

Abstract
We investigate theoretically the site occupancy of Al3+ in the fast-ion-conducting cubic-garnet Li7–3xAl3+xLa3Zr2O12 (Ia-3d) using density functional theory. By comparing calculated and measured 27Al NMR chemical shifts an analysis shows that Al3+ prefers the tetrahedrally coordinated 24d site and a distorted 4-fold coordinated 96h site. The site energies for Al3+ ions, which are slightly displaced from the exact crystallographic sites (i.e., 24d and 96h), are similar leading to a distribution of slightly different local oxygen coordination environments. Thus, broad 27Al NMR resonances result reflecting the distribution of different isotropic chemical shifts and quadrupole coupling constants. From an energetic point of view, there is evidence that Al3+ could also occupy the 48g site with its almost regular octahedral coordination sphere. Although this has been reported by neutron powder diffraction, the NMR chemical shift calculated for such an Al3+ site has not been observed experimentally.
Introduction
The fast Li-ion conductor with the nominal composition Li7La3Zr2O12 (LLZO) is receiving much scientific attention since its discovery in 2007.1 It has a garnet-based structure, and it occurs in at least two structural modifications.2,3 At room temperature, LLZO is tetragonal (I41/acd) while the cubic modification (Ia-3d) is stable above approximately 150 °C.4 Geiger et al. argued that the better conducting cubic phase can be stabilized at room temperature (RT) through the incorporation of small amounts of Al3+.4 The stabilizing effect of Al3+ has now been confirmed by a number of subsequent investigations.5−19 The exact role Al3+ plays in cubic Al-bearing LLZO is important because LLZO shows a high ionic conductivity of about 10–4 S/cm at RT. This is approximately 2 orders of magnitude higher than that for the lower symmetry Al-free tetragonal LLZO phase. LLZO also has good chemical and thermal stability, as well as a wide energy potential window making it an excellent candidate for use as an electrolyte in an all-solid-state lithium-ion battery.1,20
As has been shown recently, ionic conductivity also seems to depend on the amount of Al3+ incorporated during synthesis.6,8 Further work, however, is needed to quantify this effect. For this purpose, the chemical and physical properties governing Li+ diffusion have to be understood in detail; in particular this includes the important question as to which crystallographic sites the Al3+ ions preferably occupy in the cubic phase of LLZO.
Considerable experimental research has been undertaken to obtain information about Al3+ in LLZO, including its local coordination and site partitioning behavior. In this context, 27Al magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy is a key method. Several NMR studies have proposed that the resonance observed at a chemical shift ranging from 64 to 68 ppm corresponds to Al3+ located at the “standard garnet” tetrahedral site.4−7 There is uncertainty, however, about the interpretation and assignment of the other measured NMR lines that have chemical shifts ranging from approximately 78 to 82 ppm. The interpretations given so far include Al3+ residing at nontetrahedral Li+ sites (4-fold to possibly 6-fold coordinated) and tetrahedrally coordinated sites in the neighborhood of La3+ or Zr4+ vacancies in Al-rich LLZO.4,5 In particular, mechanochemically prepared LLZO samples with a high amount of Al3+, but reduced in La3+ and Zr4+ content, even indicate two magnetically inequivalent tetrahedral sites.6 Neutron powder diffraction (NPD) measurements were interpreted as indicating that Al3+ is located at an octahedrally coordinated site in the garnet framework.11
Summarizing the various published experimental results, there is no clear understanding as to which sites are occupied by Al3+ in cubic LLZO. This also concerns the detailed interpretation of 27Al NMR MAS spectra and diffraction results. To address the important role of Al3+ in LLZO and to obtain a better understanding of the various experimental results, we undertook a density functional theory (DFT) investigation of Al-bearing cubic LLZO with the aim to calculate (relative) 27Al NMR parameters such as chemical shifts and electric quadrupole coupling constants.
Computational Methods
In order to understand the computational models used in this investigation, we provide a short description of the crystal structure of cubic LLZO garnet (Figure 1).
Figure 1.
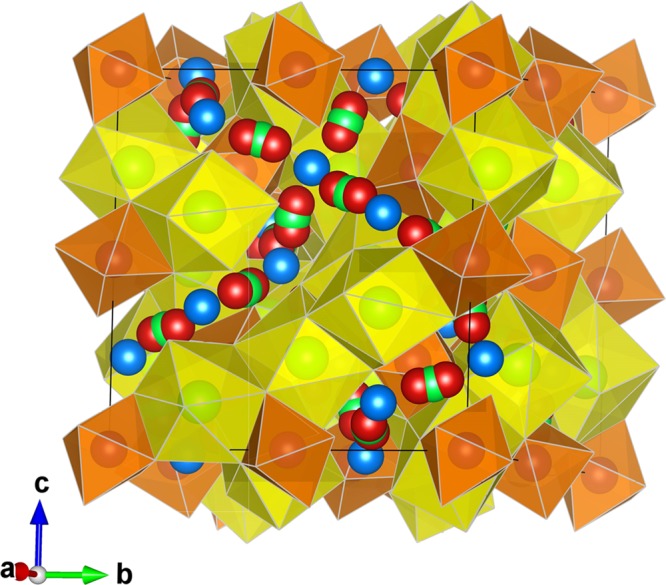
Crystal structure of cubic LLZO. The yellow dodecahedrally coordinate La3+ (at the Wyckoff position 24c) and orange octahedrally coordinate Zr4+ (16a). The blue spheres correspond to tetrahedrally coordinated (24d) Li+, green spheres to octahedrally coordinated (48g) Li+, and red ones to distorted 4-fold coordinated (96h) Li+.
“Garnet” is the common name for a large number of natural and synthetic metal oxide and fluoride phases.22 Conventional oxide garnets have the general formula A3B2C3O12 and crystallize with cubic symmetry Ia-3d. In the case of LLZO, the O2– ions, located at general crystallographic positions, 96h, form an oxygen-ion framework with interstices occupied by the A cations (La3+) at an 8-fold coordinated position 24c (point symmetry 222), by B cations (Zr4+) at a 6-fold coordinated position 16a (point symmetry −3), and by C cations (Li+) at a 4-fold coordinated 24d position (point symmetry −4). In addition to these cation sites, there are other interstices within the oxygen framework that are empty in the conventional garnet structure,23 such as the 6-fold coordinated 48g positions (point symmetry 2) and a general 96h position, sometimes described as 4-fold coordinated with two additional longer bonds greater than 2.8 Å in length,24 5-fold4 coordinated or 6-fold11 coordinated (point symmetry 1). These interstices can be filled by “extra” cations (such as Li+), giving rise to compositions with nonstandard garnet stoichiometry. An important property is the partial occupancy of the structural sites (24d, 48g, and 96h) where Li+ is located and also delocalization of Li+ ions throughout.
Several theoretical DFT studies on LLZO have been published before.25−30 They concentrate on the basic structural properties and on the Li+ diffusion, but none of them considers Al3+ ions explicitly.
For our first-principle calculations, the ions in LLZO were arranged on the basis of crystal structure descriptions in the literature.25 Three different structural models were used with all having a body centered (I-type) Bravais lattice with a = b = c = 12.972 Å and α = β = γ = 90°. Three garnet compositions were chosen, namely Li44Al4La24Zr16O96 with Al3+ at 24d, 48g and 96h, Li56Al4La20Zr16O96 with Al3+ solely at 24c, and Li60Al4La24Zr12O96 with Al3+ solely at 16a. The Li+ ions were distributed over the 24d and the 96h sites following Xu et al.26 No significant effect on the results was observed by choosing a different Li+ arrangement. The highest symmetry possible was maintained to save computational costs. The first model was used to understand the behavior of Al3+ at various possible sites occupied by Li+. The calculations were made with 17 different local Al3+ positions located among the 24d, 96h, and 48g sites. To avoid cation repulsion, all Li+ ions close to Al3+ were removed. The various Al3+ positions were fixed during relaxation and all Al3+ ions are equivalent in the unit cell.
All calculations are based on DFT methods using the all-electron full potential linearized augmented plane wave (LAPW) method as implemented in the Wien2k code.31,32 The Perdew Burke Ernzerhof (PBE) generalized gradient approximation (GGA) was employed.33 The atomic positions were optimized by minimization of the forces (below 2 mRy/au) acting on the atoms simultaneously with the self-consistent-field cycle as implemented by Marks.34 The eigenvalue problem was solved by an “iterative diagonalization” using an efficient preconditioning (inverse of H - λS) and the block-Davidson method.35 The radii of the atomic spheres (RMT) for the Li+, La3+, Zr4+, O2-, and Al3+ ions were chosen to be 1.46, 2.27, 1.93, 1.71, and 1.40 au, respectively. The cutoff for the plane wave RMTKmax = 6.0 and the maximum Fourier expansion of charge density cutoff Gmax = 12 (au)−1 were applied. The separation parameter between the valence and core states was chosen to be −6.0 Ry. We used 1 k-points in the irreducible Brillouin zone. The computational considerations were checked by increasing RMTKmax and the number of k-points, but no significant changes with respect to the energy, geometry, and the electric field gradient (EFG) around Al3+ were observed.
The behavior of Al3+ at different crystallographic sites was analyzed qualitatively by weighing the various Al3+ positions, xi, with their corresponding energy, E(xi). The various local energies for Al3+ at and around the crystallographic positions where Li can also be located, were described by fitting a polynomial, describing a possible diffusion pathway. The ocuppation probabilities for an Al3+ ion along this path are described using a normalized Boltzmann factor given by
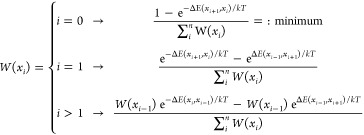 |
where k is Boltzmann’s constant, and where xi+1 indicates the states next to xi, xi–1 indicates the states before xi with an interval of 0.01 Å, and T is 298.15 K. The site preference energy, ΔE, is defined as ΔE = E(xi) – Eglobal, where Eglobal is the global energy minimum and corresponds to Al3+ located at 24d. It has the lowest total energy for all the calculated arrangements of Al in LLZO.
In NMR spectroscopy, the chemical shift, δ, of a nucleus, i, describes the nuclear shielding effect of an applied (external) static magnetic field and the locally induced magnetic field arising from the surrounding electrons with a certain probability of presence near the corresponding nuclear site i. The magnitude of the resulting effective field, Beff, is given by Beff = B0(1 – σi), where σi is a second-rank nuclear shielding tensor, 1 is the unit matrix and B0 is a uniform external field along the z-axis. The resonance NMR frequency, νi, is then given by vi = (γi/2π) Beff, where γi is the magnetogyric ratio of the nucleus under observation. The isotropic chemical shift, δiso (henceforth δ), describes the relation between the NMR resonance frequency for the nucleus of interest, νi, and the corresponding resonance frequency for a reference compound, νref, giving δ = 106 × (vi – vref)/vref. Because of the orientation in systems with periodic boundary conditions, all anisotropic interactions causing line broadening are projected onto the axis of rotation where they collapse at the magic angle.36 To compute δ, we used the relaxed geometry and applied the NMR module37−39 of the Wien2k code.32 These chemical shift calculations are based on an all-electron linear response method where one obtains the induced current density considering the perturbation of the ground state wave functions due to the external magnetic field. The resulting magnetic shielding is then obtained by integration of the all-electron current according to Biot–Savart’s law without further approximations. Because there is agreement in the literature4−7 that the 27Al NMR resonance at 66 ± 2 ppm represents tetrahedrally coordinated Al3+ at 24d in LLZO (δ24d = 68 ppm), we calculate the difference of the chemical shifts (Δδ) for Al3+ at other positions xi with a δxi as follows
Besides the chemical shift, the interaction of the quadrupole moment, Q, of the 27Al nucleus (spin-quantum number I = 5/2) with a sufficiently large electric field gradient (EFG), which is produced by a nonspherical charge distribution around the nuclear site, also affects the central NMR transition. Second order quadrupolar effects, influencing the shape of the NMR central line, cannot be (completely) eliminated by magic angle spinning. From the simulation of spectra recorded under MAS conditions, the quadrupole coupling constant
and the corresponding asymmetry parameter
can be estimated. Here, e is the positive elementary charge, h denotes Planck’s constant, and Vxx , Vyy and Vzz are the elements of the traceless EFG tensor V with |Vzz| ≥ |Vxx| ≥ |Vyy|. Vzz = eq denotes its principal component. The shape of the NMR central line depends sensitively on the asymmetry of the electronic charge density close to the nucleus. The parameter η describes the deviation of the EFG from axial symmetry and it can take values between 0 and 1. Second order quadrupole interactions also affect the position of the NMR line in terms of a quadrupolar shift. A larger external magnetic field, however, lowers the effect. Here, we compute the EFG from the all-electron charge distribution without further approximation and CQ is obtained using the nuclear quadrupole moment Q(27Al) = 1.616 × 10–29 m2 determined from the slope of the linear regression proposed by Body et al.40 The data visualization were performed using the program VESTA.41
Results
Crystal Chemistry of Al-bearing LLZO and Al3+ Partitioning Behavior
The most energetically favorable position of Al3+ in LLZO is at the tetrahedrally coordinated crystallographic special 24d position. Al3+ can be displaced toward a neighboring vacant general 96h site, which leads to a distortion of its oxygen coordination polyhedron. Calculated Al–O bond lengths for this coordination at 0 K are between 1.76 Å and 1.79 Å and the tetrahedral volume is 2.86 Å3 (see Table 1).
Table 1. Various Calculated Interatomic Al–O Distances (d) [Å] and Coordination Polyhedra Volumes (V) [Å3] in Cubic Li7La3Zr2O12.
| 24d | 96h | 48g | 16a | 24c | |
|---|---|---|---|---|---|
| Al–O | Al–O | Al–O | Al–O | Al–O | |
| d | 1.76 | 1.76 | 2.12 | 1.90 | 1.95 |
| d | 1.78 | 1.92 | 1.93 | 2.11 | 1.95 |
| d | 1.78 | 1.84 | 1.93 | 2.03 | 1.94 |
| d | 1.79 | 1.82 | 2.00 | 1.90 | 1.94 |
| d | 2.90 | 2.00 | 2.03 | 2.82 | |
| d | 3.15 | 2.12 | 2.11 | 2.82 | |
| d | 2.92 | ||||
| d | 2.92 | ||||
| ⟨d⟩ | 1.78 | 1.83a | 2.02 | 2.01 | 1.94a |
| V | 2.86 | 2.69a | 10.67 | 10.69 | 22.86b |
Based on 4-fold coordination.
Based on 8-fold coordination.
Our calculations show that Al3+ at 96h is 4-fold coordinated, and distorted from tetrahedral coordination, with Al–O bond lengths varying from 1.76 to 1.92 Å, because of the displacement of Al3+ towards a vacant 24d site. Additionally, there are two O2– ions further away that have on average approximately 1 Å longer bond distances. If they would be included in the local coordination sphere around Al3+, a distorted octahedral coordination polyhedron with a volume of 16.33 Å3 would result. The volume for the 4-fold coordinated polyhedron is 2.69 Å3. Al3+ could also possibly be located at the special 48g site with three different pairs of Al–O bonds and calculated bond lengths of 1.93 Å, 2.00 Å and 2.12 Å, respectively, yielding a coordination volume of 10.67 Å3. Any possible Al3+ at the 16a site would also be 6-fold coordinated with again three pairs of Al–O bonds with lengths of 1.90 Å, 2.03 Å, and 2.11 Å and a coordination volume of 10.69 Å3. Al3+ at the 24c site would have four shorter Al–O bond lengths equal to 1.94 Å and four longer bonds of 2.84 Å length. This would give rise to a quasi-4-fold planar coordination for Al3+. Assuming, on the other hand, 8-fold coordination around the 24c site, the resulting polyhedron would have a volume of 22.86 Å3. The various hypothetical Al3+ coordination polyhedra at 0 K, along with their associated site preference energy, are shown in Figure 2.
Figure 2.
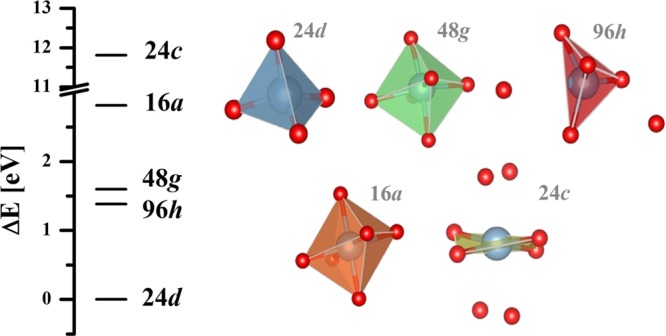
Various coordination polyhedral around Al3+ located at the 24d, 48g, 96h, 16a, and 24c sites. (Nonbonded red spheres indicate additional O2– ions not considered as next nearest neighbors in the first coordination sphere. Their distances are given in Table 1.) Their associated site preference energies in cubic LLZO.
The calculated site preference energies, ΔE, for Al3+ in LLZO are as follows: 24d > 96h > 48g > 16a ≫ 24c. Al3+ at the 24d site has the global energy minimum and is 1.38 eV more stable than Al3+ at 96h and 1.60 eV more stable than Al3+ at 48g. The latter two sites are energetically similar with an energy difference of just 0.22 eV. The site preference energy for Al3+ at 16a and 24c is 0.41 and 1.74 eV higher, respectively, when referenced to the global energy minimum. The site preference energies for Al3+ at various structural positions between 48g and 24d are shown in Figure 3.
Figure 3.

Site preference energies, ΔE, for Al3+ at various structural positions (gray crosses) at and near the 48g, 96h and 24d sites in LLZO garnet. The solid lines represent polynomial fits to the various ΔE values. The fits were used to calculate the Boltzmann distribution, W(xi) (see text), discussed below. The different distributions of Al3+ at the various crystallographic sites are normalized to 1 and indicate the room temperature probability of finding Al3+ at a distinct position.
Calculated 27Al NMR Parameters
Calculated 27Al NMR parameters for Al3+ at the different structural sites in LLZO are shown in Table 2. The corresponding polyhedra (Figure 4) around Al3+, slightly displaced from 24d and 96h (Figure 3), were used to analyze the experimental NMR spectra. The calculations show that Al3+ coordinations given by 1 to 5 (Figure 4) have Δδ values between −3.7 and 0.2 ppm, CQ values between −1.38 and 5.01 MHz and η values between 0.20 and 0.70. Al3+ for coordinations given by 9 to 13 have Δδ values between 10.0 and 14.4 ppm, CQ values between 2.4 and 3.3 MHz and η values between 0.24 and 0.70. On the other hand, Al3+ located at 48g has an extremely large calculated Δδ value of 133.5 ppm, 118.5 ppm at 24c, and 108.5 ppm at 16a. The latter three sites have δ values that are much larger than any experimentally observed NMR resonances.
Table 2. Calculated 27Al NMR Parameters for Al3+ at Various Sites in Cubic Li7La3Zr2O12 Compared to Experimental Values from the Literature.
| positiona | δb [ppm] | Δδc [ppm] | VZZd [1021 V/m2] | CQe [MHz] | ηf | assignment | |
|---|---|---|---|---|---|---|---|
| Geiger et al.4 | 68.0 | 13.0 | 5.0–5.2 | 0.0–0.1 | 24d | ||
| 81.0 | 3.3 | 0.7 | 96h | ||||
| Buschmann et al.5 and Düvel et al.6 | 64.0 | 14.0 | 24d | ||||
| 78.0 | 18.0 | ||||||
| 82.0 | |||||||
| Hubaud et al.7 | 68.4 | 4.8 | 0.40 | 24d | |||
| 73.5 | 5.1 | 6.9 | 0.13 | 24d | |||
| Our calculations | 1 | 68.0 | –1.38 | –0.8 | 0.10 | 24d | |
| 2 | 68.2 | 0.2 | 2.14 | 1.3 | 0.94 | ||
| 3 | 64.3 | –3.7 | 3.03 | 1.8 | 0.79 | ||
| 4 | 64.5 | –3.5 | 3.97 | 2.3 | 0.71 | ||
| 5 | 64.8 | –3.2 | 5.01 | 2.9 | 0.66 | ||
| 13 | 79.9 | 11.9 | 4.03 | 2.4 | 0.28 | ||
| 12 | 78.0 | 10.0 | 4.08 | 2.4 | 0.24 | 96h | |
| 11 | 79.4 | 11.4 | 4.63 | 2.7 | 0.45 | ||
| 10 | 82.4 | 14.4 | 5.03 | 3.0 | 0.54 | ||
| 9 | 82.2 | 14.2 | 5.63 | 3.3 | 0.70 |
The position corresponds to Al3+ with the coordination polyhedra in Figure 4 and the site preference energies in Figure 3.
Calculated values are referenced to the global minimum given by δ = 68 ppm at position 1.
Literature: Δδ = δ – δ24d; Calculation: Δδ = δ – 68 ppm.
Principal component of the EFG.
Quadrupole coupling constant.
Asymmetry parameter.
Figure 4.

Oxygen coordination polyhedra around Al3+ for Al–O distances less than 2.6 Å at 17 local positions starting with 24d (1) and going to 96h (12) and beyond and ending at 48g (20) in cubic LLZO.
Discussion
In this DFT study, we investigated the crystal-chemical role of Al3+ in LLZO. Specifically, we address the question of the coordination and the site distribution behavior of Al3+. The results of the calculations can be used, together with published experimental results, to achieve a better understanding of the nature of Al3+ incorporation in LLZO. We find that Al3+ at the 24d site is energetically the most stable state, followed by Al3+ at 96h and 48g, whereby the site preference energies for the latter two are similar. Al3+ at both the 16a and 24c sites is energetically unfavorable. Based on our DFT calculations, Al3+ should be exclusively located at 24d at 0 K, which would give rise to a single NMR resonance. Thus, the question arises why additional resonances are observed in the 27Al MAS NMR spectra (Figure 5).4−7
Figure 5.

Experimental 27Al NMR MAS spectra4−7 compared to calculated δ values. The gray areas represent the variation in chemical shift values, Δδ, (13 to 18 ppm) in literature. The reference point refers to Δδ = 0 given by the calculations.
Before discussing our results, we briefly outline the NMR results from the various studies with respect to the Al3+ site occupation in LLZO. In all recent studies the resonance at approximately 66 ± 2 ppm is assigned to Al3+ at the 24d site.4−7 Other NMR lines also indicate 4-fold coordinated Al3+ ions,4−7 because their chemical shift values of 78 to 82 ppm are usually indicative of tetrahedral coordination. Geiger et al. assigned the resonance at 81 ppm to Al3+ residing at the 96h sites.4 In particular, the spectra presented in ref (6) indicate that several NMR lines contribute to a signal occurring between 75 to 85 ppm. It should be noted that the latter has been reported for LLZO samples with a high amount of Al3+ and lower than stoichiometric La3+ and Zr4+ contents.6 Such samples were prepared by mechanochemical activation combined with subsequent annealing at moderate temperatures.
Düvel et al. observed a stronger NMR signal at 64 ppm and a less intense line at 78 ppm for garnets with Al3+ contents up to 0.30 per formula unit (pfu). However, at concentrations above 0.60 Al3+ pfu the resonance at 78 ppm becomes the most intense. At even higher Al3+ contents a new NMR line at 81 ppm emerges, which notably appears in La- and Zr-deficient LLZO samples.
Geiger et al. made, in addition to 27Al MAS NMR experiments, also 27Al MQMAS NMR measurements.4 The latter spectra show a resonance at δ = 81 ppm, a coupling constant, CQ, of 3.3 MHz and an asymmetry parameter, η, of 0.7. These parameters indicate a strongly distorted local coordination geometry. The second resonance observed at δ = 68 ppm shows a stronger quadrupolar interaction with CQ = 5.0 to 5.2 MHz and a lower η value between 0.0 and 0.1 that suggests axial symmetry at the site. It should be noted that the 27Al MAS NMR spectrum was recorded at 156.26 MHz, thus providing a sufficiently high resolution to simulate its line shape. Such a simulation yields a resonance with δ = 81 ppm, CQ = 3.3 MHz and η = 0.7, and for the second signal values of δ = 70 ppm, CQ = 5.5 MHz and η = 0.5.
The calculated NMR parameters (Table 2) are in good agreement with an assignment of Al3+ to 96h for the resonance at 81 ppm. The calculated NMR parameters for the NMR line at 68 ppm that is assigned to Al3+ at 24d, however, disagree with those determined from the MQMAS NMR experiments. For the latter, it has been proposed that possibly more than one Al3+ site is reflected by this broad resonance. This possibility was underlined by Hubaud et al.7 They made 27Al MQMAS NMR spectroscopic measurements to investigate this resonance at 68 ppm for an Al-doped LLZO garnet synthesized at 850 °C. Their line shape analysis yielded two NMR lines with the first having δ = 68.4 ppm and CQ = 4.8 MHz and the second δ = 73.5 ppm and CQ = 6.9 MHz. Both resonances were assigned to tetrahedrally coordinated Al3+ at 24d in two different garnet phases. The two tetrahedral sites have slightly different distortions and thus different δ and CQ values. Hubaud et al. report that this interpretation is consistent with their high-resolution XRPD results, and they proposed that slow Al3+ diffusion within the lattice is responsible for the disordering over the two sites.
Our DFT calculations permit a somewhat different interpretation of the experimental results. For the first step, we calculated the NMR parameters for Al3+ at all crystallographic sites, via 24d, 48g, 96h, 16a, and 24c. According to our results, the resonance at 80 ± 2 ppm should be assigned to Al3+ residing at the 96h sites and the resonance at 66 ± 2 ppm to Al3+ occupying 24d. These results agree with interpretations of experimental NMR spectra.4−6 There is good agreement between the calculated CQ value for Al3+ at 96h, but disagreement between CQ values for Al at 24d. Hubaud et al. proposed a large CQ value for Al3+ at 24d, but it cannot be easily explained why the symmetric 24d site should yield a CQ value approximately two times larger than Al3+ at the more distorted 96h site.7
To understand this issue better, we calculated the NMR parameters for Al3+ at all positions at and around 24d and 96h that are occupied by a certain probability. Shifting Al3+ away from 24d and 96h leads to a distribution of slightly different local oxygen coordination environments. Thereby, broad 27Al MAS NMR resonances could result, which would reflect a distribution of slightly different δ and CQ values. We note that our calculations for η for Al3+ at 24d do not agree with experiment, but η was obtained from the experimental spectra by using only one4 or two7 Al3+ resonances to simulate this broad feature at 66 ± 2 ppm.
The NMR signal located at 78 to 82 ppm is also asymmetric in shape. This is possibly due to the slightly different geometries of local Al3+ coordinations, as given by their calculated probabilities, toward the 48g minimum versus those in the direction of 24d. This situation could produce two overlapping resonances having similar values of δ and CQ (see also the agreement with experimental results and the discussion in ref (6)).
Although experiment and calculations are in broad agreement in terms of the crystal chemical role of Al3+ in LLZO, it is not clear based on the DFT calculations alone, why Al3+ is not located exclusively at 24d6 as suggested by the site energies. Al3+ at 96h and 48g must also be considered, which, however, would lead to an additional NMR line which is usually not observed experimentally. (For the sake of completeness, let us note that an additional NMR line has been observed at 93 ppm for Al-doped Li6.5La2.5Ba0.5ZrTaO12 [42] and for some of the Al-doped LLZO samples prepared via mechanosynthesis [6]; the prominent NMR line at 65 ppm, however, is absent in these cases.) Here, it must be stated that the exact thermodynamic state of LLZO, as obtained in the various sintering experiments, is not known. It is possible that metastable structural states are obtained that can depend on a number of experimental factors (dopant concentration, sintering temperature and time, heating rate, grain sizes, starting materials, etc.). Thus, Al3+ could potentially be incorporated metastably at 96h and 48g as well in cubic LLZO, because both sites have similar site energies.
The different site energies for Al3+ at 24d and 96h could provide an explanation for differences in published NMR spectra of LLZO. The relationship between Al3+ concentration and variations in the intensities of the different resonances (and thus Al3+ site occupancies), as observed by Düvel et al., can be interpreted crystal chemically. When an Al3+ ion is located at 24d, because of its large effective charge radius, it could create a larger inaccessible region around 24d compared to the situation for Al3+ at 96h. This region cannot be occupied by other ions. This leads to a reduction of entropy and a loss of energy possibly making the 96h site energetically more accessible for Al3+ with increased Al3+ contents in contrast to the 24d site. Similar considerations were made by Bernstein et al.37 They showed by using molecular dynamics simulations that the introduction of vacancies in LLZO does indeed reduce the free energy. This indirectly supports our suggestion.
Lastly, we consider the coordination geometry around the 96h site in LLZO, which has been described differently.4,11,24 It is, of course, a matter of definition as to what constitutes a bond in a first coordination sphere. According to Li et al. in their description of Al at 96h, the average Al–O bond distance is d[4] = 2.08 Å, with a difference of 0.38 Å between the length of the shortest and the longest bonds. There are two further O2– ions that give Al–O = 2.69 Å and 2.80 Å, thus being 0.46 Å and 0.57 Å longer compared to the longest bond in strict 4-fold coordination. Based on our calculations, Al3+ at 96h has a locally 4-fold distorted coordination (Figure 4). It is worth mentioning that long-range structural properties, as determined by the diffraction experiment, such as neutron powder diffraction, can differ from those measured via spectroscopy, for example, NMR, which probes structure at shorter length scales.
Conclusion
Based on our DFT results, we propose that Al3+ could have a number of slightly different local 4-fold coordinations around the crystallographic 24d and 96h sites in cubic LLZO garnet. The calculations are in general agreement with published experimental 27Al NMR spectra. It should be noted further that Li et al.,11 in a neutron diffraction study of cubic LLZO garnet, proposed that Al3+ is located at the 48g site in octahedral coordination. In terms of calculated site energies, this site could be partially occupied. However, the calculated Al NMR chemical shift value for such coordination has not been observed in experimental NMR spectra.
Acknowledgments
We thank M. Grodzicki (University Salzburg) for discussions and the CHPC Salzburg, especially Peter Zinterhof junior (University of Salzburg), and the HPC-EUROPA2 project (project number: 1212) for support within the European Commission Capacities Area, Research Infrastructures Initiative. This research was made possible by the Austrian Science Fund (FWF) through project number P25702. M.W. thanks the Austrian Federal Ministry of Economy, Family and Youth, and the Austrian National Foundation for Research, Technology, and Development for financial support. Thanks also go to John T. Vaughey and Baris Key (Argonne National Laboratory) for providing their NMR spectra (ref (7)).
The authors declare no competing financial interest.
References
- Murugan R.; Thangadurai V.; Weppner W. Angew. Chem. 2007, 119, 7925. [DOI] [PubMed] [Google Scholar]
- Awaka J.; Kijima N.; Hayakawa H.; Akimoto J. J. Solid State Chem. 2009, 182, 2046. [Google Scholar]
- Awaka J.; Takashima A.; Hayakawa H.; Kijima N.; Idemoto Y.; Akimoto J. Key Eng. Mater. 2011, 485, 99. [Google Scholar]
- Geiger C. A.; Alekseev E.; Lazic B.; Fisch M.; Armbruster T.; Langner R.; Fechtelkord M.; Kim N.; Pettke T.; Weppner W. Inorg. Chem. 2011, 50, 1089. [DOI] [PubMed] [Google Scholar]
- Buschmann H.; Dölle J.; Berendts S.; Kuhn A.; Bottke P.; Wilkening M.; Heitjans P.; Senyshyn A.; Ehrenberg H.; Lotnyk A.; Duppel V.; Kienle L.; Janek J. Phys. Chem. Chem. Phys. 2011, 13, 19378. [DOI] [PubMed] [Google Scholar]
- Düvel A.; Kuhn A.; Robben L.; Wilkening M.; Heitjans P. J. Phys. Chem. C 2012, 116, 15192. [Google Scholar]
- Hubaud A. A.; Schroeder D. J.; Key B.; Ingram B. J.; Dogan F.; Vaughey J. T. J. Mater. Chem. A 2013, 1, 8813. [Google Scholar]
- Rangasamy E.; Wolfenstine J.; Sakamoto J. Solid State Ionics 2012, 206, 28. [Google Scholar]
- Kanamura K.; Kaeriyama A.; Honda A.; Yoshida T.; Sato Y. US patent 20110053000, 2011.
- Kotobuki M.; Kanamura K.; Sato Y.; Yoshida T. J. Power Sources 2011, 196, 7750. [Google Scholar]
- Li Y.; Han J.-T.; Wang C.-A.; Vogel S. C.; Xie H.; Xu M.; Goodenough J. B. J. Power Sources 2012, 209, 278. [Google Scholar]
- Kuhn A.; Choi J.-Y.; Robben L.; Tietz F.; Wilkening M.; Heitjans P. Z. Phys. Chem. 2012, 226, 525. [Google Scholar]
- Jin Y.; McGinn P. J. Electrochim. Acta 2013, 89, 407. [Google Scholar]
- Jin Y.; McGinn P. J. J. Power Sources 2011, 196, 8683. [Google Scholar]
- Wolfenstine J.; Sakamoto J.; Allen J. L. J. Mater. Sci. 2012, 47, 4428. [Google Scholar]
- Tietz F.; Wegener T.; Gerhards M. T.; Giarola M.; Mariotto G. Solid State Ionics 2013, 230, 77. [Google Scholar]
- Matsui M.; Takahashi K.; Sakamoto K.; Hirano A.; Takeda Y.; Yamamoto O.; Imanishi N. Dalton Trans. 2014, 43, 1019. [DOI] [PubMed] [Google Scholar]
- Cheng L.; Park J. S.; Hou H.; Zorba V.; Chen G.; Richardson T.; Cabana J.; Russo R.; Doeff M. J. Mater. Chem. A 2014, 2, 172. [Google Scholar]
- Sudo R.; Nakata K.; Ishiguro K.; Matsui M.; Hirano A.; Takeda Y.; Yamamoto O.; Imanishi N.. Solid State Ionics 2013, article in press
- Cussen E. J. Mater. Chem. 2010, 20, 5167. [Google Scholar]
- Menzer G. Z. Kristallogr. 1928, 69, 300. [Google Scholar]
- Hellner E.; Gerlich R.; Koch E.; Fischer W.. Physics Data; Fachinformationszentrum Energie, Physik, Mathematik: Karlsruhe, Germany, 1979; 16–1: 1–16. [Google Scholar]
- Percival J.; Kendrick E.; Smith R. I.; Slater P. R. Dalton Trans. 2009, 26, 5177. [DOI] [PubMed] [Google Scholar]
- Xie H.; Alonso J. A.; Li Y.; Fern M. T.; Goodenough J. B Chem. Mater. 2011, 23, 3587. [Google Scholar]
- Xu M.; Park M. S.; Lee J. M.; Kim T. Y.; Park Y. S.; Ma E. Phys. Rev. B 2012, 85, 052301. [Google Scholar]
- Bernstein N.; Johannes M. D.; Hoang K. Phys. Rev. Lett. 2012, 109, 205702. [DOI] [PubMed] [Google Scholar]
- Nakayama M.; Kotobuki M.; Munakata H.; Nogami M.; Masayuki K.; Kanamura K. Phys. Chem. Chem. Phys. 2012, 14, 10008. [DOI] [PubMed] [Google Scholar]
- Jalem R.; Yamamoto Y.; Shibba H.; Nakayama M.; Munakata H.; Kasuga T.; Kanamura K. Chem. Mater. 2013, 25, 425. [Google Scholar]
- Miara J. L.; Ong S. P.; Mo Y.; Richards W. D.; Park Y.; Lee J.-M.; Lee H. S.; Ceder G. Chem. Mater. 2013, 25, 3048. [Google Scholar]
- Madsen G. K. H.; Blaha P.; Schwarz K.; Sjöstedt E.; Nordström L. Phys. Rev. B 2001, 64, 195134. [Google Scholar]
- Blaha P.; Schwarz K.; Madsen G. K. H.; Kvasnicka D.; Luitz J.. WIEN2K, Version 13.1; An Augmented Plane Wave + Local Orbital Program for Calculating Crystal Properties; Technical University: Vienna, Austria, 2001. [Google Scholar]
- Perdew J . P.; Burke K.; Ernzerhof M. Phys. Rev. Lett. 1996, 77, 3865. [DOI] [PubMed] [Google Scholar]
- Marks L. D. J. Chem. Theor. Comput. 2013, 9, 2786. [DOI] [PubMed] [Google Scholar]
- Blaha P.; Hofstätter H.; Koch R.; Laskowski R.; Schwarz K. J. Comput. Phys. 2009, 229, 453. [Google Scholar]
- Maricq M. M.; Waugh J. S. J. Chem. Phys. 1979, 70, 33003316. [Google Scholar]
- Laskowski R.; Blaha P. Phys. Rev. B 2012, 85, 035132. [Google Scholar]
- Laskowski R.; Blaha P.; Tran F. Phys. Rev. B 2013, 87, 195130. [Google Scholar]
- Laskowski R.; Blaha P. Phys. Rev. B 2014, 89, 014402. [Google Scholar]
- Body M.; Legein C.; Buzare J.-Y.; Silly G.; Blaha P.; Martineau C.; Calvayrac F. J. Phys. Chem. A 2007, 111, 1187. [DOI] [PubMed] [Google Scholar]
- Momma K.; Izumi F. J. Appl. Crystallogr. 2011, 44, 1272. [Google Scholar]
- Narayanan S.; Epp V.; Wilkening M.; Thangadurai V. RSC Adv. 2012, 2, 2553. [DOI] [PubMed] [Google Scholar]