h at 37°C and 5% CO2. Following incubation, R. gracilis DAAO+Opti-MEM is removed and replaced with serum-containing culture media. Astrocytes are ready for experiments 4 days after adenovirus treatment. Use the empty vector adenovirus and no adenovirus in the same treatment as a negative control. An MOI of 15 was determined using adenoviral GFP (adGFP) and anti-flag immunostaining for the flag-tagged R. gracilis DAAO, which had an ~80% transduction efficiency with no astrocyte toxicity (Haskew-Layton et al., 2010). We recommend first treating cells with adGFP at varying MOIs to determine optimal (70–80%) efficiency, prior to using adenoviral R. gracilis DAAO. The adenoviral R. gracilis DAAO construct expresses DAAO in the cytoplasm; we will discuss other site targeted DAAO in Section 2.5.
2.3. Induction of H2O2 production in cells
The R. gracilis DAAO enzyme oxidatively deaminates D-amino acids using FAD as an electron acceptor. Concomitantly, R. gracilis DAAO uses molecular O2 to oxidize FAD and H2O2 is produced as a by-product (Fig. 14.1). Using this model and D-ala as a limiting substrate, we manipulated endogenous H2O2 production as described below. As demonstrated in Haskew-Layton et al. (2010), R. gracilis DAAO expression in the absence of substrate (D-ala+FAD) administration was insufficient in generating H2O2 production. As well, D-ala treatment in cells not expressing R. gracilis DAAO failed to generate measurable H2O2 levels. This highlights that the R. gracilis DAAO system is ideal for the precise experimental control of intracellular H2O2 production. Some cell types fail to transport alanine very effectively, and for these cell types, we have tested esterified forms of D-amino acids which can diffuse through the membrane and be trapped by nonspecific esterases. These esterified D-amino acids are available through Sigma-Aldrich. We have found it useful to evaluate a panel of esterified amino acids for each cell type to establish which amino acid works best.
Figure 14.1.
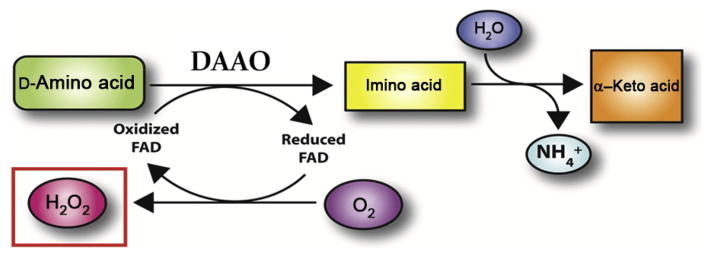
DAAO enzymatic function DAAO deaminates D-amino acid (in this case D-ala) with the cofactor FAD to produce imino acid. In the process, FAD is reduced which is then quickly oxidized by O2 to produce oxidized FAD and H2O2. Imino acid eventually reacts with H2O to produce NH4 + and α-keto acid. Figure modified from Haskew-Layton et al. (2010).
2.3.1 D-Alanine and FAD concentrations for treatment of DAAO-transfected cells
D-Alanine (Cat.: A7377; Sigma-Aldrich) at concentrations of 0–4 mM is diluted into basal media containing 135 mM NaCl, 3.8 mM KCl, 1.2 mM MgSO4, 1.3 mM CaCl2, 1.2 mM KH2PO4, 10 mM D-glucose, 10 mM HEPES (Sigma-Aldrich), at a pH 7.4. Experimental data showed that in rgDAAO transduced astrocytes, D-ala in the absence of FAD is insufficient in maintaining continuous H2O2 production beyond 10 min (Haskew-Layton et al., 2010). To maintain H2O2 production, basal media+ D-ala must be supplemented with the cofactor FAD (Cat.: F6625; Sigma-Aldrich) at a concentration of 2.5 μM, which was found to be sufficient in maintaining H2O2 production beyond 10 min (Fig. 14.2). Media lacking both D-ala and FAD was also used as negative controls to measure baseline H2O2 production in cells.
Figure 14.2.

Amplitude of H2O2 production by D-ala. Application of D-ala+ FAD in R. gracilis DAAO-transfected astrocytes causes a significant dose dependent production of H2O2 (shown in red (light gray in the print version); ***p <0.001). Production of H2O2 is observed at concentrations as low as 0.02 mM of D-ala. Empty vector (blue; dark gray in the print version) and nontransfected (black) controls had no change in peroxide production with D-ala treatment. Figure modified from Haskew-Layton et al. (2010).
2.3.2 H2O2 production and measurement
The HRP/Amplex Red substrate assay (Cat.: A22188; Life Technologies) is used to initially confirm extracellular H2O2 levels released from live cells. This method assumes that H2O2 is freely diffusible through cell membranes. In this assay, HRP uses peroxide as a cofactor to convert Amplex Red into resorufin, which is a fluorescent or colorimetric indicator. The net rate of H2O2 production by R. gracilis DAAO equals the rate of its consumption by HRP, as monitored by the accumulation of resorufin (ext coefficient = 54,000 M−1 S−1).
To measure kinetic H2O2 production, cells are washed once with serum-free basal media to remove serum. After wash, prewarmed (37 °C) D-ala+FAD in basal media is added over cell cultures and is incubated for approx. 30 s at room temperature (RT). After 30 s, prewarmed reaction mixture containing 50 μM Amplex Red and 0.1 U/mL HRP (from life technologies kit) in basal media is added on top of the D-ala+FAD containing media. Note that HRP is not a limiting factor, being shown to have similar rate of resorufin production up to 0.8 U/mL. After adding the reaction mixture, resorufin is measured every 30 s for 1 h using a spectrophotometer at 560 nm (Spectramax Plus 384; Molecular Devices, Sunnyvale, CA). Alternatively, resorufin can be measured using a fluorescent plate reader at 560 nm excitation and 590 nm emission detection.
For end-point H2O2 measurements, cultures are washed once with basal media and treated with D-ala+FAD in basal media for 30 min in the cell culture incubator. Following D-ala +FAD treatment, bathing media is collected and HRP/Amplex Red reaction buffer is added to the bathing media. Bathing media+reaction buffer solution is incubated for 5 min at RT and then final resorufin concentration is measured using a spectrophotometer. We also recommend using known concentrations of H2O2 as a control to determine relative measurements of peroxide concentration.
2.4. Heterologous cell-type culture
One advantage of the R. gracilis DAAO model over other models of oxidative stress is the ability to manipulate H2O2 in a single cell type in a heterogeneous population of cells. We have previously used this method to produce H2O2 in astrocytes when cocultured with neurons. As described in Section 2.2.1, astrocyte primary cultures are transduced with R. gracilis DAAO for 4 days. Then, astrocytes are treated with desired concentration of D-ala and FAD for 4–24 h. D-Ala is then washed off and cortical neurons from E1 rat embryos are plated, using methods described in Haskew-Layton et al. (2010).
Although D-ala is known to be an N-methyl-D-aspartate (NMDA) receptor coagonist at the glycine-binding site (McBain, Kleckner, Wyrick, & Dingledine, 1989; Tsai, Yang, Chang, & Chong, 2006), it is important to note that in the coculture model, the D-ala is completely washed off of the astrocytes prior to neuronal plating to avoid direct neuronal exposure to D-ala. Use of the R. gracilis DAAO model in mature neurons would require the use of a D-amino acid substrate that does not act as an NMDA receptor coagonist. This model is useful in understanding how kinetic parameters of H2O2 production in one cell type can influence another.
2.5. Differentiating pro-survival and pro-death ROS concentrations
ROS are known to be involved in both cell survival and cell death mechanisms. Pretreatment with low concentrations of exogenous H2O2 has been shown to cause neuroprotective preconditioning, while high concentrations of ROS lead to cell death (Chang, Jiang, Zhao, Lee, & Ferriero, 2008; Furuichi, Liu, Shi, Miyake, & Liu, 2005; McLaughlin et al., 2003). In order to finely modulate between physiological/survival and pathological ROS levels, we use our R. gracilis DAAO model to generate a broad spectrum of H2O2 levels in astrocytes coupled with the glutathione depletion model to promote oxidative stress-induced neuronal death. By titrating the level of H2O2 production in the R. gracilis DAAO expressing astrocytes with different concentrations of D-ala, we can determine what level of astrocytic H2O2 will protect neurons from oxidative stress-induced death. The glutathione depletion model uses glutamate/HCA to inhibit the Xc(−) transporter, which in turn inhibits production of the antioxidant glutathione. The level of neuronal survival following glutamate/HCA treatment of R. gracilis DAAO expressing cells determines what concentrations of endogenously produced H2O2 in astrocytes is protective. In Haskew-Layton et al. (2010), we demonstrated that the R. gracilis DAAO model can be used to reveal both the physiological and pathological roles of H2O2. We showed that 16 μM D-ala+FAD stimulated an intracellular mechanism in R. gracilis DAAO astrocytes that protected neurons from oxidative stress, while 2 mM D-ala+FAD produced levels of H2O2 from R. gracilis DAAO astrocytes that were lethal to neighboring neurons (when neurons were directly exposed to H2O2 being released from the astrocytes). Although we have used this method in a coculture model to determine H2O2’s effect on noncell autonomous neuronal protection, the R. gracilis DAAO model could also be employed in a cell autonomous system to determine H2O2’s physiological parameters using cultures of a single cell type.
2.5.1 Glutamate/HCA model with R. Gracilis DAAO
In the astrocyte-neuron coculture model, R. gracilis DAAO expressing astrocytes are treated with a broad range of D-ala concentrations (from 0.016 to 4 mM) supplemented with 2.5 μM FAD for 4–24 h. D-Ala+FAD is then washed off and immature E18 rat neurons are plated at a density of 500,000 cells/mL immediately in the presence or absence of 5 mM HCA (Cat.: H9633; Sigma-Aldrich) for 48 h prior to determining neuronal viability. As shown in Haskew-Layton et al. (2010), although 5 mM HCA depletes glutathione in both the astrocytes and the neurons, only the neurons die as a consequence of the glutathione depletion. To determine how astrocytic H2O2 affects the survival of neurons subjected to HCA-induced oxidative stress, it is essential to distinguish between neuronal and astrocyte viability. Control treatments lacking HCA are used to determine baseline neuronal death in cultures. To measure specific neuronal viability when cocultured with astrocytes, we monitor levels of the neuronal specific marker MAP2 using an immunoassay. Forty-eight hours following HCA treatment, the cocultures are fixed in 4% paraformaldehyde and then incubated with primary MAP2 antibodies (as described in Haskew-Layton et al., 2010). Secondary antibodies conjugated with HRP are then employed to bind the primary MAP2 antibodies as a means to quantify MAP2 levels. To measure HRP activity an Amplex Red assay is used. In brief, after removal of secondary antibodies with a wash, the fixed cocultures are exposed to basal media containing 100 μM Amplex Red and 400 μM H2O2. In the presence of H2O2, HRP converts Amplex Red to the colorimetric/fluorescent product resorufin, the levels of which are quantified with a spectrophotometer at 560 nM. Alternatively, a fluorescent plate reader can be used to monitor resorufin as well.
In a single cell-type culture model, D-ala with 2.5 μM of FAD is applied to cells expressing R. gracilis DAAO for 24 h. D-Ala+FAD is then washed with PBS and replaced with growth media supplemented with 5 mM of glutamate or HCA for 24 h. Cell death is measured using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cell viability assay (Cat.: G4100; Promega, Fitchburg, WI), a colorimetric assay where live cells reduce tetrazolium dye MTT into insoluble formazan. In a 96-well plate, 5 μL of MTT is added to each well of 100 μL of media and incubated for 30 min–2 h (depending on cell type). After incubations, wells are treated with 20 μL of stop solution, which stops the reduction of MTT and solubilizes the formazan so that it can be detected. Plates are shaken at 65 rpm in the dark for ~1 h, after which formazan levels are read using a colorimetric plate reader. For a positive control, treat cells with a known antioxidant (such as N-acetylcysteine or catalase) to ensure that it is oxidative stress that is killing the cells. Sometimes, antioxidants can directly reduce MTT, so we recommend replacing the antioxidant rich media with media lacking antioxidants, waiting 2 h, rinsing again with media lacking antioxidants and proceeding with the MTT assay. Also cell death can be visually confirmed using a light microscope and observing cell density. Incubation times with MTT will vary according to the celltype. To obtain a linear relationship between cell number and MTT reduction, incubate cells treated with and without HCA in MTT at varying times and measure which time point gives the largest colorimetric difference between live and dead cultures.
2.6. Targeted H2O2 production in cells
Intracellular ROS is primarily generated at subcellular compartments, specifically the mitochondria, peroxisomes, and cellular membrane. Most commonly used oxidative stress models, as well as our R. gracilis DAAO model, are limited to cytoplasmic modulation of ROS and are unable to regulate ROS production in a site-specific manner. Since the R. gracilis DAAO model uses an expressed enzyme to manipulate ROS production and enzymes can be trafficked to different subcellular compartments, we have been developing R. gracilis DAAO constructs that contain subcellular targeting sequences. The following targeting sequences have been N-terminally attached to R. gracilis DAAO: myristoylated DAAO (Myr-DAAO: MGSSKSKPK) which targets membrane expression; mitochondrial matrix-targeted DAAO (mito DAAO; protein sequence: MLSRAVCGTSRQLAPALGYLGSRQ) which targets the mitochondria; nuclear localization sequence DAAO (NLS-DAAO; protein sequence: DPKKKRKV) which targets inside the nucleus; and nuclear exclusion sequence DAAO (NES-DAAO; protein sequence: LPPLERLTL) which traffics R. gracilis DAAO outside the nucleus into the cytoplasm. We verified that R. gracilis DAAO targeted to the mitochondria can generate peroxide. Experimental studies using targeted R. gracilis DAAO must confirm that D-amino acid substrates can get distributed to distinct sites equally in the cell.
2.6.1 R. gracilis DAAO expression confirmation
Prior to experiments, R. gracilis DAAO expression at their intended target is verified using immunofluorescence against flag-tag sequence in R. gracilis DAAO constructs. As described in Sections 2.2 and 2.3, cells are transduced with one of the site-directed R. gracilis DAAO constructs. Cultures are then fixed in 4% paraformaldehyde for 15 min, washed with PBS, and then blocked with 1% bovine serum albumin (BSA; Cat.: A3059; Sigma-Aldrich) in PBS for 1 h. Fixed cultures are then incubated with primary FLAG antibodies in 1% BSA solution overnight. Primary antibody is washed with PBS and fluorescent secondary antibodies (red) in 1% BSA are then employed to bind the primary antibodies to determine R. gracilis DAAO expression localization. The fluorescent nuclear probe, Hoechst 33342 (Cat.: H1399; Life Technologies), is added with secondary antibody solution to stain (blue) nucleus of the cell (Fig. 14.3). Empty vector adenovirus is used as negative controls to ensure that viral transfection alone has no effect on immunofluorescence and antibodies are specific to the expression of the flag-tag sequence.
Figure 14.3.
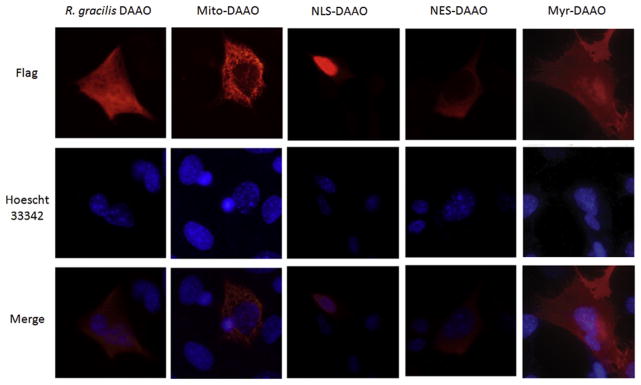
Expression location of R. gracilis DAAO targeted sequences. Immunofluorescence staining of cells transduced with adenoviral R. gracilis DAAO (no targeting, expressed primarily in cytoplasm), Mito-DAAO (targeting mitochondria), NLS-DAAO (targeting nucleus), NES-DAAO (targeting outside nucleus), and Myr-DAAO (targetting cell membrane) all shown in red. Hoechst 33342 staining in blue to localize nucleus. Immunofluorescence confirms that adding targeting sequence to the R. gracilis DAAO construct causes DAAO expression in specific subcellular compartments.
2.6.2 Cytoplasmic and mitochondrial H2O2 production in cell
Protocols to transduce cytoplasmic R. gracilis DAAO and mito DAAO constructs into cells and manipulation of H2O2 are similar to those described in Sections 2.2 and 2.3. For targeted generation of H2O2 in cytoplasm and mitochondria, R. gracilis DAAO and mito DAAO-transduced cells are washed once with PBS and then incubated for 45 min with 25 μM of fluorescent probe dichlorofluorescein (DCF; Cat.: ab113851; Abcam, Cambridge, MA) in PBS at 37 °C. Following incubation, cells are washed once with 1× buffer and location of peroxide production is visualized using an inverted fluorescent microscope system (Zeiss Axiovert 200 M) with excitation 485 nm and emission 535 nm (Fig. 14.4). DCF is at best a semi-quantitative indication of a redox change. To verify that DCF differences between groups actually relate to changes in the cellular redox state, we perform several controls. First, we show that the maximum fluorescence achieved by adding 1 mM peroxide to cultures is similar between distinct experimental groups. Second, we monitor pH levels using BCECF AM (Cat.: B-1170; Life Technologies) to exclude potentially confounding changes in pH. Third, we ideally overexpress an antioxidant enzyme to demonstrate that DCF fluorescence can be reduced by reducing the oxidant of interest.
Figure 14.4.
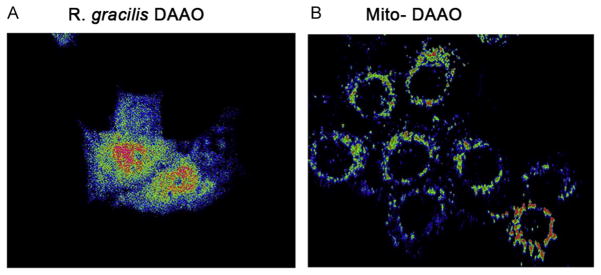
Fluorescent imaging of ROS expression in DAAO targeted expression cells DCF fluorescent images of HT22 cells transduced with either (A) R. gracilis DAAO or (B) mito DAAO, that produce H2O2 in either the cytoplasm or mitochondria. Fluorescent intensity was converted to red to black color gradient, where red indicates high fluorescent intensity and blue represents low.
3. TWO-PHOTON FLUORESCENCE IMAGING OF H2O2
In addition to manipulating intracellular ROS generation, a substantial challenge remains in developing validation methods to determine the spatial and temporal dynamics of specific ROS in living systems. The use of conventional confocal microscopy to visualize fluorescence has limitations for real-time in vivo H2O2 imaging, specifically phototoxicity, photo-bleaching, and limited imaging depth (Chen et al., 2013; Guo et al., 2013). In addition, prolonged light exposure can cause artifact ROS generation and signal amplification (Hockberger et al., 1999; Squirrell, Wokosin, White, & Bavister, 1999). To overcome these limitations, two-photon imaging of H2O2 offers an attractive alternative, since it detects fluorescence inside the tissue in real time and does not require long exposure to light resulting in H2O2 artifacts (Chung, Srikun, Lim, Chang, & Cho, 2011).
3.1. Two-photon microscopy
The first TPM system was developed by Denk, Strickler, and Webb (1990). This method provides high-resolution (submicron) imaging with lower phototoxicity and deeper tissue penetration than confocal imaging (Chen et al., 2013; Guo et al., 2013). In the two-photon excitation model, a molecule simultaneously absorbs two photons whose individual energy is only half of the energy needed to excite that molecule, and then releases the energy to a fluorescence photon. TPM generally uses a near infrared excitation wavelength laser that reduces the tissue autofluorescence and optical scattering. Therefore, it can provide deeper penetration depth than regular confocal microscopy. Two-photon fluorescence (TPF) can only occur at the focus when the laser power is high enough for excitation. In other words, TPM can perform “optical sectioning” without using the physical pinhole that is used in confocal microscopy, since there is no two-photon signal from either above or below the focal plane. As a result, TPM can collect signals more efficiently than confocal microscopy. TPM imaging can be achieved from two-photon excitation of conventional fluorophores such as fluorescent dyes, fluorescent proteins, and nanoparticles. With fluorescent labeling, TPM has been used for intracellular imaging of molecules such as calcium, oxygen partial pressure (pO2), and ROS (Chen et al., 2013; Guo et al., 2013; Kwan & Dan, 2012; Sakadzic et al., 2010). It can also image endogenous fluorescence molecules such as the reduced nicotinamide adenine dinucleotide (NADH), FAD, and keratin (Chen et al., 2013; Zipfel et al., 2003).
We have published the use of next-generation boronate-based probes in combination with TPF imaging for the detection of intracellular H2O2 in the cytoplasm and mitochondria (Guo et al., 2013). In this section, we describe this method to validate and confirm in vitro manipulation of peroxide in cells transduced by R. gracilis DAAO and treated with D-ala.
3.2. Chemoselective fluorescent probes
Current strategies of detecting ROS production in vitro have relied upon several fluorescent probes that are based on small molecules, fluorescent proteins, and nanoparticles (Belousov et al., 2006; Crow, 1997; Dickinson & Chang, 2008; Lee et al., 2007; Miller, Tulyathan, Isacoff, & Chang, 2007; Wu, Zhang, & Ju, 2007; Zhao et al., 2010). Among these technologies, small-molecule probes offer an attractive approach to ROS detection due to their ability to detect intracellular H2O2 and their general compatibility with an array of biological systems without requiring external activating enzymes or genetic manipulation. However, limitations to small-molecule probes, such as DCF derivatives, include lack of specific ROS detection (DCF detects a number of ROS and RNS) and lack of identification of the intracellular ROS source (Belousov et al., 2006; Crow, 1997). To overcome these disadvantages, new chemoselective fluorescent indicators with a boronate-based molecular detection mechanism have been developed (Miller et al., 2007). These indicators provide improved selectivity for H2O2 in comparison to related ROS, such as superoxide, nitric oxide, and hydroxyl radical. Some probes also come in different colors and are specifically directed to subcelluar compartments, allowing for simultaneous H2O2 measurement at multiple intracellular locations. These probes include peroxyfluor-2 (PF2), peroxy yellow 1 (PY1), peroxy orange 1 (PO1), peroxyfluor-6 acetoxymethyl ester (PF6-AM), and mitochondrial peroxy yellow 1 (MitoPY1) (Chan, Dodani, & Chang, 2012; Dickinson & Chang, 2008; Dickinson, Huynh, & Chang, 2010; Dickinson, Peltier, Stone, Schaffer, & Chang, 2011; Lippert, Van de Bittner, & Chang, 2011; Miller et al., 2007).
We describe here two previously reported chemoselective probes with two useful colors: peroxyfluor-6 acetoxymethyl ester, PF6-AM (green), and mitochondria peroxy yellow 1, MitoPY1 (yellow). These probes contain an aryl boronate group that is selectively switched to phenol by H2O2 over other ROS. Upon reaction with H2O2, a highly fluorescent product is released, which can be measured by fluorescence imaging. All fluorescence probes are derivatives of fluorescein/rhodamine. PF6-AM is a modified PF6 with acetoxymethyl ester groups that has improved cell membrane permeability in comparison to PF6. Upon penetration of the cell membrane, PF6-AM is hydrolyzed (deprotected) by intracellular esterases, releasing the dianionic PF6 that is then trapped in the cytosol (Dickinson et al., 2011). MitoPY1 is derived from PY1 to include both a boronate-based switch and a mitochondrial-targeting phosphonium moiety for the detection of H2O2 localized to cellular mitochondria (Dickinson & Chang, 2008). Prior papers have extensively reviewed chemoselective fluorescent probes (Dickinson et al., 2010; Lin, Dickinson, & Chang, 2013). These probes were designed and synthesized by Chris Chang’s lab at the University of California Berkeley.
3.2.1 Optical parameters for probes
The two-photon activation times, single-photon absorption, and emission peaks of the probes are shown in Table 14.1, originally published in Guo et al. (2013). Activation time is related to the deprotection efficiency, specifically the average time when TPF intensity increases to saturation intensity. Compared with the nonspecific probe DCF, the chemoselective probes demonstrate much faster responses to H2O2.
Table 14.1.
Optical parameters for probes
| Probes | Absorption peak (nm) | Emission peak (nm) | Activation time (min) |
|---|---|---|---|
| DCF | 500 | 525 | 48±1 |
| PF6-AM | 460 | 520 | 14±1 |
| MitoPY1 | 510 | 530 | 24±5 |
3.3. Measurement of cellular H2O2 concentration
3.3.1 Microscope setup
Prior to H2O2 imaging we set up our microscope as follows: We used a commercial laser scanning inverted microscope system (Zeiss 710NLO) configured for both confocal microscopy and TPM. A Ti:sapphire laser (Coherent Chameleon Vision II) at 770 nm was used to coexcite fluorescent probes PF6-AM, MitoPY1, and Hoechst 33342, a fluorescent nuclear probe. A 20×/0.80 NA objective was used to focus the excitation laser beam onto cells, as well as collect emitted fluorescence into the photo-multiplier tube. A prism-based 34-channel QUASAR detection unit was used for tunable spectral bandwidth collection without traditional band-pass filters. Live cells were maintained in a 5% CO2 circulation and 37 °C thermal chamber during imaging.
3.3.2 TPF imaging of intracellular H2O2
For exogenous manipulation of H2O2, 20 μM of fluorescent probes, Pf6-AM (green) and MitoPY1 (yellow/green), are individually diluted in PBS. Hoechst 33342 (blue) is also added in each fluorescent probe mixture at 20 μM to stain the nucleus. Then, HT22 hippocampal neuroblasts at 70–80% confluence are first washed once with PBS to remove media and then incubated with fluorescent probe solution for either 20 min (for PF6-AM solution) or 10 min (for MitoPY1 solution) at 37 °C with 5% CO2. After incubation, the cells plated on a glass bottom culture dish are transferred to the microscope chamber and 50 μM of H2O2 (or desired concentration) is continuously bath applied. TPM time lapse can monitor real-time changes in H2O2 concentrations in the cytoplasm (for PF6-AM solution; Fig. 14.5A) and mitochondria (for MitoPY1 solution; Fig. 14.5B). To confirm mitochondria localization use the MitoTracker Red assay (Life Technologies) which stains for mitochondria in live cells. In this assay, HT22 cells are first incubated with 5 μM of MitoPY1 for 25 min. Then H2O2 is applied either exogenously or is endogenously produced. After peroxide is produced, 1 mM of MitoTracker Red is added for 60 min to ensure full staining of mitochondria. Both MitoPY1 and MitoTracker Red are coexcited at 770 nm excitation.
Figure 14.5.

TPM of H2O2 presence in cytoplasm and mitochondria of HT22 cells (A) PF6-AM (green; white in the print version) staining peroxide in the cytoplasm of HT22 cells with exogenously applied 50 μM H2O2 for 3 min (left panel) and 38 min (right panel). Hoechst 33342 (blue; light gray in the print version) shows nuclear staining of live cells. (B) MitoPY1 (green; white in the print version) staining of H2O2 in mitochondria. Here, cells were either untreated (left panel) or treated with 50 nM rotenone, a chemical which interferes with the mitochondrial electron transport chain and produces ROS. Figure modified from Guo et al. (2013).
For endogenous H2O2 manipulation by DAAO, astrocytes transfected with R. gracilis DAAO are first treated with D-ala+FAD to induce H2O2 production, as described in Section 2. Various incubation periods of D-ala+FAD can be used to observe kinetic H2O2 production. Then, 5 μM of PF6-AM and Hoechst 33342 solution is added to the media for 30 min before use. PF6-AM and Hoechst 33342 are coexcited at 770 nM and visualized using TPF (Fig. 14.6). Visualization of cells treated with targeted R. gracilis DAAO expression (described in Section 2.6.1) is yet to be done by our lab.
Figure 14.6.
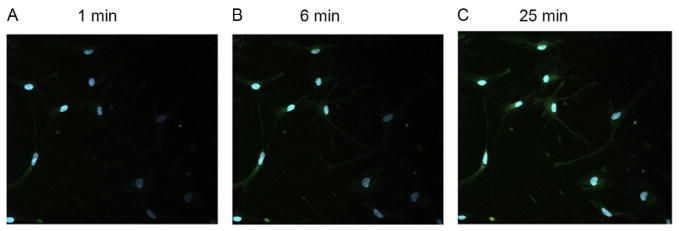
TPM of cytoplasmic H2O2 stained with PF6-AM in astrocytes. Astrocytes are incubated with 2 mM D-ala +FAD for (A) 1 min, (B) 6 min, and (C) 25 min before staining with PF6-AM (green; gray in the print version) and Hoechst 33342 (blue; white in the print version). Cytoplasmic H2O2 concentration accumulates the longer the D-ala + FAD treatment as shown in greater green intensity after 25 min compared to 1 min. Figure modified from Guo et al. (2013).
4. SUMMARY
Accumulation of ROS is known to be a key trigger of cell death in ischemia and neurodegenerative disorders. However, completely removing ROS can interfere with ROS-dependent physiological pathways. Differentiating the role of ROS in pathological and physiological conditions requires a finely tuned method of temporally and spatially manipulating and measuring specific ROS. Herein we described methods using R. gracilis DAAO to endogenously manipulate H2O2 production by modulating concentrations of the substrate D-ala. In addition, we also describe how to use TPF and chemoselective fluorescent probes to visualize ROS accumulation in real time in the mitochondria and cytoplasm. Studies exploring the full potential of these methods are ongoing, which will allow us to manipulate site-specific production of H2O2 and observe in real time how this effects cellular functions both in vivo and in vitro.
Acknowledgments
Research reported in this chapter was supported by the National Institute of Health Grants NS04059, NS39170, and 2P01AG014930, The Hartman Foundation and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to R. R. R.). Authors would like to thank Dr. Jose M. Garcia-Manteiga for his technical expertise in DCF fluorescence images.
References
- Bao L, Avshalumov MV, Patel JC, Lee CR, Miller EW, Chang CJ, et al. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2009;29:9002–9010. doi: 10.1523/JNEUROSCI.1706-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nature Reviews Drug Discovery. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- Chan J, Dodani SC, Chang CJ. Reaction-based small-molecule fluorescent probes for chemoselective bioimaging. Nature Chemistry. 2012;4:973–984. doi: 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Jiang X, Zhao C, Lee C, Ferriero DM. Exogenous low dose hydrogen peroxide increases hypoxia-inducible factor-1alpha protein expression and induces preconditioning protection against ischemia in primary cortical neurons. Neuroscience Letters. 2008;441:134–138. doi: 10.1016/j.neulet.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Guo HC, Gong W, Qin LY, Aleyasin H, Ratan RR, et al. Recent advances in two-photon imaging: Technology developments and biomedical applications. Chinese Optics Letters. 2013;11:011703. [Google Scholar]
- Chung C, Srikun D, Lim CS, Chang CJ, Cho BR. A two-photon fluorescent probe for ratiometric imaging of hydrogen peroxide in live tissue. Chemical Communications (Cambridge, England) 2011;47:9618–9620. doi: 10.1039/c1cc13583j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell RM, Russell JW. Nitrosative injury and antioxidant therapy in the management of diabetic neuropathy. Journal of Investigative Medicine. 2004;52:33–44. doi: 10.1136/jim-52-01-24. [DOI] [PubMed] [Google Scholar]
- Crow JP. Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: Implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide. 1997;1:145–157. doi: 10.1006/niox.1996.0113. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. Journal of the American Chemical Society. 2008;130:9638–9639. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Huynh C, Chang CJ. A palette of fluorescent probes with varying emission colors for imaging hydrogen peroxide signaling in living cells. Journal of the American Chemical Society. 2010;132:5906–5915. doi: 10.1021/ja1014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Peltier J, Stone D, Schaffer DV, Chang CJ. Nox2 redox signaling maintains essential cell populations in the brain. Nature Chemical Biology. 2011;7:106–112. doi: 10.1038/nchembio.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MA, Weijzen S, Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. The EMBO Journal. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi T, Liu W, Shi H, Miyake M, Liu KJ. Generation of hydrogen peroxide during brief oxygen-glucose deprivation induces preconditioning neuronal protection in primary cultured neurons. Journal of Neuroscience Research. 2005;79:816–824. doi: 10.1002/jnr.20402. [DOI] [PubMed] [Google Scholar]
- Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Molecular and Cellular Biology. 2005;25:6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille JJ, Joenje H. Cell culture models for oxidative stress: Superoxide and hydrogen peroxide versus normobaric hyperoxia. Mutation Research. 1992;275:405–414. doi: 10.1016/0921-8734(92)90043-o. [DOI] [PubMed] [Google Scholar]
- Guo H, Aleyasin H, Howard SS, Dickinson BC, Lin VS, Haskew-Layton RE, et al. Two-photon fluorescence imaging of intracellular hydrogen peroxide with chemoselective fluorescent probes. Journal of Biomedical Optics. 2013;18:106002. doi: 10.1117/1.JBO.18.10.106002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskew-Layton RE, Payappilly JB, Smirnova NA, Ma TC, Chan KK, Murphy TH, et al. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17385–17390. doi: 10.1073/pnas.1003996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockberger PE, Skimina TA, Centonze VE, Lavin C, Chu S, Dadras S, et al. Activation of flavin-containing oxidases underlies light-induced production of H2O2 in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6255–6260. doi: 10.1073/pnas.96.11.6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: Linking basic science to clinical practice. Cardiovascular Diabetology. 2005;4:5. doi: 10.1186/1475-2840-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan AC, Dan Y. Dissection of cortical microcircuits by single-neuron stimulation in vivo. Current Biology. 2012;22:1459–1467. doi: 10.1016/j.cub.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, et al. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. The Journal of Experimental Medicine. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. 2011;8:59–71. doi: 10.1016/j.stem.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Khaja S, Velasquez-Castano JC, Dasari M, Sun C, Petros J, et al. In vivo imaging of hydrogen peroxide with chemiluminescent nanoparticles. Nature Materials. 2007;6:765–769. doi: 10.1038/nmat1983. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Alzheimer’s APP mangles mitochondria. Nature Medicine. 2006;12:1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- Lin VS, Dickinson BC, Chang CJ. Boronate-based fluorescent probes: Imaging hydrogen peroxide in living systems. Methods in Enzymology. 2013;526:19–43. doi: 10.1016/B978-0-12-405883-5.00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert AR, Van de Bittner GC, Chang CJ. Boronate oxidation as a bio-orthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Accounts of Chemical Research. 2011;44:793–804. doi: 10.1021/ar200126t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, Kleckner NW, Wyrick S, Dingledine R. Structural requirements for activation of the glycine coagonist site of N-methyl-D-aspartate receptors expressed in xenopus oocytes. Molecular Pharmacology. 1989;36:556–565. [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, et al. Caspase 3 activation is essential for neuroprotection in preconditioning. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EW, Tulyathan O, Isacoff EY, Chang CJ. Molecular imaging of hydrogen peroxide produced for cell signaling. Nature Chemical Biology. 2007;3:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Dave KR, Raval AP. Role of reactive oxygen species and protein kinase C in ischemic tolerance in the brain. Antioxidants & Redox Signaling. 2005;7:1150–1157. doi: 10.1089/ars.2005.7.1150. [DOI] [PubMed] [Google Scholar]
- Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G. Physiological functions of D-amino acid oxidases: From yeast to humans. Cellular and Molecular Life Sciences. 2007;64:1373–1394. doi: 10.1007/s00018-007-6558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR, Baraban JM. Apoptotic death in an in vitro model of neuronal oxidative stress. Clinical and Experimental Pharmacology & Physiology. 1995;22:309–310. doi: 10.1111/j.1440-1681.1995.tb02006.x. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. Journal of Neurochemistry. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. Journal of the American Society of Nephrology. 2003;14:S211–S215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- Rieger S, Sagasti A. Hydrogen peroxide promotes injury-induced peripheral sensory axon regeneration in the zebrafish skin. PLoS Biology. 2011;9:e1000621. doi: 10.1371/journal.pbio.1000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, et al. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2003;23:3597–3606. doi: 10.1523/JNEUROSCI.23-09-03597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakadzic S, Roussakis E, Yaseen MA, Mandeville ET, Srinivasan VJ, Arai K, et al. Two-photon high-resolution measurement of partial pressure of oxygen in cerebral vasculature and tissue. Nature Methods. 2010;7:755–759. doi: 10.1038/nmeth.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. The EMBO Journal. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squirrell JM, Wokosin DL, White JG, Bavister BD. Long-term two-photon fluorescence imaging of mammalian embryos without compromising viability. Nature Biotechnology. 1999;17:763–767. doi: 10.1038/11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegman LD, Zheng H, Neal ER, Ben Yoseph O, Pollegioni L, Pilone MS, et al. Induction of cytotoxic oxidative stress by D-alanine in brain tumor cells expressing rhodotorula gracilis D-amino acid oxidase: A cancer gene therapy strategy. Human Gene Therapy. 1998;9:185–193. doi: 10.1089/hum.1998.9.2-185. [DOI] [PubMed] [Google Scholar]
- Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- Terman JR, Mao T, Pasterkamp RJ, Yu HH, Kolodkin AL. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell. 2002;109:887–900. doi: 10.1016/s0092-8674(02)00794-8. [DOI] [PubMed] [Google Scholar]
- Thompson JW, Narayanan SV, Perez-Pinzon MA. Redox signaling pathways involved in neuronal ischemic preconditioning. Current Neuropharmacology. 2012;10:354–369. doi: 10.2174/157015912804143577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai GE, Yang P, Chang YC, Chong MY. D-alanine added to antipsychotics for the treatment of schizophrenia. Biological Psychiatry. 2006;59:230–234. doi: 10.1016/j.biopsych.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Wu L, Zhang X, Ju H. Highly sensitive flow injection detection of hydrogen peroxide with high throughput using a carbon nanofiber-modified electrode. Analyst. 2007;132:406–408. doi: 10.1039/b701933e. [DOI] [PubMed] [Google Scholar]
- Yin Y, Terauchi Y, Solomon GG, Aizawa S, Rangarajan PN, Yazaki Y, et al. Involvement of p85 in p53-dependent apoptotic response to oxidative stress. Nature. 1998;391:707–710. doi: 10.1038/35648. [DOI] [PubMed] [Google Scholar]
- Zhao BS, Liang Y, Song Y, Zheng C, Hao Z, Chen PR. A highly selective fluorescent probe for visualization of organic hydroperoxides in living cells. Journal of the American Chemical Society. 2010;132:17065–17067. doi: 10.1021/ja1071114. [DOI] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7075–7080. doi: 10.1073/pnas.0832308100. [DOI] [PMC free article] [PubMed] [Google Scholar]