

Abstract
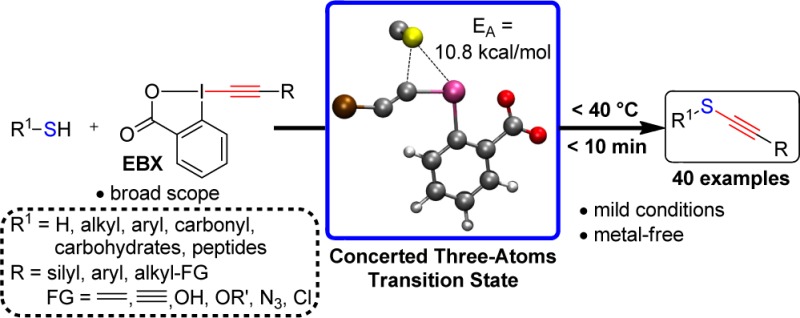
Among all functional groups, alkynes occupy a privileged position in synthetic and medicinal chemistry, chemical biology, and materials science. Thioalkynes, in particular, are highly useful, as they combine the enhanced reactivity of the triple bond with a sulfur atom frequently encountered in bioactive compounds and materials. Nevertheless, general methods to access these compounds are lacking. In this article, we describe the mechanism and full scope of the alkynylation of thiols using ethynyl benziodoxolone (EBX) hypervalent iodine reagents. Computations led to the discovery of a new, three-atom concerted transition state with a very low energy barrier, which rationalizes the high reaction rate. On the basis of this result, the scope of the reaction was extended to the synthesis of aryl- and alkyl-substituted alkynes containing a broad range of functional groups. New sulfur nucleophiles such as thioglycosides, thioacids, and sodium hydrogen sulfide were also alkynylated successfully to lead to the most general and practical method yet reported for the synthesis of thioalkynes.
Introduction
One of the most important tasks of organic chemistry is discovering practical and general methods for introducing functional groups into molecules to modify their properties and to serve as a platform for further modifications. At a time when research in neighboring fields such as medicine, biology, or materials science is becoming increasingly molecular, easy-to-perform transformative reactions that do not require highly specialized synthetic skills have a particularly broad impact. Among all functional groups in organic chemistry, alkynes occupy a privileged position in this respect.1 Despite being one of the simplest functional groups with only two carbon atoms, the reactivity of the triple bond makes alkynes exceptionally useful in organic chemistry. They have found applications in bulk chemical synthesis based on acetylene gas2 as well as in fine chemistry for the stereoselective construction of the carbon backbone of complex natural products3 and in a myriad of complexity-enhancing metal-catalyzed cyclization reactions to access carbo- and heterocycles.4 Most importantly, the utility of alkynes has now crossed the boundaries of organic chemistry: their electronic properties have led to widespread applications in organic materials and dyes5 and the [3 + 2] cycloaddition with azides is now recognized as one of the best biorthogonal conjugation method to modify biomolecules and polymers.6 The latter transformation is particularly representative of how discoveries in fundamental organic reactivity can strongly impact neighboring research areas. When considering the importance of alkynes for progress in numerous fields of molecular sciences, the development of new methods to access them efficiently under user-friendly conditions is highly desirable.
Among the different classes of alkynes, those directly substituted by a heteroatom are especially interesting for two reasons:7 (1) the electron-rich heteroatom makes the triple bond more reactive, allowing new chemical transformations and (2) they constitute value-added building blocks, as heteroatoms are essential for the physical and biological properties of small molecules. In short, they bring together the new properties conferred by heteroatoms with the exceptionally rich chemistry of alkynes (Scheme 1). Nevertheless, the synthetic potential of heteroatom-substituted alkynes has long remained underdeveloped due to the absence of convenient methods to access these often sensitive compounds. The coupling of heteroatoms with acetylides is indeed not favorable, as both fragments are inherently nucleophilic and an Umpolung of the reactivity is required.
Scheme 1. Heteroatoms-Substituted Alkynes: The Best of Two Worlds, but How To Access Them?
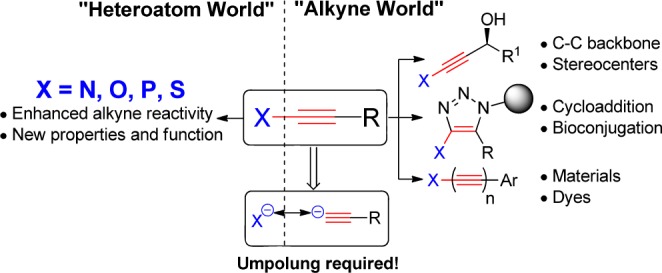
In the specific case of nitrogen-substituted alkynes (ynamines and ynamides), however, the situation has changed dramatically in the last two decades, when new synthetic methods for the alkynylation of nitrogen with either hypervalent iodine reagents or terminal and halogeno alkynes in the presence of copper catalysts were developed.8 Ynamides in particular are now intensively used in modern synthetic chemistry.7c,7d In contrast, the chemistry of thioalkynes is still underdeveloped. This is surprising considering the importance of sulfur in drugs, materials and biomolecules.9 In fact, the high nucleophilicity of thiols has led to the development of very efficient methods for the formation of S–S (oxidative disulfide formation), S–C(sp3) (alkylation, thiol–ene) and S–C(sp2) (thiol–yne) bonds.10 All of these reactions are routinely used for important transformations in chemical biology and materials science. Until very recently, the direct synthesis of thioalkynes from thiols, on the other hand, has been limited to addition–elimination reactions on alkynyl or alkenyl halides under strongly basic conditions (Scheme 2).11 The most commonly used methods permitting access have been based on the reaction of terminal alkynes with activated sulfur derivatives bearing a leaving group, such as chloride, tosyl or cyano, or disulfides.12 This lack of efficient synthetic methods under mild conditions has clearly limited the applications of potentially very useful thioalkynes in synthetic and medicinal chemistry.
Scheme 2. Method To Access Thioalkynes Based on Umpolung of Sulfur or Alkyne.
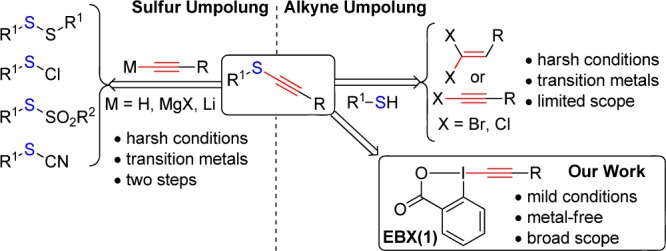
In 2013, the first efficient metal-catalyzed examples of alkynylation of thiols appeared using copper, palladium or nickel catalysts.13 Nevertheless, these transformations still required the use of a transition metal catalyst, and the scope of thiols described in these works remained limited to very simple thiophenols and aliphatic thiols13a or to thioglycosides,13b,13c respectively. The same year, our group reported the first method for the alkynylation of thiols using 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX, 1a, R = SiiPr3).14 In contrast to previously published methods, the reaction was efficient and user-friendly, leading to the complete alkynylation of aromatic and aliphatic thiols in less than 1 min at room temperature in an open flask. Furthermore, selective alkynylation of thiols was possible in the presence of numerous functional groups such as halogens, alcohols, carboxylic acids, electron-rich aromatic groups or free amines. Nevertheless, two aspects of the developed methodology were not fully satisfying: (1) The mechanistic basis of the extreme efficiency of the reaction could not be rationalized. An in-depth understanding of the alkynylation would be highly useful for further development. (2) The reaction was limited to the transfer of silyl acetylenes on thiols as nucleophiles. Although we demonstrated that the obtained products could easily be deprotected and functionalized, this made our approach less convergent and attractive if the introduction of functionalized alkyne groups is desired. Furthermore, the alkynylation of other sulfur nucleophiles such as thioacetals, thioacids or sulfides salts would also be important in extending the range of accessible thioalkynes.
In this article, we address both issues. On the basis of the isolated side products and computational studies, we propose a mechanism for the reaction. In particular, computations showed that a concerted transition state between the deprotonated thiol and the iodine reagent was possible, leading to an exceptionally low (10.8 kcal/mol) activation energy for the alkynylation using silylated EBX reagents. This type of mechanism has never been proposed for the alkynylation of nucleophiles using hypervalent iodine reagents and is expected to change the way many researchers think about these transformations. We then report an extension of the alkynylation reaction to the use of aryl and functionalized alkyl acetylenes and further extend the scope of thiols to thioglycosides, thioacids and sufide salts, resulting in the most general and practical thiol alkynylation method reported to date.
Results and Discussion
Mechanism and Computational Studies
Investigating the reaction mechanism of the alkynylation with TIPS-EBX (1a) is particularly challenging because of the fast rate: even 10 s after addition of the reagent, full conversion of thiol 2 was already observed when using 1,1,3,3-tetramethyl guanidine (TMG) as a base (eq 1). Furthermore, low-temperature experiments are difficult because of low solubility of the hypervalent iodine reagent below 0 °C. Nevertheless, control experiments showed that no or very little alkynylation was observed in the absence of a base. In this case, oxidative dimerization to form disulfide 4 was the major process. In addition, no reaction or interaction was observed by NMR between reagent 1a and TMG. These results permit the reasonable assumption that deprotonation of thiol 2 to form thiolate 2′ is required for the reaction to occur (Scheme 3).
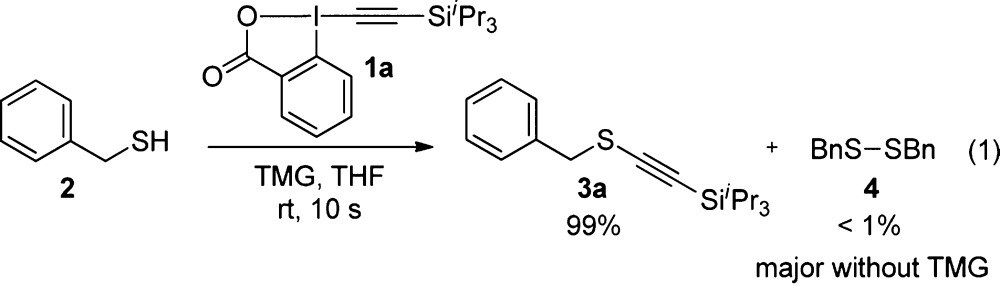 |
1 |
Scheme 3. Initial Speculative Mechanism for the Alkynylation Reaction.
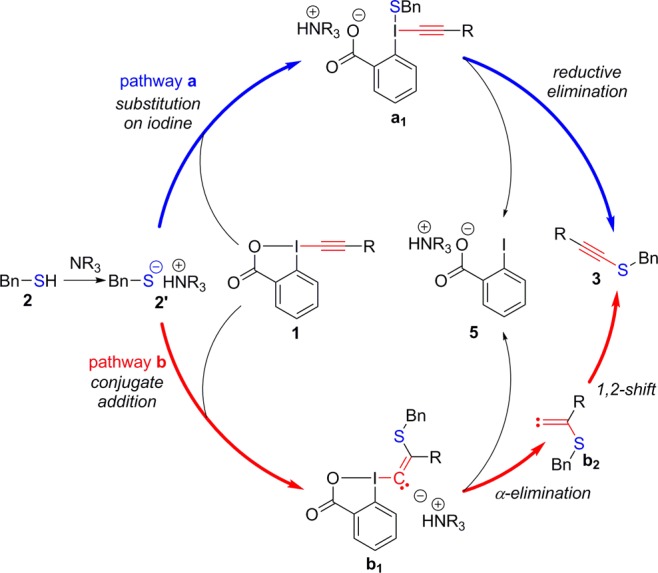
From this point forward, a first possible mechanism would be substitution on iodine to give intermediate a1, followed by reductive elimination (pathway a, blue in Scheme 3). This mechanism is well-established in hypervalent iodine chemistry, especially with aryliodonium salts.15
Alternatively, a single electron transfer mechanism (SET) has also been proposed.16 Nevertheless, in the case of the alkynylation reaction, no radical intermediate could be trapped when using TEMPO as a reagent and the reaction occurred with the same yield and reaction time. Although this experiment is naturally not sufficient to exclude a SET pathway, it is also important to note that most reactions of hypervalent iodine reagents occurring via a SET pathway require activation of the reagent by a Lewis or Brønsted acid to accelerate electron transfer,16 and the alkynylation reaction occurs only under basic conditions.
Nevertheless, alkynyliodonium salts constitute a unique class of hypervalent iodine reagents, as the β-carbon of the alkyne has very strong electrophilic character.17 In this case, an alternative mechanism is possible: conjugate addition of the thiolate 2′ on EBX 1 to give a vinyl benziodoxolone intermediate b1 (pathway b, red in Scheme 3). From b1, α-elimination of iodobenzoate 5 followed by a 1,2-shift of either the sulfur or silicium substituent gave thioalkyne 3. In fact, this type of mechanism was proposed by Ochiai and co-workers in the case of carbon nucleophiles based on the isolation of C–H insertion products originating from carbene intermediate b2.18 Importantly, such insertion products could be observed only when carbon-substituted alkynyliodonium salts were used in the reaction, as the 1,2-shift of silyl groups was faster than insertion reactions.
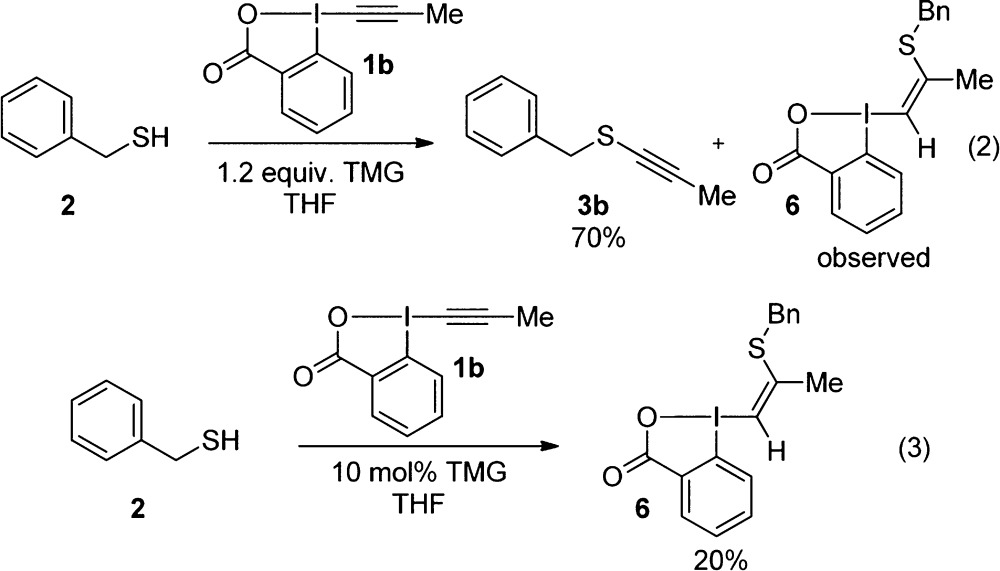
To establish if Ochiai’s mechanism was also correct in the case of sulfur nucleophiles, we decided to investigate Me-EBX (1b) as a reagent, as the methyl group was expected to have a very low migrating aptitude. This reagent was easily synthesized using the one-step protocol recently developed by Olofsson and co-workers.19 Surprisingly, the desired alkynylation product 3b could still be isolated in 70% yield, although the reaction was less clean than usual (eq 2). In particular, a polar side product with NMR signals in accordance with vinyl benziodoxolone 6 could be observed, although the small quantities of 6 formed did not allow us to isolate this product in pure form at this stage. Compound 6 most likely originated from protonation of intermediate b1. Consequently, we hypothesized that its formation could be favored if only a catalytic amount of base was used. Indeed, this was the case and we were pleased to see that 6 precipitated directly from the reaction mixture in 20% yield as a single Z isomer when only 10 mol % of TMG was used (eq 3). The isolation of 6 constituted strong support for the conjugate-addition/α-elimination/1,2-shift mechanism. Furthermore, the relatively good yield of isolated alkynylation product was in accordance with a shift of the sulfur atom, as the migration of the methyl group was expected to be too slow to be productive.
 |
2 |
Nevertheless, no intermediate could be observed in the case of TIPS-EBX (1a). To gain a more comprehensive understanding of the mechanistic details leading to thioalkyne formation, density functional theory (DFT) computations were undertaken using benzyl thiol (2) as a substrate. As described above, in the most likely scenario, a deprotonated thiol directly attacks TIPS-EBX (1a). This occurs either by direct attack onto the hypervalent iodine atom (pathway a in Scheme 3) or through a conjugate addition (pathway b in Scheme 3). Computations (at the PBE0-dDsC/TZ2P//M06-2X/def2-SVP theoretical level, see Computational Details for additional information) designed to probe the potential energy surface revealed two low-energy van der Waals complexes that roughly correspond to the entry points into the two mechanistic pathways a and b. The first, a0 in Figure 1, is a lower energy conformation in which the sulfur atom and the iodine atom are in close proximity (2.913 Å). However, the sulfur atom is not exactly opposite to the aryl ring as generally expected for interaction of nucleophiles with hypervalent iodine reagents, but instead lies in a position roughly equidistant between the iodine (2.913 Å) and α-carbon (3.128 Å) atoms of the acetylene. A second complex (b0 in Figure 1), lying ∼15.8 kcal/mol higher in energy, was found to correspond to the aforementioned conjugate addition pathway b. Here, the sulfur atom of the deprotonated thiol can clearly participate in a direct attack on the β-carbon of the acetylene unit.
Figure 1.
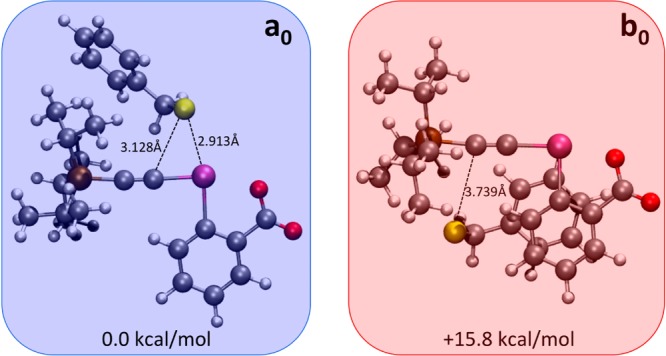
Computed geometries (M06-2X/def2-SVP level) of the van der Waals complexes a0 and b0 for TIPS-EBX (1c) and thiolate 2′. Free energies computed at the PBE0-dDsC/TZ2P//M06-2X/def2-SVP level and include solvation correction in THF determined using COSMO-RS.
Our initial assumption was that complex a0 (depicted in blue) is the reactant species leading to formation of a S–I bond, as represented by intermediate a1. To our surprise, no minima containing a formal S–I bond could be located on the potential energy surface (Figure 2, blue pathway). Instead, the quasi-triangular atomic arrangement between the sulfur, iodine, and α-carbon atoms results in a direct addition of the sulfur onto the α-carbon atom of the acetylene unit with simultaneous breaking of the C–I bond.20 This process is associated with a quite modest aTS1 barrier height of only 10.8 kcal/mol.21 Upon formation of the new S–C bond (Figure 3, aTS1 in blue), the thioalkyne-iodobenzoic acid complex a3•5 is spontaneously formed in a highly exergonic process. Importantly, we find no computational evidence predicting formation of a stable intermediate, which is in agreement with of our failure to isolate any side products with TIPS-EBX (1a) as reagent.
Figure 2.

Reaction free energy profile [PBE0-dDsC/TZ2P//M06-2X/def2-SVP level in implicit THF solvent (COSMO-RS)] for the two possible mechanistic pathways a (blue) and b (red) for the reaction of TIPS-EBX (1a) with thiolate 2′. *Positive deltaE at the M06-2X/def2-SVP level.
Figure 3.
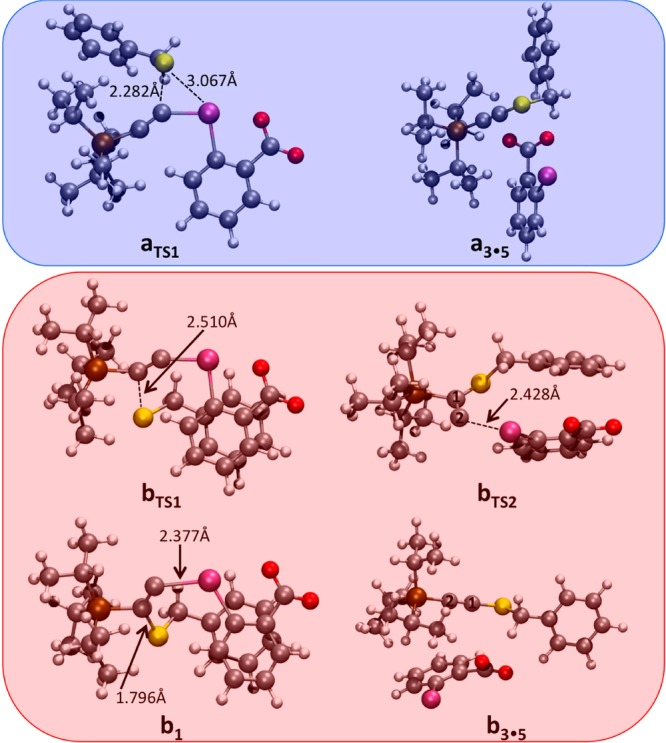
Computed geometries (M06-2X/def2-SVP level) of the relevant structures.
The conjugate addition pathway b (Figure 2, red pathway), begins from the higher energy (+15.8 kcal/mol, red) van der Waals complex b0. Here, the reaction proceeds as originally proposed by Ochiai.18 Formation of the new S–C bond occurs in a facile process, requiring only ∼7.2 kcal/mol of energy (bTS1). In contrast to the previously discussed pathway, the β-carbon conjugate addition route b does lead to the formation of the expected intermediate species b1. However, the TS barrier associated with cleavage of the I–C bond (bTS2, Figure 3) is negligible (ΔG = −0.7 kcal/mol at the PBE0-dDsC/TZ2P//M06-2X/def2-SVP level, ΔE = +0.3 kcal/mol at the M06-2X/def2-TZVP//M06-2X/def2-SVP level), meaning the species is likely short-lived, making experimental characterization, observation or even protonation highly unlikely. As in pathway a, formation of the final product complex is highly exergonic, proceeding via a 1,2-shift of the silicium atom. No minima corresponding to a free carbene b2 could be located on the potential energy surface.
From the reaction free energy profile in Figure 2, it is clear that both the a (blue) and b pathway (red) mechanisms are easily accessible at room temperature. While the β-conjugate addition pathway b involves a slightly lower TS barrier bTS1 corresponding to S–C bond formation, the lower relative energy of the α-addition van der Waals complex a0 (blue, Figure 1) results in pathway a being the overall energetically preferred reaction mechanism by 12.2 kcal/mol. This is highly interesting, considering that a mechanism involving addition to the α-carbon atom concerted with elimination of the iodine has never seriously been considered for alkynyl hypervalent iodine reagents.
To ensure that other mechanisms that include either direct attack by a protonated thiol or explicit involvement of the base (TMG) are not more energetically favorable, we computed a number of additional pathways (see SI for details). However, none were found to be energetically competitive with the direct attack of thiolate mechanism described above. As an example, direct attack by the protonated thiol species involves a TS barrier of 32.6 kcal/mol for S–C bond formation. This compares quite unfavorably to the 10.8 kcal/mol barrier seen when the deprotonated thiol is involved in a direct attack type mechanism.22
The computational results obtained for the reaction of TIPS-EBX (1a) are highly interesting. Nevertheless, they seemed to be in contradiction with the isolation of side product 6 when Me-EBX (1b) was used (eq 3), as 6 would be generated from very short-lived intermediate b1 via the least favored pathway b. To see if this result could be rationalized by computation, we also examined the reaction free energy profile for Me-EBX (1b) (Figure 4). Like for TIPS-EBX (1a) (Figure 2), a mechanism featuring attack on the α-carbon is favored based on enhanced stability of the prereaction van der Waals complex a0 and b0. In contrast, however, the TS barrier associated with formation of the S–C bond (aTS1 and bTS1, Figure 4) lie relatively close to each other (within 5 kcal/mol at the PBE0-dDsC/TZ2P level and within 2 kcal/mol at the M06-2X/def2-TZVP level).23 Since this first transition state represents the highest point along the reaction pathway for both mechanisms, it can be envisaged that a small percentage of reactions proceed via the red pathway a, as opposed to the more energetically favorable blue pathway b. Furthermore, a significant bTS2 barrier height (+5.7 kcal/mol) is obtained, in contrast to the reaction with silylated reagent 1c. This enhanced barrier, probably due to the less favorable shift of the sulfur group in this case, likely equates to a longer-lived intermediate that can be protonated and observed as 6 (eq 3).
Figure 4.

Reaction free energy profile [PBE0-dDsC/TZ2P//M06-2X/def2-SVP level in implicit THF solvent (COSMO-RS)] for the two possible mechanistic pathways a (blue) and b (red) for the reaction of Me-EBX (1b) with thiolate 2′.
When comparing the lowest energy pathway a for the reaction of TIPS-EBX (1a) and Me-EBX (1b), a strong accelerating effect of the silicium atom (about 5 kcal/mol) is apparent. Although understanding fully this effect will require more in-depth studies, some insights can already be gained by looking closely at the unsymmetrical structure of transition state aTS1. Indeed, the sulfur atom is already in close proximity to the α carbon atom of the alkyne, and the geometry around the β carbon is strongly distorted from sp to sp2. This would lead to the formation of a partial charge on the β carbon. Indeed, calculations indicated an iterative Hirshfeld charge of −0.82 at this position in the case of TIPS-EBX (1a) (Figure 2). In contrast, no strong charge transfer was observed in the case of Me-EBX (1b) (charges of −0.27 and −0.12 at the α and β positions, respectively, Figure 4). Silicium is well-known to stabilize negative charges on adjacent carbons (α-silicium effect).24 Consequently, a lower activation barrier could be expected in this case through transition state stabilization.
Scope Extension
The previous method developed in our group was very efficient using TIPS-EBX (1a) as a reagent. However, to introduce a fluorophore or other functional groups on the alkyne, two further steps were required (silyl deprotection and cycloaddition), making the approach less convergent.14a Furthermore, sensitive substrates could potentially not resist in the desilylation step using basic TBAF. Consequently, it would be highly desirable to install the desired functionality on the alkyne on the cyclic hypervalent iodine reagent before carrying out the thioalkynylation reaction. On the basis of the side reactions observed by Ochiai and co-workers with alkyl-substituted alkynyliodonium salts and carbon nucleophiles,18 a generalization of the scope appeared highly challenging to us when we started this project. However, the surprisingly good results obtained with Me-EBX (1b) during our mechanistic investigations and the very low activation energies obtained by computation indicated that sulfur nucleophiles should behave differently and have a much broader scope in the alkynylation reaction. To test this hypothesis, benziodoxolone hypervalent iodine reagents were first synthesized via Olofsson’s one-step method starting from alkynyl boronic esters (eq 4).19 This protocol turned out to be very efficient and easily scalable giving the EBX reagents in 71–95% yield in up to 9 g scale in the case of Me-EBX (1b). Nevertheless, in case of more functionalized alkynes, it was not possible to access the very sensitive alkynyl boronic esters. In this case, the use of more stable silylated alkynes gave the desired products in 27–47% yield. Importantly, EBX reagents bearing functional groups such as alkenes, alkynes, halogens, azides or alcohols were synthesized for the first time. Although the yield was moderate, the procedure was easily scalable to the multigram scale and the enhanced stability of benziodoxolone compared to alkynyliodonium salts allowed easy purification by column chromatography to obtain pure reagents 1. A significant amount of silyl acetylenes could also be recovered.25
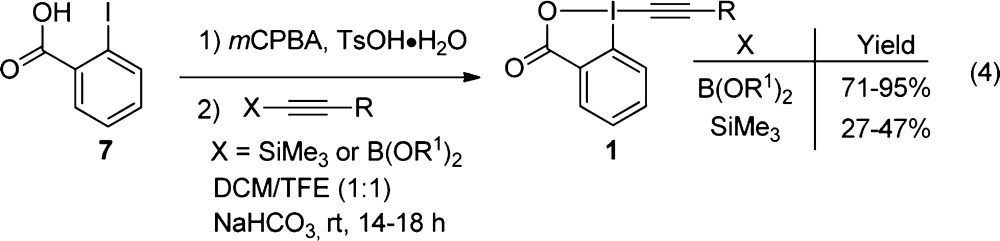 |
4 |
Due to the significance of aromatic thiols as important structural motifs in the synthesis of pharmaceutical, natural, and medicinal compounds and the excellent results obtained in our previous work, we decided to start our investigation with this class of substrates. o-Bromo thiophenol 8 was chosen as a substrate for studying the different EBX reagents, as the bromo group constitutes an ideal handle for further functionalization. In the case of aliphatic EBX reagents, we found that TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene) was superior to TMG as a base and THF was still the optimal solvent. When EBX reagent 1b was added to the mixture consisting of o-bromo thiophenol (8), TBD, and THF at room temperature and stirred for 5 min in an open flask, the corresponding thioalkynylated product 9a was obtained in 93% yield (Table 1, entry 1). Noteworthy, the reaction was not affected by the water which was added to increase the solubility of the formed thiolate salts in certain cases. The alkynylation with longer alkyl chains (entries 2 and 3) or a tert-butyl group (entry 4) proceeded in quantitative yields. At this point, the use of functionalized alkyl-EBX reagents was investigated for the first time (entries 5–9).
Table 1. Scope of EBX Reagents with o-Bromo Thiophenola.
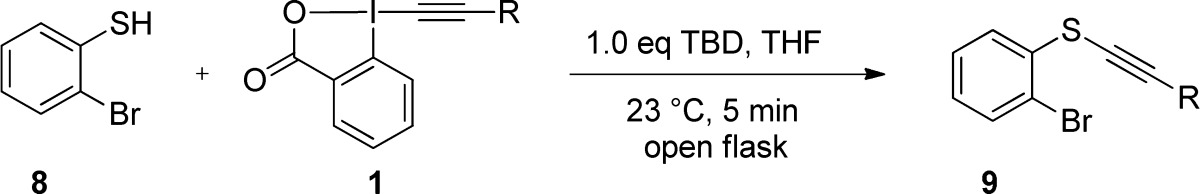
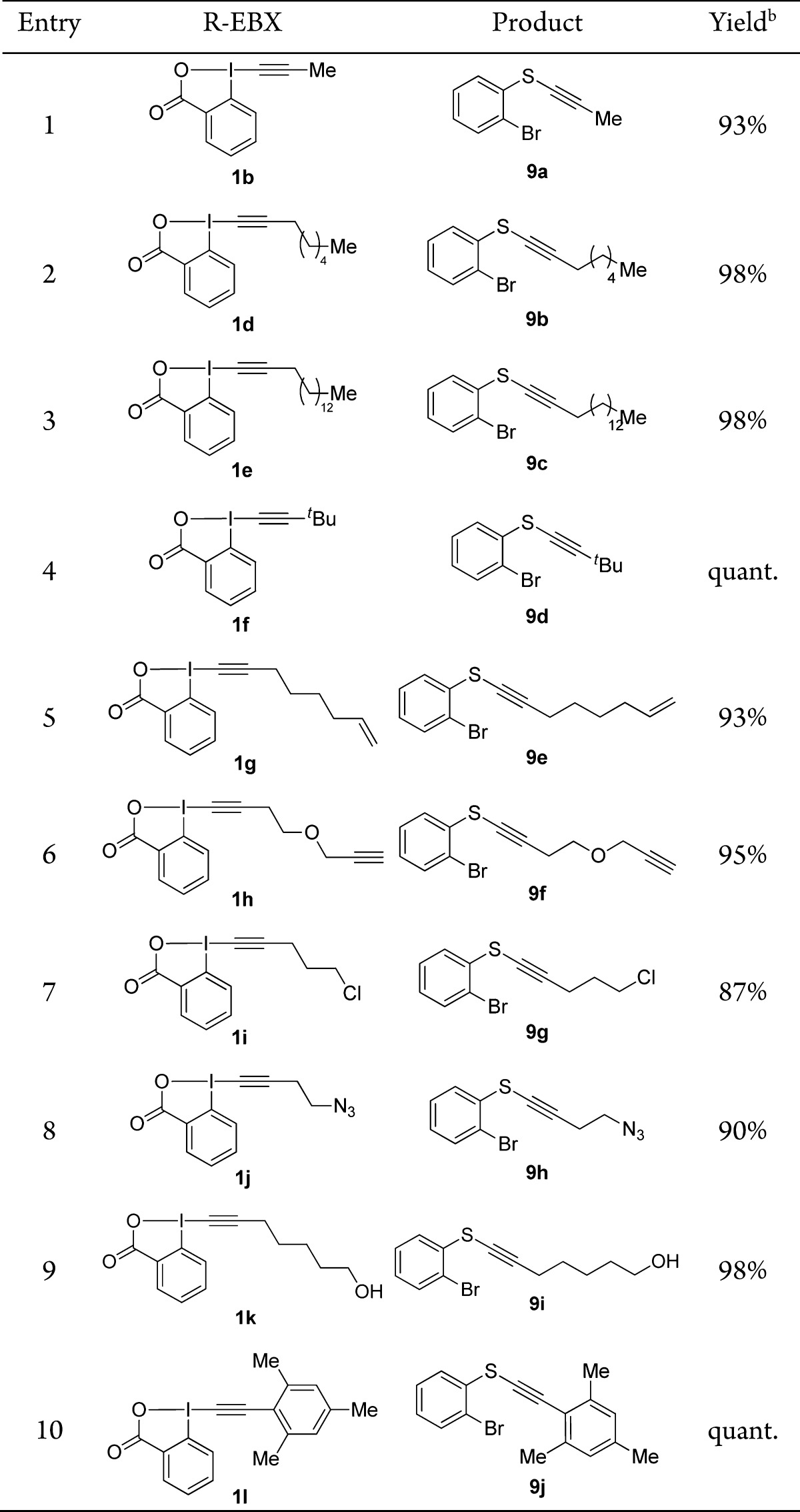
O-Bromo thiophenol (8, 0.30–0.80 mmol), alkyne transfer reagent (1, 0.33–0.88 mmol), base (0.30–0.80 mmol), THF (3.75–10.0 mL), 23 °C, 5 min, open flask.
Isolated yield after purification by column chromatography.
We were pleased to see that the alkynylation with reagents bearing an alkene, an alkyne, a chloride, an azide or a free alcohol proceeded in 87–98% yield. The fact that functional groups are tolerated both on the thiol and alkyne reagent makes the method more attractive for the synthesis of thioalkynes. Finally, the alkynylation was not limited to the transfer of aliphatic alkynes: mesityl-substituted product 9j was also obtained in quantitative yield (entry 10).
Inspired by the efficiency of the thioalkynylation reaction (Table 1), we further studied the use of EBX reagents for the multiple alkynylation of benzene-1,3,5-trithiol (10) (eq 5). In this context, TIPS-EBX (1a) and EBX reagents 1g and 1k were examined. We were able to isolate the corresponding triple-alkynylated products 11a–c in 96%, 87% and 88% yield, respectively. Due to the efficiency of the reaction and the versatility of the introduced functional groups, the method is highly useful for the synthesis of dendrimers with potential applications in materials science or drug delivery.
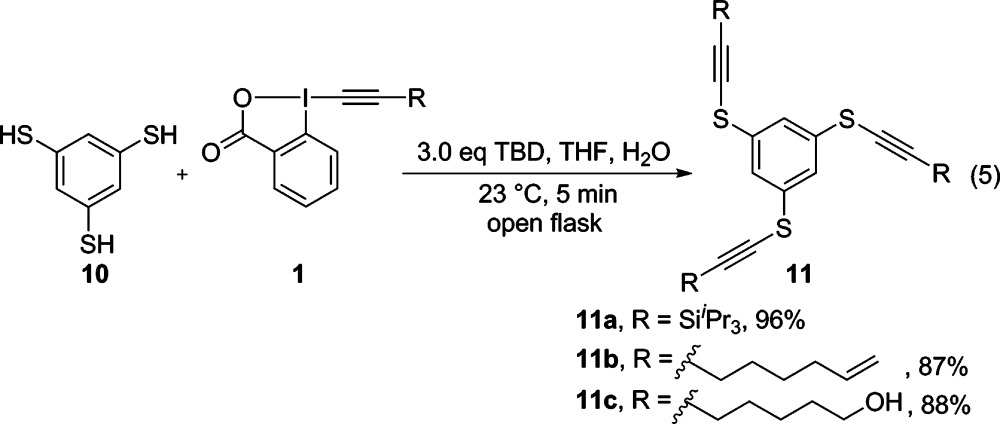 |
5 |
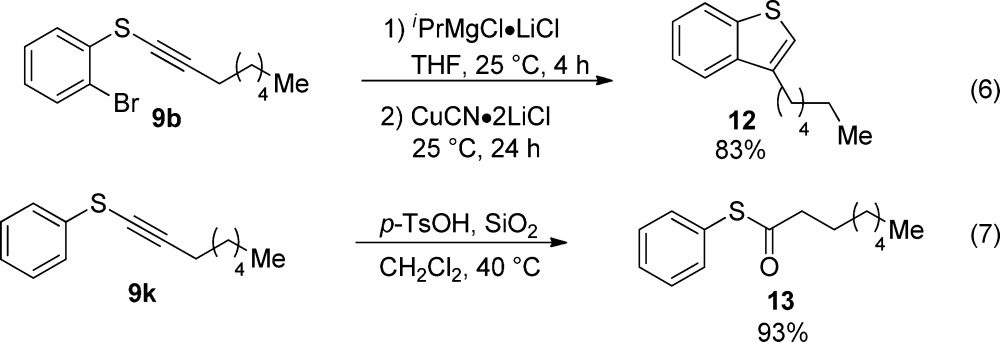
To further demonstrate the utility of the synthesized thioalkyne products, we investigated their transformation into other useful building blocks. We first examined the transformation of thioalkyne 9b into the corresponding benzothiophene 12 using a protocol developed by Knochel and co-workers (eq 6).26 The addition of iPrMgCl·LiCl in THF to 9b followed by CuCN·2LiCl afforded the 3-alkyl benzothiophene 12 in 83% yield after 24 h. Thiolakyne 9k, easily obtained from the alkynylation of thiophenol with reagent 1d, was subjected to acid hydrolysis to furnish the corresponding thioester 13 in 93% yield (eq 7).27
 |
6 |
After having successfully extended the scope of the alkynylation to alkyl- and aryl-substituted EBX reagents in the case of thiophenols, we turned to aliphatic thiols (Scheme 4). (4-Methoxyphenyl)methanethiol (14) was alkynylated successfully, furnishing alkynylation products 17a–d in 59–84% yield (Scheme 4A).28 Again, long and short alkyl chains, as well as a chloride and an alcohol, were well tolerated on the reagent, although the yields were lower than in the case of thiophenols. As a second important class of aliphatic thiols, we decided to turn to thioglycosides (Scheme 4B). These compounds are key building blocks in carbohydrate synthesis and exhibit important biological activities.29 Recently, Messaoudi and co-workers reported the alkynylation of protected and unprotected thioglycosides using transition metal catalysts.13b,13c Our metal free thioalkynylation method provides a valuable alternative that avoids the use of expensive or toxic metal catalysts, ligands, base and higher temperature. Furthermore, only the synthesis of aromatic alkynes had been reported to date. Using our method, protected thioglycoside 15a was alkynylated with TIPS-EBX (1b) and reagents 1j and 1k in a few minutes at room temperature to afford the corresponding products 18a–c in 45–84% yield. Protection of the hydroxy groups of the carbohydrate was not required: products 18d and 18e bearing four free hydroxy groups were also obtained in 81% and 60% yields, respectively.
Scheme 4. Scope of the Alkynylation for Benzylic Thiol 14 (A), Thioglycosides 15 (B), and Amino Acid Derivative 16 (C).

See Supporting Information for experimental details (solvents, base).
After demonstrating that the reaction protocol worked well with simple aliphatic thiols and thioglycosides, we studied the alkynylation of a cysteine containing dipeptide 16a (TrpCys). In fact, cysteine is one of the natural amino acids and plays a key role in the activity and structure of proteins. It is also an ideal entry for bioconjugation reactions.9,30 As shown in Scheme 4C, the reaction worked efficiently for a variety of EBX reagents. Substituents containing an alkyl chain without (product 19a) or with additional functional groups (products 19b–e) were tolerated including a chloro, an ether,31 an azido and a hydroxy group. Finally, a mesityl-substituted alkyne group could also be introduced in 80% yield (product 19f). The selective alkynylation of dipeptide 16a with different functionalized alkynes is an important preliminary result in view of the modification of more complex molecules, such as peptides and proteins. In addition, the alkynylation of N-unprotected cysteine ester 16b with tBu-EBX (1f) gave exclusively thiol alkynylation in 94% yield.
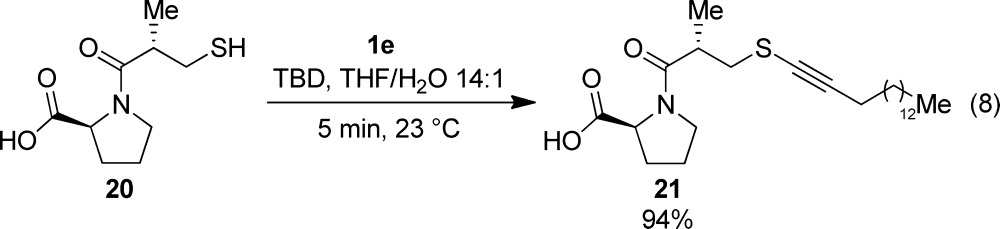
As a last example of more complex aliphatic thiol, angiotensin-converting enzyme (ACE) inhibitor Captopril (20), which is used as a drug to treat hypertension,32 was successfully alkynylated with 1e to give the corresponding thioalkyne 21 in 94% yield (eq 8). This result demonstrated that free carboxylic acid groups were tolerated in the alkynylation reaction.
 |
8 |
To further demonstrate the generality of the alkynylation reaction, we studied two classes of sulfur nucleophiles which were not included in our previous studies: thiocarboxylic acids and sodium hydrogen sulfide, the simplest of all sulfur nucleophiles (Scheme 5). Thiocarboxylates are less nucleophilic than thiolates. Nevertheless, TIPS-EBX (1a) was again an excellent reagent for the alkynylation of thiobenzoic acid (22a) giving the desired product 24a in 94% yield. Electron-donating and -withdrawing groups were well tolerated on the benzene ring (products 24b–d). On the other hand, thioesters derived from aliphatic alkynes were unstable under the reaction conditions, and ketone product 26, probably resulting from the hydration of the triple bond, was instead isolated in 45% yield. Only very low yields were obtained in the case of aliphatic thioacids (results not shown). Unlike other thiols, the alkynylation of thioacids did not require the use of base, probably due to the higher acidity of these substrates.
Scheme 5. Alkynylation of Thioacids 22 (A) and Sodium Hydrogen Sulfide (23) (B).

Due to the significant properties of diethynyl sulfides in material and interstellar chemistry,33 we then attempted their synthesis through the alkynylation of sodium hydrogen sulfide. The resulting one-step protocol would be unprecedentedly fast to access this class of compounds. The double alkynylation worked for all three classes of alkynes: silyl (product 25a), alkyl (product 25b) and aryl (product 25c). The lower yield of the latter is probably due to the low solubility of the reagent Ph-EBX (1o). The reaction also worked for the introduction of a propargylic ether (product 25d) or an alcohol (product 25e).34
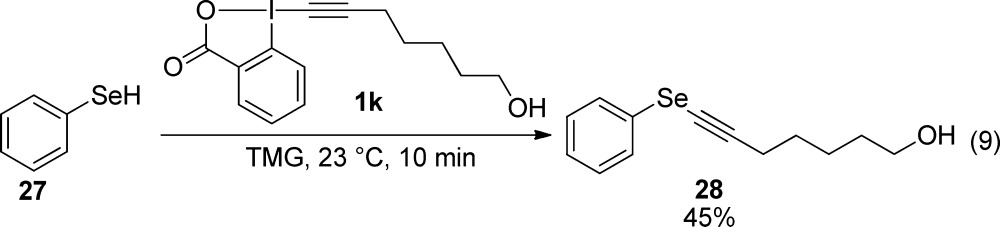
We then examined if the alkynylation method could be extended to selenium nucleophiles. In fact, alkynyl selenium compound 28 was obtained in 45% yield from phenylselenol (27) without further optimization of the reaction conditions. This preliminary result is promising for the development of a more general method for the alkynylation of selenols.
 |
9 |
Conclusion
In summary, in this manuscript we described the first computational studies of the alkynylation of thiols using ethynyl benziodoxolone (EBX) reagents. An unprecedented concerted mechanism involving the sulfur, iodine and α-carbon atoms of the alkyne was discovered by computation, leading directly to the alkynylation products with a low activation barrier. In case of silyl-substituted EBX reagents, the activation energy was exceptionally low (10.8 kcal/mol), which made this pathway the most favorable according to the computations and may explain the high rate observed for the reaction. For the case of alkyl-substituted reagents, a second mechanism involving conjugate addition, followed by simultaneous α elimination of iodobenzoic acid and sulfur 1,2-shift was also found to be competitive. This latter result rationalizes the isolation of small amounts of vinyl benziodoxolone intermediate 6 resulting from conjugate addition of benzylthiol (2) on Me-EBX (1b) followed by protonation.
The unique properties of thiolates in reaction with EBX reagents led us to anticipate that the transformation may have a broader scope than reactions involving carbon nucleophiles. This was indeed the case, and we could use, for the first time, functionalized alkyl- and aryl-substituted EBX reagents for the alkynylation of both aromatic and aliphatic thiols. Functional groups such as alkenes, alkynes, ethers, chlorides, azides and alcohols were tolerated on the alkynes. In addition to simple thiophenols and benzylic thiols, the alkynylation of cysteine in a dipeptide, thioglycosides, thiobenzoic acid derivatives and sodium hydrogen sulfide was also successful. The practical and user-friendly character of the method (5 min reaction time, open-flask, water tolerance, room temperature) and a deeper understanding of the reaction mechanism have set the stage for a broader application in the functionalization of materials and biomolecules in the future.
Computational Details
All geometries were optimized using Truhlar’s M06-2X35 density functional with the def2-SVP basis set in Gaussian09.36 M06-2X computations employed the “Ultrafine” grid to remove known problems with the size of the integration grid for this functional family.37 To obtain refined energy estimations that explicitly account for nonbonded interactions, a density dependent dispersion correction38 was used appended to the PBE039 functional (PBE0-dDsC). PBE0-dDsC single point computations made use of the slater-type orbital 3-ζ basis set, TZ2P, as implemented in ADF.40 To confirm the accuracy of the PBE0-dDsC computations, a second set of single point energies was obtained at the M06-2X/def2-TZVP level. All reported free energies include the effects of solvation (in THF) using the implicit continuum model for realistic solvents41 (COSMO-RS), also as implemented in ADF, as well as free energy correction derived from M06-2X/def2-SVP computations. Iterative Hirshfeld charges42 were computed using Q-Chem43 at the M06-2X/def2-SVP level.
Acknowledgments
We thank EPFL and F. Hoffmann-La Roche Ltd for financial support. The work of R.F. was further supported by a Marie Curie International Incoming Fellowship (Grant Number 331631) of the 7th Framework Program of the European Union and the work of M.D.W. by ERC (European Research Council, Starting Grant iTools4MC, number 334840). M.D.W. thanks Prof. Clémence Corminboeuf (EPFL) for helpful suggestions and comments. The Laboratory for Computational Molecular Design at EPFL is acknowledged for providing computational resources. Dr. Gergely Tolnai from Eötvös Loránd University in Hungary is acknowledged for the synthesis of reagent 1n during his stay in our laboratory.
Supporting Information Available
Experimental procedures and analytical data for all new compounds. Additional computational, data including Cartesian coordinates of relevant compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Bern University of Applied Sciences, Solothurnstrasse 102, CH-2504 Biel, Switzerland.
The authors declare no competing financial interest.
Supplementary Material
References
- Acetylene Chemistry: Chemistry, Biology and Material Science; Diederich F., Stang P. J., Tykwinski R. R., Eds.; Wiley-VCH: Weinheim, 2005. [Google Scholar]
- a Schobert H. Chem. Rev. 2014, 114, 1743. [DOI] [PubMed] [Google Scholar]; b Trotus I. T.; Zimmermann T.; Schuth F. Chem. Rev. 2014, 114, 1761. [DOI] [PubMed] [Google Scholar]
- a Frantz D. E.; Fassler R.; Carreira E. M. J. Am. Chem. Soc. 2000, 122, 1806. [Google Scholar]; b Trost B. M.; Weiss A. H. Adv. Synth. Catal. 2009, 351, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zeni G.; Larock R. C. Chem. Rev. 2004, 104, 2285. [DOI] [PubMed] [Google Scholar]; b Michelet V.; Toullec P. Y.; Genet J. P. Angew. Chem., Int. Ed. 2008, 47, 4268. [DOI] [PubMed] [Google Scholar]; c Jimenez-Nunez E.; Echavarren A. M. Chem. Rev. 2008, 108, 3326. [DOI] [PubMed] [Google Scholar]; d Godoi B.; Schumacher R. F.; Zeni G. Chem. Rev. 2011, 111, 2937. [DOI] [PubMed] [Google Scholar]
- a Bunz U. H. F. Chem. Rev. 2000, 100, 1605. [DOI] [PubMed] [Google Scholar]; b Liu J. Z.; Lam J. W. Y.; Tang B. Z. Chem. Rev. 2009, 109, 5799. [DOI] [PubMed] [Google Scholar]; c Yella A.; Lee H. W.; Tsao H. N.; Yi C. Y.; Chandiran A. K.; Nazeeruddin M. K.; Diau E. W. G.; Yeh C. Y.; Zakeeruddin S. M.; Gratzel M. Science 2011, 334, 629. [DOI] [PubMed] [Google Scholar]
- a Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. [DOI] [PubMed] [Google Scholar]; b Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596. [DOI] [PubMed] [Google Scholar]; c Tornoe C. W.; Christensen C.; Meldal M. J. Org. Chem. 2002, 67, 3057. [DOI] [PubMed] [Google Scholar]; d Meldal M.; Tornoe C. W. Chem. Rev. 2008, 108, 2952. [DOI] [PubMed] [Google Scholar]; e Agard N. J.; Prescher J. A.; Bertozzi C. R. J. Am. Chem. Soc. 2004, 126, 15046. [DOI] [PubMed] [Google Scholar]; f Sletten E. M.; Bertozzi C. R. Angew. Chem., Int. Ed. 2009, 48, 6974. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Thirumurugan P.; Matosiuk D.; Jozwiak K. Chem. Rev. 2013, 113, 4905. [DOI] [PubMed] [Google Scholar]
- a Shindo M. Tetrahedron 2007, 63, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kondoh A.; Yorimitsu H.; Oshima K. Chem.—Asian J. 2010, 5, 398. [DOI] [PubMed] [Google Scholar]; c Evano G.; Coste A.; Jouvin K. Angew. Chem., Int. Ed. 2010, 49, 2840. [DOI] [PubMed] [Google Scholar]; d DeKorver K. A.; Li H. Y.; Lohse A. G.; Hayashi R.; Lu Z. J.; Zhang Y.; Hsung R. P. Chem. Rev. 2010, 110, 5064. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wang X. N.; Yeom H. S.; Fang L. C.; He S. H.; Ma Z. X.; Kedrowski B. L.; Hsung R. P. Acc. Chem. Res. 2014, 47, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Murch P.; Williamson B. L.; Stang P. J. Synthesis 1994, 1255. [Google Scholar]; b Witulski B.; Stengel T. Angew. Chem., Int. Ed. 1998, 37, 489. [DOI] [PubMed] [Google Scholar]; c Frederick M. O.; Mulder J. A.; Tracey M. R.; Hsung R. P.; Huang J.; Kurtz K. C. M.; Shen L. C.; Douglas C. J. J. Am. Chem. Soc. 2003, 125, 2368. [DOI] [PubMed] [Google Scholar]; d Dunetz J. R.; Danheiser R. L. Org. Lett. 2003, 5, 4011. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hamada T.; Ye X.; Stahl S. S. J. Am. Chem. Soc. 2008, 130, 833. [DOI] [PubMed] [Google Scholar]; f Evano G.; Jouvin K.; Coste A. Synthesis 2013, 45, 17. [Google Scholar]; g Evano G.; Gaumont A. C.; Alayrac C.; Wrona I. E.; Giguere J. R.; Delacroix O.; Bayle A.; Jouvin K.; Theunissen C.; Gatignol J.; Silvanus A. C. Tetrahedron 2014, 70, 1529. [Google Scholar]
- a Zambrowicz B. P.; Sands A. T. Nat. Rev. Drug Discovery 2003, 2, 38. [DOI] [PubMed] [Google Scholar]; b Paquette L. A.Sulfur-Containing Reagents; Wiley: Chichester, 2009. [Google Scholar]; c Masella R.Glutathione and Sulfur Amino Acids in Human Health and Disease; Wiley: Hoboken, 2009. [Google Scholar]; d Hoyle C. E.; Lowe A. B.; Bowman C. N. Chem. Soc. Rev. 2010, 39, 1355. [DOI] [PubMed] [Google Scholar]
- a Hoyle C. E.; Bowman C. N. Angew. Chem., Int. Ed. 2010, 49, 1540. [DOI] [PubMed] [Google Scholar]; b Hoogenboom R. Angew. Chem., Int. Ed. 2010, 49, 3415. [DOI] [PubMed] [Google Scholar]; c Lowe A. B.; Hoyle C. E.; Bowman C. N. J. Mater. Chem. 2010, 20, 4745. [Google Scholar]; d Stephanopoulos N.; Francis M. B. Nat. Chem. Biol. 2011, 7, 876. [DOI] [PubMed] [Google Scholar]; e Stenzel M. H. ACS Macro Lett. 2012, 2, 14. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Ziegler G. R.; Welch C. A.; Orzech C. E.; Kikkawa S.; Miller S. I. J. Am. Chem. Soc. 1963, 85, 1648. [Google Scholar]; b Marchueta I.; Montenegro E.; Panov D.; Poch M.; Verdaguer X.; Moyano A.; Pericas M. A.; Riera A. J. Org. Chem. 2001, 66, 6400. [DOI] [PubMed] [Google Scholar]; c Ni Z.; Wang S.; Mao H.; Pan Y. Tetrahedron Lett. 2012, 53, 3907. [Google Scholar]; In addition, an interesting example of the reaction of thioethers with alkynyliodonium salts to give thioalkynes has been reported by Ochiai and co-workers:; d Ochiai M.; Nagaoka T.; Sueda T.; Yan J.; Chen D. W.; Miyamoto K. Org. Biomol. Chem. 2003, 1, 1517. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Braga A. L.; Reckziegel A.; Menezes P. H.; Stefani H. A. Tetrahedron Lett. 1993, 34, 393. [Google Scholar]; b Bieber L. W.; da Silva M. F.; Menezes P. H. Tetrahedron Lett. 2004, 45, 2735. [Google Scholar]; c Arisawa M.; Fujimoto K.; Morinaka S.; Yamaguchi M. J. Am. Chem. Soc. 2005, 127, 12226. [DOI] [PubMed] [Google Scholar]; d Godoi B.; Speranca A.; Back D. F.; Brandao R.; Nogueira C. W.; Zeni G. J. Org. Chem. 2009, 74, 3469. [DOI] [PubMed] [Google Scholar]; e Chandanshive J. Z.; Bonini B. F.; Gentili D.; Fochi M.; Bernardi L.; Franchini M. C. Eur. J. Org. Chem. 2010, 6440. [Google Scholar]
- a Yang Y.; Dong W.; Guo Y.; Rioux R. M. Green Chem. 2013, 15, 3170. [Google Scholar]; b Brachet E.; Brion J.-D.; Alami M.; Messaoudi S. Adv. Synth. Catal. 2013, 355, 2627. [Google Scholar]; c Brachet E.; Brion J.-D.; Alami M.; Messaoudi S. Chem.—Eur. J. 2013, 19, 15276. [DOI] [PubMed] [Google Scholar]
- a Frei R.; Waser J. J. Am. Chem. Soc. 2013, 135, 9620. [DOI] [PubMed] [Google Scholar]; First synthesis of EBX reagents:; b Ochiai M.; Masaki Y.; Shiro M. J. Org. Chem. 1991, 56, 5511. [Google Scholar]; c Zhdankin V. V.; Kuehl C. J.; Krasutsky A. P.; Bolz J. T.; Simonsen A. J. J. Org. Chem. 1996, 61, 6547. [DOI] [PubMed] [Google Scholar]; Selected other examples of reactions with EBX reagents:; d Brand J. P.; Charpentier J.; Waser J. Angew. Chem., Int. Ed. 2009, 48, 9346. [DOI] [PubMed] [Google Scholar]; e Fernandez Gonzalez D.; Brand J. P.; Waser J. Chem.—Eur. J. 2010, 16, 9457. [DOI] [PubMed] [Google Scholar]; f Nicolai S.; Piemontesi C.; Waser J. Angew. Chem., Int. Ed. 2011, 50, 4680. [DOI] [PubMed] [Google Scholar]; g Li Y.; Brand J. P.; Waser J. Angew. Chem., Int. Ed. 2013, 52, 6743. [DOI] [PubMed] [Google Scholar]; h Chen C. C.; Waser J. Chem. Commun. 2014, 50, 12923. [DOI] [PubMed] [Google Scholar]; i Liu X.; Wang Z.; Cheng X.; Li C. J. Am. Chem. Soc. 2012, 134, 14330. [DOI] [PubMed] [Google Scholar]; j Wang Z.; Li X.; Huang Y. Angew. Chem., Int. Ed. 2013, 52, 14219. [DOI] [PubMed] [Google Scholar]; k Collins K. D.; Lied F.; Glorius F. Chem. Commun. 2014, 50, 4459. [DOI] [PubMed] [Google Scholar]; l Feng C.; Loh T.-P. Angew. Chem., Int. Ed. 2014, 53, 2722. [DOI] [PubMed] [Google Scholar]; m Huang H.; Zhang G.; Gong L.; Zhang S.; Chen Y. J. Am. Chem. Soc. 2014, 136, 2280. [DOI] [PubMed] [Google Scholar]; n Nierth A.; Marletta M. A. Angew. Chem., Int. Ed. 2014, 53, 2611. [DOI] [PubMed] [Google Scholar]; o Xie F.; Qi Z.; Yu S.; Li X. J. Am. Chem. Soc. 2014, 136, 4780. [DOI] [PubMed] [Google Scholar]; p Silva L. F. Jr; Utaka A.; Calvalcanti L. Chem. Commun. 2014, 50, 3810. [DOI] [PubMed] [Google Scholar]; q Wu X.; Shirakawa S.; Maruoka K. Org. Biomol. Chem. 2014, 12, 5388. [DOI] [PubMed] [Google Scholar]; r Ariafard A. ACS Catal. 2014, 4, 2896. [Google Scholar]; s Jeong J.; Patel P.; Hwang H.; Chang S. Org. Lett. 2014, 16, 4598. [DOI] [PubMed] [Google Scholar]; t Lu B.; Wu J.; Yoshikai N. J. Am. Chem. Soc. 2014, 136, 11598. [DOI] [PubMed] [Google Scholar]; u Wang Z.; Li L.; Huang Y. J. Am. Chem. Soc. 2014, 136, 12233. [DOI] [PubMed] [Google Scholar]
- a Lancer K. M.; Wiegand G. H. J. Org. Chem. 1976, 41, 3360. [Google Scholar]; b Wang B. J.; Graskemper J. W.; Qin L. L.; DiMagno S. G. Angew. Chem., Int. Ed. 2010, 49, 4079. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pinto de Magalhães H.; Lüthi H. P.; Togni A. Org. Lett. 2012, 14, 3830. [DOI] [PubMed] [Google Scholar]; d Malmgren J.; Santoro S.; Jalalian N.; Himo F.; Olofsson B. Chem.—Eur. J. 2013, 19, 10334. [DOI] [PMC free article] [PubMed] [Google Scholar]; General reviews on hypervalent iodine:; e Kita Y.; Koser G. F.; Ochiai M.; Tohma H.; Varvoglis A.; Wirth T.; Zhdankin V. V. In Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis; Topics in Current Chemistry; Springer: Berlin, 2002; Vol. 224. [Google Scholar]; f Zhdankin V. V.; Stang P. J. Chem. Rev. 2008, 108, 5299. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Zhdankin V. V.Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds. Wiley: Weinheim, 2013. [Google Scholar]
- Dohi T.; Ito M.; Yamaoka N.; Morimoto K.; Fujioka H.; Kita Y. Tetrahedron 2009, 65, 10797. [Google Scholar]
- Zhdankin V. V.; Stang P. J. Tetrahedron 1998, 54, 10927. [Google Scholar]
- a Ochiai M.; Kunishima M.; Nagao Y.; Fuji K.; Shiro M.; Fujita E. J. Am. Chem. Soc. 1986, 108, 8281. [Google Scholar]; b Ochiai M.; Ito T.; Takaoka Y.; Masaki Y.; Kunishima M.; Tani S.; Nagao Y. J. Chem. Soc., Chem. Commun. 1990, 118. [Google Scholar]
- Bouma M. J.; Olofsson B. Chem.—Eur. J. 2012, 18, 14242. [DOI] [PubMed] [Google Scholar]
- The concerted nature of this mechanism can clearly be seen via examination of the intrinsic reaction coordinate (IRC). Selected IRC structures are provided in the Supporting Information.
- Computed free energies at the M06-2X/def2-TZVP//M06-2X/def2-SVP level in implicit THF solvent (COSMO-RS) yielded the same trends, albeit with a slightly lower energy difference between the two pathways (Energy aTS1, 11.8 kcal/mol; bTS1, 20.1 kcal/mol; difference, 8.3 kcal/mol. See Supporting Information for details.
- Computation of the transition state aTS1 for other nucleophiles was also done in order to better understand the observed chemioselectivity. With methanol and dimethyl phosphite, no transition state could be located. Higher energies were observed for methylamine and acetate as nucleophiles (+30.8 and +18.1 kcal/mol, respectively). In contrast, a low energy transition state (+12.2 kcal/mol) was observed in the case of deprotonated dimethyl phosphite. Indeed, the facile alkynylation of this class of nucleophiles was recently reported by our group (ref (14h)). See Supporting Information for details.
- Computed free energies at the M06-2X/def2-TZVP//M06-2X/def2-SVP level in implicit THF solvent (COSMO-RS): aTS1, 18.9 kcal/mol; bTS1, 20.8 kcal/mol; difference, 1.9 kcal/mol. In addition, the difference in free energies obtained using B3LYP-dDsC and B3LYP-D3 methods was situated in between the other results (4.3 and 3.3 kcal/mol, respectively). Nevertheless, the importance of the three-atom concerted pathway should not be overestimated at this stage. See Supporting Information for details.
- a Zhang S.; Zhang X.-M.; Bordwell F. G. J. Am. Chem. Soc. 1995, 117, 602. [Google Scholar]; b Römer B.; Gatev G. G.; Zhong M.; Brauman J. I. J. Am. Chem. Soc. 1998, 120, 2919. [Google Scholar]
- See Supporting Information for further details.
- Kunz T.; Knochel P. Angew. Chem., Int. Ed. 2012, 51, 1958. [DOI] [PubMed] [Google Scholar]
- Braga A. L.; Martins T. L. C.; Silveira C. C.; Rodrigues O. E. D. Tetrahedron 2001, 57, 3297. [Google Scholar]
- Thiol 14 was chosen as starting material due to its higher boiling point and simpler NMR spectrum when compared to benzyl thiol (2). Comppound 17d was obtained using the corresponding EBX reagent 1m bearing a free alcohol.
- a Driguez H.Thiooligosaccharides in Glycobiology. In Glycoscience Synthesis of Substrate Analogs and Mimetics; Springer Verlag: Berlin, 1997; Vol. 187, pp 85–116. [Google Scholar]; b Pachamuthu K.; Schmidt R. R. Chem. Rev. 2005, 106, 160. [DOI] [PubMed] [Google Scholar]
- a Lundblad R. L.Chemical Reagents for Protein Modification; 3rd ed.; CRC Press: Boca Raton, FL, 2005. [Google Scholar]; b Hermanson G. T.Bioconjugate Techniques; 2nd ed.; Academic Press: San Diego, CA, 2008. [Google Scholar]
- Alkyne 19c was obtained using the corresponding EBX reagent 1n bearing a protected propargylic alcohol.
- Migdalof B. H.; Antonaccio M. J.; McKinstry D. N.; Singhvi S. M.; Lan S. J.; Egli P.; Kripalani K. J. Drug Metab. Rev. 1984, 15, 841. [DOI] [PubMed] [Google Scholar]
- a Lee A. W. M.; Yeung A. B. W.; Yuen M. S. M.; Zhang H.; Zhao X.; Wong W. Y. Chem. Commun. 2000, 75. [Google Scholar]; b Matzger A. J.; Lewis K. D.; Nathan C. E.; Peebles S. A.; Peebles R. A.; Kuczkowski R. L.; Stanton J. F.; Oh J. J. J. Phys. Chem. A 2002, 106, 12110. [Google Scholar]; c Lewis K. D.; Wenzler D. L.; Matzger A. J. Org. Lett. 2003, 5, 2195. [DOI] [PubMed] [Google Scholar]; d Lewis K. D.; Rowe M. P.; Matzger A. J. Tetrahedron 2004, 60, 7191. [Google Scholar]; e Gleiter R.; Werz D. B. Chem. Rev. 2010, 110, 4447. [DOI] [PubMed] [Google Scholar]; f Su Q.; Zhao Z. J.; Xu F.; Lou P. C.; Zhang K.; Xie D. X.; Shi L.; Cai Q. Y.; Peng Z. H.; An D. L. Eur. J. Org. Chem. 2013, 2013, 1551. [Google Scholar]
- At the current stage of development, the reaction is limited to the synthesis of symmetrical diynes.
- a Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]; b Zhao Y.; Truhlar D. G. Acc. Chem. Res. 2008, 41, 15. [DOI] [PubMed] [Google Scholar]
- Gaussian 09, Revision D.01; Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam M. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian, Inc.: Wallingford CT, 2009.
- Wheeler S. E.; Houk K. N. J. Chem. Theory Comput. 2010, 6, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Steinmann S. N.; Corminboeuf C. J. Chem. Theory Comput. 2011, 7, 3567. [DOI] [PubMed] [Google Scholar]; b Steinmann S. N.; Corminboeuf C. J. Chem. Phys. 2011, 134, 044117. [DOI] [PubMed] [Google Scholar]; c Steinmann S. N.; Corminboeuf C. Chimia 2011, 65, 240. [DOI] [PubMed] [Google Scholar]; d Steinmann S. N.; Corminboeuf C. J. Chem. Theory Comput. 2010, 6, 1990. [DOI] [PubMed] [Google Scholar]
- a Perdew J. P.; Burke K.; Ernzerhof M. Phys. Rev. Lett. 1996, 77, 3865. [DOI] [PubMed] [Google Scholar]; b Adamo C.; Barone V. J. Chem. Phys. 1999, 110, 6158. [Google Scholar]
- a te Velde G.; Bickelhaupt F. M.; van Gisbergen S. J. A.; Fonseca Guerra C.; Baerends E. J.; Snijders J. G.; Ziegler T. J. Comput. Chem. 2001, 22, 931. [Google Scholar]; b Fonseca Guerra C.; Snijders J. G.; te Velde G.; Baerends E. J. Theor. Chem. Acc. 1998, 99, 391. [Google Scholar]
- Klamt A. WIREs Comput. Mol. Sci. 2011, 1, 699. [Google Scholar]
- a Hirshfeld F. L. Theor. Chim. Acta 1977, 44, 129. [Google Scholar]; b Bultinck P.; Van Alsenoy C.; Ayers P. W.; Carbo-Dorca R. J. Chem. Phys. 2007, 126, 144111. [DOI] [PubMed] [Google Scholar]; c For a discussion on the advantages of iterative Hirshfeld charges see:Gonthier J. F.; Steinmann S. N.; Wodrich M. D.; Corminboeuf C. Chem. Soc. Rev. 2012, 41, 4671. [DOI] [PubMed] [Google Scholar]
- Shao Y.; Molnar L. F.; Jung Y.; Kussmann J.; Ochsenfeld C.; Brown S. T.; Gilbert A. T. B.; Slipchenko L. V.; Levchenko S. V.; O’Neill D. P.; DiStasio R. A.; Lochan R. C.; Wang T.; Beran G. J. O.; Besley N. A.; Herbert J. M.; Lin C. Y.; Van Voorhis T.; Chien S. H.; Sodt A.; Steele R. P.; Rassolov V. A.; Maslen P. E.; Korambath P. P.; Adamson R. D.; Austin B.; Baker J.; Byrd E. F. C.; Dachsel H.; Doerksen R. J.; Dreuw A.; Dunietz B. D.; Dutoi A. D.; Furlani T. R.; Gwaltney S. R.; Heyden A.; Hirata S.; Hsu C. P.; Kedziora G.; Khalliulin R. Z.; Klunzinger P.; Lee A. M.; Lee M. S.; Liang W.; Lotan I.; Nair N.; Peters B.; Proynov E. I.; Pieniazek P. A.; Rhee Y. M.; Ritchie J.; Rosta E.; Sherrill C. D.; Simmonett A. C.; Subotnik J. E.; Woodcock H. L.; Zhang W.; Bell A. T.; Chakraborty A. K.; Chipman D. M.; Keil F. J.; Warshel A.; Hehre W. J.; Schaefer H. F.; Kong J.; Krylov A. I.; Gill P. M. W.; Head-Gordon M. Phys. Chem. Chem. Phys. 2006, 8, 3172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.