

Abstract
Using a combination of complementary in situ X-ray photoelectron spectroscopy and X-ray diffraction, we study the fundamental mechanisms underlying the chemical vapor deposition (CVD) of hexagonal boron nitride (h-BN) on polycrystalline Cu. The nucleation and growth of h-BN layers is found to occur isothermally, i.e., at constant elevated temperature, on the Cu surface during exposure to borazine. A Cu lattice expansion during borazine exposure and B precipitation from Cu upon cooling highlight that B is incorporated into the Cu bulk, i.e., that growth is not just surface-mediated. On this basis we suggest that B is taken up in the Cu catalyst while N is not (by relative amounts), indicating element-specific feeding mechanisms including the bulk of the catalyst. We further show that oxygen intercalation readily occurs under as-grown h-BN during ambient air exposure, as is common in further processing, and that this negatively affects the stability of h-BN on the catalyst. For extended air exposure Cu oxidation is observed, and upon re-heating in vacuum an oxygen-mediated disintegration of the h-BN film via volatile boron oxides occurs. Importantly, this disintegration is catalyst mediated, i.e., occurs at the catalyst/h-BN interface and depends on the level of oxygen fed to this interface. In turn, however, deliberate feeding of oxygen during h-BN deposition can positively affect control over film morphology. We discuss the implications of these observations in the context of corrosion protection and relate them to challenges in process integration and heterostructure CVD.
Introduction
The development of controllable and scalable growth, processing, and interfacing techniques remains the largest challenge in realizing the application potential of two-dimensional (2D) materials.1,2 Chemical vapor deposition (CVD) has emerged as the most viable route for high-quality graphene synthesis, and a lot of progress has also been made in developing CVD processes for other 2D materials, such as hexagonal boron nitride (h-BN)1,2 and related 2D heterostructures.3−11 Essential to the development of process rationales for 2D material CVD is an understanding of the underlying growth mechanisms,12−14 in particular the role of typically used process catalysts. While graphene CVD relies on the interactions of carbon with the surface, sub-surface, and bulk of the catalyst,12,15,16 the growth mechanisms for compound 2D materials such as h-BN are inherently more complex, as two elements need to be fed and incorporated into the growing nanostructure and bond polarity can affect the structural formation.1,2 There is typically a range of poly-types for compound materials, and hence selective growth becomes increasingly challenging.1,2
Monolayer and few-layer h-BN has received considerable attention recently, reflecting an ever increasing demand for ultrathin dielectric, support, or barrier layers in electronics, photonics, and many other applications.17−19 While progress has been made in synthesizing h-BN on several catalyst metals,5,9,20−39 the focus has remained limited to catalyst surface pre-treatment and tuning of precursors and/or exposure conditions.5,9,20−39 Further progress and, for instance, control of layer texture, grain structure, and number of layers require a more detailed understanding, in particular of the basic interactions of B and N with the catalyst. The interaction of h-BN with the catalyst also affects h-BN transfer and an increasing number of applications, such as oxidation barriers and corrosion protection, that utilize h-BN as-grown on top of the catalyst metal.40 Unlike bulk h-BN which is typically thermally and chemically very stable, the use of few-layer h-BN as a high-temperature oxidation barrier on Ni, Cu, and stainless steel has recently highlighted40 that the stability and maximum possible temperatures of these ultrathin films are critically linked to their support and environment.
Here, using a combination of in situ X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) during actual synthesis conditions, we fingerprint the entire h-BN CVD growth process on polycrystalline Cu using borazine, (HBNH)3, as the precursor. We focus on the important questions of how the precursor and constituent B and N as well as the as-grown h-BN interact with the catalyst during and after growth. Polycrystalline Cu foil catalysts are currently most widely used for graphene CVD, motivated by the rationale that the low carbon solubility of Cu offers a relatively error-tolerant growth window for monolayer graphene formation via a mainly Cu surface-based mechanism.12,41 Based on the ease of processing, polycrystalline Cu foils are now also used for h-BN CVD.5,20−26 Our data show that, at the typically used growth temperatures, the borazine molecule decomposes and B incorporates into the bulk of the Cu catalyst. During h-BN growth the Cu is in the metallic face-centered-cubic (fcc) phase; whereby, we can measure a lattice dilation due to the dissolved B. Upon post-growth cooling we observe precipitation of B from the fcc Cu. Compared to graphene CVD, the role of the surface, sub-surface, and bulk of the catalyst is more complex, considering that N has a considerably lower solubility in Cu42,43 and its incorporation into the Cu bulk is not detected. We invoke a comparison to catalytically mediated III–V nanowire growth, i.e., the CVD of 1D nanomaterials, where the growth kinetics crucially depend on the different solubilities of the group III and V constituents in the catalyst and where atomically sharp and controlled heterostructures can be grown despite a comparably high bulk solubility of the group III element.44,45 We further show that after h-BN CVD and during commonly used further processing of h-BN on the Cu catalyst in ambient air, oxygen intercalation readily occurs under as-grown h-BN and negatively affects the stability of h-BN on the catalyst. For extended air exposure Cu oxidation is observed, and upon reheating in vacuum an oxygen-mediated disintegration of the h-BN film via volatile boron oxides occurs. Of key importance is that this disintegration is catalyst mediated, i.e., occurs at the catalyst/h-BN interface and depends on the level of oxygen present at this interface. In turn, however, we demonstrate that pre- and co-exposure with oxygen species can be used to influence h-BN growth morphologies. We discuss the implications of these observations in the context of corrosion protection40,46 and relate them to challenges for process integration and heterostructure growth.
Results
We adopt a simple CVD process, and Figure 1 outlines the nomenclature for the process steps that will be used throughout this study. CVD of h-BN was performed at ∼950–1000 °C in customized cold-wall CVD reactors12,14 using borazine, (HBNH)3, as precursor on commercially available cold-rolled polycrystalline Cu foils as catalyst (see Methods).
Figure 1.
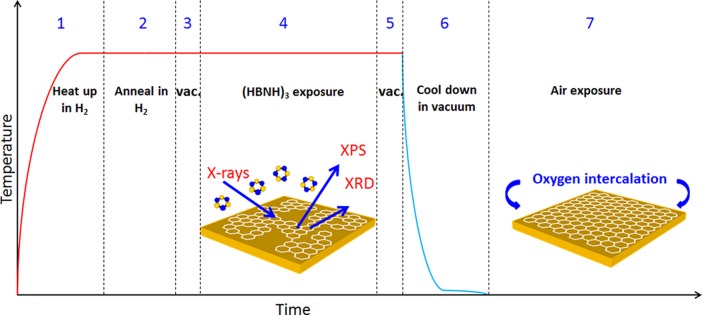
Process diagram illustrating the salient stages of h-BN CVD on polycrystalline Cu.
Figure 2 highlights via various ex situ characterization techniques, the representative h-BN film quality for the CVD recipe used throughout this study. Transmission electron microscopy images at high magnification show a hexagonal lattice structure (Figure 2a), and selected area diffraction (Figure 2b) shows the typical hexagonal spot pattern of h-BN, confirming the high crystalline quality at the microscopic level.47 Figure 2c shows an optical microscopy image of a continuous h-BN film transferred to a SiO2(300 nm)/Si wafer. The image shows large areas of uniform contrast indicating the macroscopic uniformity of the h-BN films along with specs of residual Polymethylmethacrylate (PMMA) typically seen for such transferred films. The PMMA contamination toward the edge of the layer helps to guide the eye to distinguish the film from the substrate in spite of the low optical contrast of h-BN placed on SiO2(300 nm)/Si wafer support.22,48 Raman spectra (Figure 2d) taken on the transferred h-BN film on SiO2 (300 nm)/Si (see Figure 2c) show a single peak at ∼1368 cm–1 consistent with the signature for h-BN.21,24,25,48−50 The corresponding ex situ XPS core-level signatures [i.e., post ambient air exposure (step 7, Figure 1)] acquired on the h-BN films on Cu show in the B1s region a main component at ∼190.6 eV (Figure 2e, inset additionally shows the π bond shake up satellite at 200 eV) and in the N1s region a main component at ∼398.1 eV (Figure 2f). From the intensities we estimate a B:N ratio of ∼1, again consistent with h-BN. We also note that our measured binding energies (BEs) are consistent with previous XPS characterization studies of h-BN.20,23,24,51−53 Further additional fitted components are seen in the ex situ XPS spectra at B1s ∼191.1, 191.9 eV and in the N1s at ∼398.6, 399.3 eV, respectively, and we will discuss the assignment of these peaks in the sections below.
Figure 2.
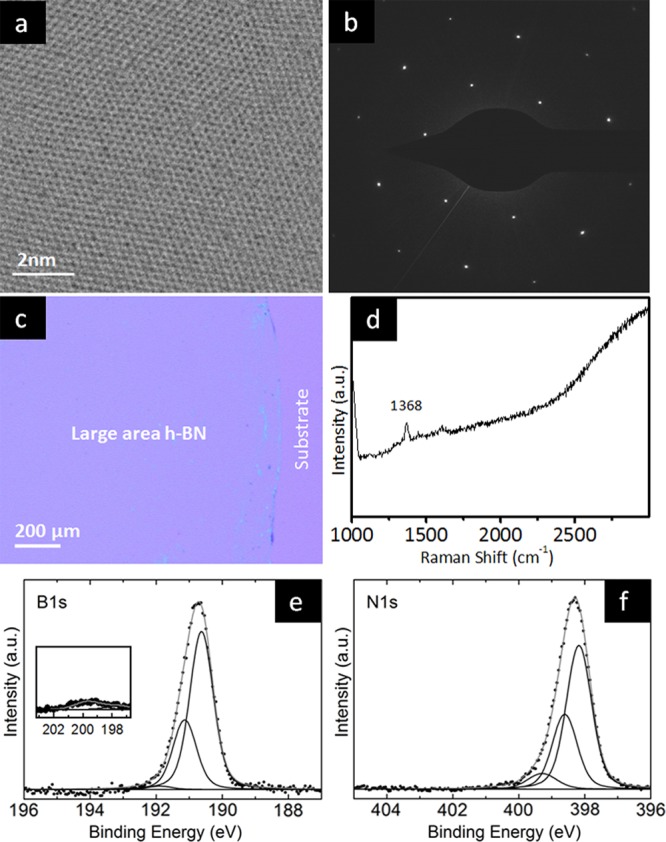
Ex situ characterization of the synthesized h-BN films. (a) High-magnification TEM image showing the hexagonal lattice structure of h-BN. (b) Selected area diffraction pattern for the h-BN film. (c) Optical microscopy image for h-BN film transferred onto a SiO2(300 nm)/Si wafer. The h-BN layer can be clearly distinguished in spite of the low contrast due to the edge where PMMA residue accumulates. (d) Raman spectrum for the transferred h-BN film in (c). (e,f) XPS B1s (e, inset shows the π bond shake-up satellite at 200 eV) and N1s (f) core-level signatures for h-BN on the Cu foil.
Figure 3a and Figure S1 in the Supporting Information show post-growth (step 7, Figure 1) scanning electron microscopy (SEM) images of as-grown h-BN on polycrystalline Cu foil for ∼5, 15, and 30 min of borazine exposure. While the SEM contrast mechanisms of such 2D materials supported on metallic foils/films is still an area of active research, in Figures 3a and S1 the h-BN appears darker than the uncovered Cu. Figure 3a and S1 highlight that a typical h-BN CVD film grows via multiple nucleation points within each facet of the polycrystalline Cu foil (see Figure S1) which then expand as isolated h-BN domains and eventually merge to form a continuous polycrystalline h-BN film. In contrast to graphene,12 the isolated h-BN domains often adopt more strict geometrical shapes (Figure 3a, 300 s and Figure S1a), such as triangles or other simple polygons, due to the bond polarity and related larger energetic differences between different edge terminations.20−22 Longer exposures lead here to the formation of more irregular-shaped domains (Figure 3a, 900 s), which eventually cover the entire Cu surface, as indirectly confirmed by the wrinkles in the resulting h-BN film (Figure 3a, 1800 s, and Figure S1c). Analogous to graphene,12,14 extended exposure times also lead here to the nucleation of further h-BN layers, i.e., multilayer formation via secondary nucleation,5,20−26 as highlighted by the darker contrast regions (Figure 3a, 1800 s). Atomic force microscopy (AFM) with step heights measured on the edges of such a h-BN film transferred to SiO2 (300 nm)/Si wafer (see Supporting Information, Figure S2) indeed confirms the growth of few-layer h-BN films for extended (∼30 min) exposure times under our experimental conditions. Having confirmed the growth of atomically thin h-BN (monolayer islands for short exposures; full-coverage few-layer films for longer exposure times) under the given CVD conditions we now turn to study the growth process using in situ experiments.
Figure 3.

(a) Ex situ high-magnification SEM images for experiments with ∼300, 900, and 1800 s of borazine exposure. Corresponding low-magnification images are presented in the Supporting Information, Figure S1. (b,c) In situ XPS confirming isothermal h-BN growth on Cu: in situ CVD process-resolved XPS B1s (b) and N1s (c) core-level scans at ∼975 °C before (step 3), during (step 4), and after borazine exposure (step 5), after cooling (step 6), and after air exposure (step7).
Figure 3b,c shows CVD process-resolved XPS B1s and N1s core-level signatures measured in situ at steps 2–7 (Figure 1), respectively. To simplify assignments, we refer to pairs of B1s/N1s components for B–N compound species while giving separate respective B1s and N1s BEs for other species. During annealing in H2 (step 2) the flat lines in the respective B1s and N1s spectra (Figure 3b,c) indicate a clean Cu surface as baseline for the subsequent borazine exposure. The time-resolved B1s spectra during borazine exposure (inset in Figure 3b, step 4) show that upon borazine introduction a broad peak in the B1s spectra emerges after ∼290 s. This is consistent with the time scale of the nucleation and growth of h-BN domains observed ex situ by SEM (Figure 3a). At ∼880 s the broad B1s peak can be resolved into two components, which are also reflected in the matching N1s scans with BE pairings of ∼191.1/398.6 eV and ∼190.6/398.1 eV. Around this exposure time the islands have grown in size but have not coalesced yet (Figure 3a). For continued exposures (up to 2270 s) the component at ∼190.6/398.1 eV starts to dominate the spectrum, while the ∼191.1/398.6 eV peak strongly decreases in relative intensity. Concurrently, the ex situ SEM now shows coalescence of the islands into a closed few-layer h-BN film.
The measured 190.6/398.1 eV component is consistent with formation of h-BN.20,23,24,52,53 The 191.1/398.6 eV component could be attributed to cubic boron nitride (c-BN),53−55 but is also consistent with a slightly higher BE of monolayer h-BN (as compared to few-layer h-BN at 190.6/398.1 eV).40 We exclude significant homogeneous c-BN formation based on the ex situ characterization of our films (Figure 2) and also based on the observation of a π bond shake-up satellite at ∼200 eV in the B1s spectra (see inset of Figure 2e).54 Therefore, we interpret the observed initial emergence of the two peaks at 190.6/398.1 and 191.1/398.6 eV at equal intensities followed by domination of the spectrum by the 190.6/398.1 eV component for extended exposure times as evidence for isothermal growth of monolayer h-BN that interacts with the Cu substrate (191.1/398.6 eV) and some fraction of multilayer h-BN nuclei (190.6/398.1 eV), where the initial monolayer domains further evolve to multilayers with extended exposure time (hence the decrease of 191.1/398.6 eV contribution relative to the 190.6/398.1 eV peaks). We suggest that the observed peak shift between 191.1/398.6 and 190.6/398.1 eV is caused by interactions of h-BN with the Cu substrate,23,56,57 similar to monolayer graphene i.e., charge transfer between the substrate and the growing 2D material.12,58 In turn this interaction is not present for multilayer h-BN, where the top layer (which contributes most significantly to the XPS signal) is supported by the other h-BN layers and thus shows the BE at 190.6/398.1 eV.58 Further evidence for this interpretation of h-BN/Cu interactions for monolayer h-BN is provided below.
In addition to the assignment of the monolayer h-BN (191.1/398.6 eV) and multilayer h-BN (190.6/398.1 eV) XPS components, we note that, due to the overlap of BE positions for multiple species, part of the signal at 191.1/398.6 eV may also be assigned to local cubic-like sp3 bonding arrangements54,55,59 at h-BN island edges or defects. Also such a cubic-like sp3 contribution from nuclei edges would similarly decrease in relative intensity with exposure time as the islands increase in area and therefore exhibit a decreasing fraction of signal from edges. Our ex situ characterization however excludes significant cubic-like BN contributions.
Besides the 191.1/398.6 and 190.6/398.1 eV components, we also observe small XPS fitted components at ∼191.9/399.3 and ∼189.8/397.5 eV. We note that borazine is known to dissociate on metal catalyst surfaces at the given growth temperature,60 meaning that these remaining unassigned B–N-related components during growth cannot be ascribed to adsorption of intact borazine molecules on the catalyst surface. Instead we find that the 191.9/399.3 eV component also emerges when we sputter ex situ grown h-BN on Cu post air exposure (see Supporting Information, Figure S3). As sputtering is known to induce defects and dangling bonds, we assign the very small components at B1s/N1s ∼191.9/399.3 eV to h-BN defect species saturated with residual oxygen. Similarly, we also assign the ∼189.8/397.5 eV components to defects in the h-BN.61
Our observations so far indicate isothermal growth of h-BN on Cu with initial monolayer nucleation (which then evolve toward few-layer h-BN) with a small amount of defects incorporated in the h-BN films (lattice, edges, grain boundaries). Such an isothermal growth scenario is consistent with ultra-high vacuum (UHV)/single crystal studies, where the precursor pressure however remained below the threshold for multilayer nucleation.9,34−38
The in situ XPS B1s and N1s fingerprints of growing h-BN are preserved upon removal of borazine at temperature (step 5, Figure 1), but interestingly one small additional component emerges upon cooling of the sample in vacuum in the B1s at ∼188.1 eV (step 6, Figure 1). This BE is known to correspond to atomic B diffused to the catalyst surface or sub-surface.60,62 This assignment of atomic B (non-nitrogen bonded) is also corroborated by the lack of a concurrently emerging N1s component upon cooling, as well as by the disappearance of the 188.1 eV signal upon subsequent air exposure, since atomic boron is known to quickly oxidize in ambient air and desorb as highly volatile boron oxides.63 The emergence of atomic B on the catalyst surface upon cooling strongly implies B precipitation from the catalyst bulk and in turn suggests dissolution of B in the polycrystalline Cu subsurface/bulk during the high-temperature borazine exposure.
To confirm this B dissolution, we employ in situ XRD to characterize the crystalline structure of the catalyst bulk during CVD. Figure 4a shows XRD patterns recorded during CVD, starting with as loaded Cu catalyst (before step 1, Figure 1), after H2 anneal (step 3, Figure 1), during borazine exposure (step 4, Figure 1), and after ambient air exposure (step 7, Figure 1). The growing h-BN layers are not seen in the XRD patterns due to the setup geometry/sensitivity used here (see methods). From the XRD patterns we observe metallic fcc Cu as the state of the catalyst bulk during the entire growth process. Importantly, no (non-equilibrium) boride or borate phases are observed. Rietveld refinement of the patterns in Figure 4a reveals that when borazine is introduced the Cu lattice constant expands by ∼0.0006 Å (Figure 4b) compared to a vacuum baseline. This lattice expansion is indicative of interstitial uptake of a borazine-derived component into the Cu lattice. We note that this lattice constant change remains after borazine exposure with the sample still at temperature (step 5, Figure 1). In order to get some indication of the nature of the dissolving species, we characterized by XRD the Cu catalyst exposed to ammonia (NH3, i.e., a nitrogen and hydrogen source without B) instead of borazine under similar pressures. For this ammonia exposure, no expansion in the Cu lattice constant is found. As ammonia is known to dissociate on Cu in this temperature regime,64,65 our data imply that N and H uptake is not responsible for the increase in the Cu lattice constant, and instead B is dissolved into the Cu during growth. Therefore, our combined XPS and XRD data suggest that the h-BN growth mechanism on Cu is not limited to a pure surface mechanism as previously suggested.52 Rather our data indicate element-specific feeding mechanisms including the bulk of the catalyst. We suggest that B is taken up in the Cu catalyst while N is not (by relative amounts), which is also in agreement with thermodynamic phase diagrams.42,43,66 We will further discuss the implications of this observation below.
Figure 4.
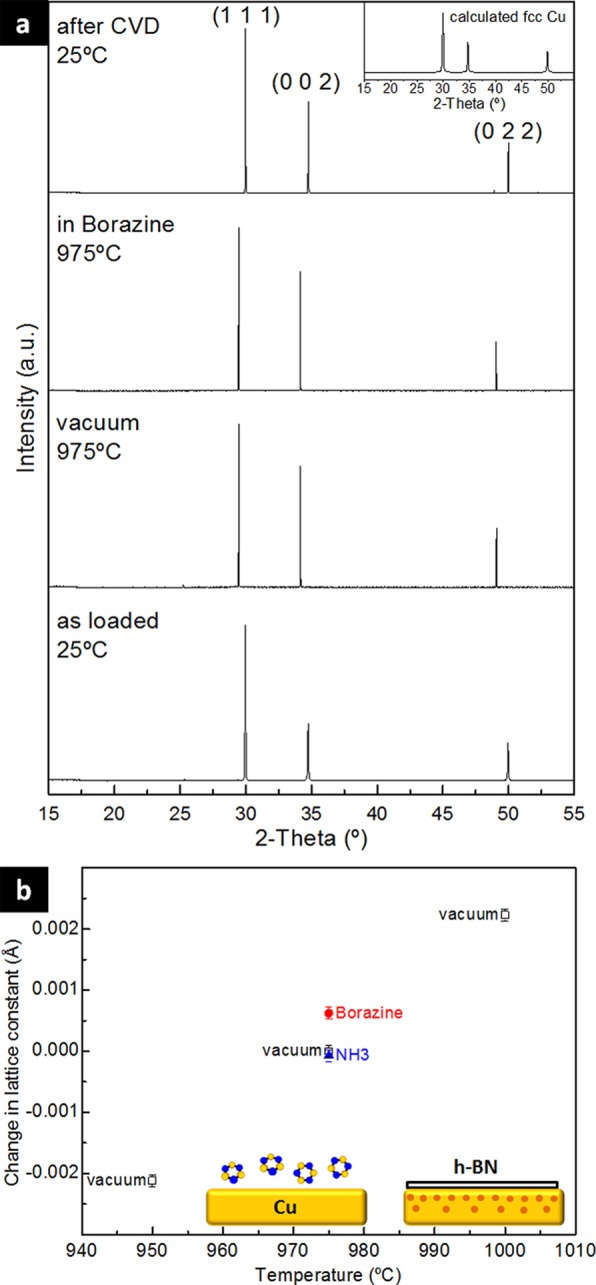
Bulk crystallography of the Cu catalyst during isothermal growth of h-BN by CVD measured using in situ XRD. (a) XRD patterns during salient stages of CVD as loaded (before step 1), after H2 anneal (step 3), during borazine exposure (step 4), and after ambient air exposure (step 7). (b) Rietveld refinement derived change in fcc Cu lattice parameter during vacuum and borazine and NH3 exposure (corrected for thermal expansion via measuring a baseline temperature series in vacuum).
Returning to the XPS data in Figure 3b,c, we find that after extended air exposure the N1s spectra show an additional small component at ∼400.5 eV that does not have a B1s counterpart and is attributed to the formation of N–O bonds.63,67 Further details on the effect of oxygen exposure of samples is provided below. We note that the final in situ XPS fingerprint after air exposure (step 7) in Figure 3b,c resembles our X-ray photoelectron spectra of the ex situ grown few-layer h-BN in Figure 2e,f which highlights the validity and relevance of our in situ measurements for scalable high-quality h-BN growth under realistic process conditions.
In addition to following the evolution of B and N species during h-BN CVD, in situ XPS also allows us (within its detection limits) to observe the surface chemistry of the Cu catalyst by simultaneously monitoring the Cu2p, CuLMM, O1s (Figure 5), and valence band (Supporting Information, Figure S4) regions.12 We find that, before CVD, the as-loaded Cu foil surface is heavily oxidized due to storage and transportation in ambient air (before step 1).12 Following an anneal (step 2) in H2 the Cu surface is reduced to metallic Cu (step 3) and remains metallic during growth (step 4) and cooling post-growth (step 6). This is in good agreement with the XRD data in Figure 4a that suggested the Cu bulk to be metallic during h-BN growth. Upon air exposure (step 7) the h-BN covered Cu surface oxidizes again. The oxidation of the Cu upon air exposure implies that the inherent polycrystallinity of CVD grown h-BN film provides diffusion pathways for species to reach the Cu/h-BN interface. This suggests that h-BN films as-grown on Cu per se are not as perfect oxidation barriers as recently reported.40,46 We emphasize here that the O1s and CuLMM are more sensitive to measure the oxidation of the Cu than the Cu2p.12 The observation of oxidation upon air exposure is also consistent with the disappearance of the elemental B peak (desorbed as volatile Boron-oxides, Figure 3b) and the formation of N–O bonds (Figure 3c) upon air exposure.
Figure 5.

Surface chemistry of the Cu catalyst during isothermal growth of h-BN by CVD: In situ XPS (a) Cu2p3/2, (b) CuLMM Auger, and (c) O1s spectra after H2 anneal in vacuum (step 3), during borazine exposure (step 4), after exposure (step 5), after cooling in vacuum (step 6), and after ambient air exposure (step 7).
To further probe this evolution of h-BN on Cu catalysts during ambient air exposure (step 7, Figure 1), we undertake complementary experiments with ex situ grown h-BN islands on Cu, for which the borazine exposure was limited to ∼5 min to obtain islands of monolayer h-BN. This significantly increases the direct contribution of the Cu/h-BN interface. We measure XPS on these ex situ grown h-BN islands on Cu exposed to ambient air for 4 days after CVD (Figure 6). The as-loaded sample shows a B1s peak centered at ∼190.6 eV and a N1s peak centered at ∼398.1 eV, and the O1s and CuLMM peaks indicate an oxidized Cu surface.12 The BEs of the B1s and N1s peaks are surprising, as we established above that monolayer h-BN should exhibit a BE of 191.1/398.6 eV. Interestingly, however, upon annealing the sample in vacuum (10–6 mbar) we observe a shift of the XPS peak centers to the expected positions for monolayer h-BN, i.e., the B1s to ∼191.1 eV and N1s to ∼398.6 eV. Concurrently, we see a reduction of the Cu in the O1s and CuLMM spectra during vacuum annealing and the appearance of a defect related component at 191.9/399.3 eV. In turn, after cooling and upon subsequent room temperature re-exposure to air for 2 days, the initial as-loaded positions of the B1s and N1s are recovered (190.6/398.1 eV) along with a re-oxidation of Cu.
Figure 6.

Re-heating of triangular h-BN nuclei on Cu in ∼10–6 mbar vacuum, after sample was stored in ambient air for ∼4 days. In situ X-ray photoelectron spectra of (a) B1s, (b) N1s, (c) Cu Auger (LMM), and (d) O1s region of as-loaded sample, during subsequent heating in vacuum up to 650 °C, post-cooling, and after re-exposure to air (from bottom to top).
This behavior, where changes in the XPS BEs of h-BN are linked to reversible support oxidation/reduction cycles, closely resembles previous observations of oxygen intercalation phenomena under graphene on metals.12,68−71 To the best of our knowledge oxygen intercalation has not been reported yet for h-BN on metals, but previous studies of Au intercalated under h-BN on Ru,72 Ni,73 and hydrogen intercalated under h-BN on Rh74 showed XPS peak shifts consistent with our interpretation. Therefore, our data strongly suggest that air exposure of h-BN on Cu leads to oxygen intercalation under the h-BN, i.e., at the h-BN/Cu interface. In this intercalated state monolayer h-BN has a XPS fingerprint of 190.6/398.1 eV, resembling the BE of few-layer h-BN but being different to the 191.1/398.6 eV measured in Figure 3b,c for monolayer h-BN during growth directly on Cu. Upon vacuum annealing the intercalated oxygen is de-intercalated and desorbed, and thus direct contact between h-BN and Cu is (re-)established albeit with a small number of defects introduced into the h-BN layer. The weak but nevertheless present electronic interaction of Cu and monolayer h-BN23,56,57,75,76 leads to a small shift in the B1s/N1s BEs to 191.1/398.6 eV (same as during growth). Thus, our data in Figure 6 evidence the “decoupling” (upon air exposure) and “recoupling” (upon vacuum annealing) of monolayer h-BN and the Cu support. We emphasize here that coupling does not necessarily imply a strong interaction with the substrate. In the case of few-layer h-BN only the “decoupled” BEs 190.6/398.1 eV are observed, as for the top h-BN layers (which mainly contribute to the XPS signal) the interaction with the Cu support is screened by the h-BN layers below.58 Therefore, in our in situ growth experiments in Figure 3b,c where few-layer h-BN was eventually grown, no B1s/N1s peak shifts were observed upon air exposure.
While our data strongly suggest intercalation of oxygen, we however note that a second possible scenario could also explain the B1s/N1s peak shifts, where oxygen species decorate edges (which are present in large number for h-BN islands) or defect sites on top of the h-BN.77 Oxygen in this scenario is not intercalated under the h-BN but rather adsorbed onto it and then removed by heating. We note that the already slightly back shifting B1s/N1s BEs during cooling in vacuum in Figure 6 could point to fast oxygen decoration of edge/defects sites from residual oxygen groups in the vacuum chamber accompanied by a small amount of degradation/defect generation in h-BN due to atmospheric contaminants. Thus, a combination of fast oxygen decoration of defects on top of h-BN and oxygen intercalation under h-BN at higher oxygen pressures is also conceivable as a combined source of the observed B1s/N1s BE shifts upon air exposure/vacuum annealing cycles.
Irrespective of the detailed configuration of the oxygen causing the B1s/N1s shifts, we find that the evolution upon vacuum-annealing of ex situ grown h-BN samples on Cu is highly dependent on their length of exposure to ambient air. In contrast to the short-term air-exposed monolayer sample in Figure 6 (4 days, termed “fresh” sample for the remainder of the text), Figure 7 shows B1s, N1s, and O1s XPS core-level signatures for an ex situ grown, full-coverage few-layer h-BN film on Cu foil stored in ambient air for ∼10 months (termed “aged” sample). The as-loaded B1s and N1s spectra show peaks corresponding to h-BN, and the O1s spectrum shows the presence of copper oxide species (Cu2O). For this “aged” sample, upon heating in vacuum (∼10–6 mbar) to 500 °C, the intensity of the B1s and N1s increases which is attributed to desorption of adsorbed atmospheric dirt. The B1s and N1s BEs remain consistent with few-layer h-BN as expected. Concurrently, the O1s spectrum shows the onset of reduction of the Cu2O and reaches a state that resembles a sub-stoichiometric oxide or adsorbed O.12 When the sample is further heated to 700 °C all three spectra change drastically. The B1s and N1s peaks reduce dramatically in intensity, and the B1s shows predominantly features of boron oxide (BxOy) species at >191.5 eV.62,67 This is accompanied by a corresponding increase in the O1s intensity, also indicating an increase in boron oxide species. This points toward oxygen-mediated disintegration of the h-BN film on the Cu foil upon vacuum annealing, where the oxygen was fed from the Cu oxide underneath the h-BN. The formed boron oxides are largely volatile63 resulting in loss of B and N into the gas phase. This is in stark contrast to the higher stability of the “fresh” h-BN islands in Figure 6 which under similar treatment only showed signs of minor degradation. We suggest that the higher degree of oxidation of the “aged” Cu foil provided a larger oxygen “reservoir” where instead of simple de-intercalation under the h-BN, catalytic oxygen-mediated etching of h-BN occurs (which in the case of the “fresh” h-BN on Cu is much milder due to the smaller oxygen reservoir in the less oxidized Cu).
Figure 7.

Reheating of full-coverage, few-layer h-BN films on Cu in vacuum (∼10–6 mbar), after sample was stored in ambient air for ∼10 months: (a) B1s, (b) N1s, and (c) O1s. From top to bottom: as loaded, at 500 °C, at 700 °C, and post-cooling.
To cross-check this hypothesis, we return to annealing similarly “fresh” h-BN islands as in Figure 6 but now anneal in the presence of 1 × 10–4 mbar oxygen (Figure 8). These experimental conditions can be considered as heating the “fresh” h-BN sample in an infinite oxygen reservoir. Figure 8 shows that in the presence of oxygen the islands quickly disintegrate on heating (confirmed by SEM in the inset), similar to the “aged” h-BN film in Figure 7. This confirms our assertion that when sufficient amounts of oxygen are provided (either from a heavily oxidized Cu as in the “aged” sample or via the gas phase as in Figure 8) h-BN on Cu is etched via a reaction with oxygen. We emphasize that control experiments with h-BN that was transferred to SiO2, i.e., removed from the Cu catalyst, did not lead to such a drastic disintegration during annealing in air up to 700 °C. This highlights the role of the Cu catalyst in the proposed oxygen-mediated etching of h-BN. Such catalytically mediated oxygen-based etching of h-BN is consistent with recent UHV-based reports of etching of h-BN on Ir67 and Ru.78
Figure 8.

Re-heating of triangular h-BN nuclei on Cu in 1 × 10–4 mbar O2, after sample was stored in ambient air for ∼15 days: (a) B1s, (b) N1s, (c) O1s, and (d) CuLMM as loaded and during subsequent heating in 1 × 10–4 mbar O2 (from top to bottom). The insets show low- and high-magnification SEM images before (left) and after (right) re-heating in O2. Heating in O2 causes the h-BN on Cu to disintegrate in contrast to Figure 6 but similar to the observations in Figure 7.
Having established that oxygen can have a drastic detrimental influence on the stability of h-BN on Cu during ambient air processing after CVD, we now study whether this impact of oxygen can also be used advantageously during h-BN growth to controllably influence h-BN characteristics by controlled etching. In analogy to graphene CVD12,79 we expect that oxygen feeding prior/during Borazine exposure will have an impact on nucleation80 and might change the balance between growth and etching reactions, and thus strongly modify the resulting h-BN film morphologies.81 Confirming this hypothesis, Figure 9a,b demonstrates a reduction in nucleation density of h-BN on Cu by dosing 1 × 10–4 mbar of air after annealing in hydrogen and before borazine exposure, i.e., between steps 2 and 4. In the case of air-dosing (Figure 9b), the nucleation density is reduced by an order of magnitude compared to the standard oxygen-free growth conditions (Figure 9a). The short exposure to air/oxygen after H2 pre-treatment partly re-oxidized the Cu surface, and subsequently, upon borazine exposure, boro-thermal reduction (see Figure 5c) of these surface copper oxides to active metallic Cu (confirmed by XRD measurements) takes place, which significantly reduces catalyst activity and thus the resulting nucleation density. In Figure 9c we show the effect of co-exposure of air and borazine: co-dosing a small air partial pressure alongside the borazine changes the h-BN domain morphology toward an flower-like shape (Figure 9c). We suggest that this change of growth morphology is due to increased selective etching reactions occurring concurrent with the growth reactions which might also contribute to a less defined h-BN termination. These observations introduce oxygen pre-treatment and co-feeding as highly relevant parameters toward optimizing h-BN CVD.
Figure 9.

SEM images of h-BN nuclei on Cu after 5 min growth, (a) similar to Figure 3a (Pborazine ≈ 1 × 10–3 mbar) and (b) with 5 × 10–4 mbar air exposure prior to step 3. Air exposure caused an order of magnitude reduction in nucleation density of h-BN on Cu for identical processing conditions. (c) Co-exposure of air (1 × 10–4 mbar) and Pborazine ≈ 1 × 10–3 mbar for 30 min leads to nuclei that are irregular in shape.
Discussion
In order to put these results in context, we start by outlining the growth models introduced for graphene. Without going into the details of the specific chemistry for each catalyst material, graphene nucleation requires a certain carbon surface concentration which is fed by the precursor dissociation and can be balanced by carbon diffusion in to and out from the catalyst subsurface and bulk.15 The growth kinetics thereby strongly depend on any possible phase changes of the catalyst surface/bulk, if the carbon incorporates from the catalyst surface or via the bulk, and how the graphene layer edges are anchored on the catalyst surface.15
In the case of h-BN the growth mechanism is expected to be more complex than for graphene since a balance of two elements, B and N, are needed to form h-BN on the catalyst. While a stoichiometric balance can be introduced into the CVD reactor by selecting a precursor with a pre-defined stoichiometry (i.e., borazine in this case with a B:N = 1), the interaction of the constituent elements with the catalyst will dictate the actual supply of the elements during CVD. In this context, diffusion rates and solubility of B, N, or both into the catalyst sub-surface or bulk, metastable or stable boride/nitride formation, and the phase of the catalyst will critically affect the supply of the constituent elements. Further, the rate of supply of the constituent elements based on dissociation kinetics, impingement flux of the precursor on the catalyst surface, sticking coefficients, and any related change in interaction between the growing h-BN and the catalyst82 need to be carefully considered.
Figure 10a schematically puts forward key points of a kinetic growth model that we propose for h-BN CVD on the basis of a consistent interpretation of the experimental data. Our data show no signatures of adsorbed borazine molecules on Cu, indicating that at the given conditions borazine dissociates on the reduced Cu surface. We also observe a lattice expansion during borazine exposure which suggests uptake of Boron into the Cu bulk, in agreement with our conjectures from the XPS B1s data, where upon post-growth cooling we observe B precipitation from Cu. We further see no evidence of bulk nitrogen dissolution or surface nitrogen species in bonding configurations other than those related to BN compounds during exposure. Further, the Cu catalyst remains in its metallic state during the entire CVD process, and no borides or nitride phases are seen. These findings are consistent with Cu–B and Cu–N phase diagrams42,43,66 as well as with previous UHV measurements of h-BN CVD on Rh and Ir62,83 catalysts and the general metallurgy literature.84,85 The simplified schematic of Figure 10a neglects a certain level of Cu sublimation under low-pressure CVD conditions,12,14 but this can be minimized12,14 or indeed might be significantly suppressed with h-BN coverage, and hence this complexity is not further discussed here.
Figure 10.
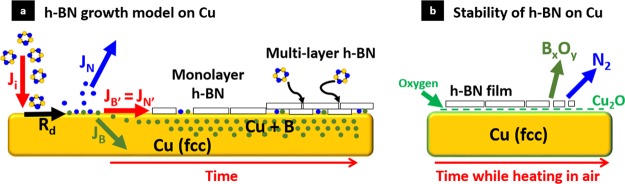
(a) Schematic diagram for h-BN growth mechanism on Cu, where Ji is the impingement flux of the precursor (borazine), Rd is the rate of dissociation of the precursor, JB is the diffusion flux of B into the Cu subsurface/bulk, JN is the diffusion flux of N away from the catalyst surface, and JB′ = JN′ is the flux of B and N required for the formation of h-BN on the catalyst surface. We propose that multilayer h-BN grows beneath the first layer in contact with the catalyst surface as seen for graphene. The schematic neglects a certain level of Cu sublimation under low-pressure CVD conditions.12,14 (b) Schematic diagram for stability of h-BN on Cu and the oxygen-mediated catalytic dissociation mechanism while heating in air or an oxygen reservoir.
The nucleation and growth of h-BN layers is found to occur isothermally, i.e., at a constant elevated temperature. Although a stoichiometrically balanced precursor is used, our data show that the boron and nitrogen supply routes for h-BN growth can be different. The boron dissolution could leave a nitrogen excess at the Cu surface which might be balanced by nitrogen desorption, as indicated in Figure 10a. Whereas boron can enter the growing h-BN either via the bulk or the catalyst surface, the nitrogen is fed only via the catalyst surface, and nitrogen desorption can lead to a situation where the h-BN growth is limited by the nitrogen supply. This is reminiscent of the situation for catalytic III–V nanowire growth, where the group V constituent typically has a low solubility in the conventionally used Au catalyst, and despite a high bulk solubility of the group III element atomically sharp heterostructures can be obtained.44,45 The focus of prior literature on h-BN CVD has been on the balance of gaseous precursors. Our data here highlight that, for further process optimization, it is crucial to consider and understand the catalyst-mediated flux balance as highlighted for Cu in Figure 10a.
Considering the growth of 2D multilayer films and heterostructures, the complexity of the kinetics of catalyst mediated flux balances increases for h-BN. Growth of h-BN multilayers can be seen analogous to graphene CVD, where additional layers nucleate and grow only in contact with the metal catalyst, i.e., at the interface between the catalyst and an existing graphene layer.14,86 The feeding of such additional layer will depend on the leakage rate of both (B,N) constituent species through an existing 2D layer.14 Further, for the growth of h-BN/graphene heterostructures via, e.g., sequential exposure to carbon and boron/nitrogen precursors, complex interactions can be expected not only for the catalytic precursor dissociation on the catalyst surface but also for the catalyst-mediated flux balances. Sutter et al.9 have studied potential smear-out effects during h-BN-graphene heterostructures synthesis and reported techniques such a pulsed oxygen etch to address reservoir effects and improve the sharpness of transitions, albeit this has been very specific so far to the exposure sequence and catalyst material (Ru). We see our insights here as being highly relevant as a framework for the understanding and further optimization of a range of compound 2D materials and more complex CVD grown heterostructures.
The catalyst support is relevant not only to the CVD growth process but, as our data show, also to the post-growth stability of as-grown h-BN, which is crucial to post-growth processing and numerous applications. Practically all current use and processing of CVD grown h-BN involves some stage of ambient air exposure when the material is still on the catalyst. Figure 10b schematically summarizes the key points regarding catalyst- and oxygen-mediated thermal h-BN stability. We find that post growth ambient air exposure leads to intercalation of oxygen under h-BN on Cu. While oxygen can reach the interface through inherent defects in the h-BN layer (including point defects,87−91 vacancy defects,89,92 Stone Wales defects,89 or out-of-plane bonds93) and domain boundaries,89 even when the catalyst surface is completely covered with monolayer and few-layer h-BN, the rate of arrival at the interface is expected to reduce significantly with increasing number of h-BN layers.94 This is analogous to what we showed for graphene, which “weakly” interacts with Cu.12 For extended air exposure Cu oxidation is observed, and upon re-heating in vacuum an oxygen-mediated disintegration of the h-BN film occurs via volatile boron oxides. Important thereby is that this disintegration is catalyst mediated. i.e., occurs at the catalyst/h-BN interface and depends on the catalyst and the level of oxygen fed to this interface (Figure 10b). The remaining nitrogen leaves the Cu catalyst surface by forming N2. This mechanism is in conceptual agreement with UHV-based reports of h-BN etching on Ir67 and Ru.78 It is important to note that annealing of h-BN films transferred to a 300 nm SiO2 /Si wafer support in air up to 700 °C did not lead to such a drastic disintegration consistent with recent reports.95 Hence our data highlight that key to the thermal stability are the catalyst interaction and the level of oxygen fed to the catalyst interface.
For few-layer and multilayer h-BN this means that the layer most likely to be etched is the one in contact with the catalyst as long as an oxygen leakage pathway is provided. We note that for certain catalysts, in particular Ni, which more strongly interact with the 2D layer on its surface, graphene82,96 has been shown to passivate the catalyst surface and prevent oxygen from reaching the catalyst/graphene interface.97 This further highlights the importance of the catalyst interaction, and the resultant different levels of oxygen intercalation can explain previous reports in literature for instance on the stability of few-layer h-BN on Cu being limited to 500 °C compared to 1100 °C for Ni.40
Combining the discussion of the h-BN growth kinetics (Figure 10a) and thermal stability (Figure 10b), it is obvious that the presence of oxygen during CVD can affect the h-BN formation in multiple ways. A more oxidized catalyst surface is likely to reduce the h-BN nucleation density, assuming a catalyst surface reduction is required. The presence of oxygen might also change the catalyst/h-BN interaction and change the B and N flux balance, hence affecting the shape of growing h-BN domains and their growth rate. This is fully consistent with our data in Figure 9. Similar to graphene CVD, we note oxygen (co-)exposure should be considered an important aspect of the parameter space for h-BN CVD.80 Although a detailed study of the effect of gaseous additions to the CVD atmosphere is beyond the scope of this paper, we emphasize that the insights provided here give a fundamental framework and highlight multiple avenues toward more advanced integrated growth and integration of h-BN and other related compound 2D materials.
Conclusions
In summary, we have used a combination of complementary in situ XPS and XRD during h-BN CVD to gain a fundamental understanding of the mechanisms underlying the growth of h-BN on polycrystalline Cu under scalable CVD conditions. h-BN was shown to grow isothermally on Cu during exposure to borazine at elevated temperature. The growth, however, is not limited to a surface-mediated growth mechanism as previously reported. Expansion of the Cu lattice during borazine exposure at high temperature and elemental B precipitation from Cu upon cooling confirm B incorporation into the bulk of the catalyst. Cu maintains its metallic state during h-BN CVD, and no additional boride or borate phases are seen. Our in situ insights into the mechanisms of h-BN growth provide guidelines for future rational CVD process engineering towards controlled h-BN film characteristics such as film quality, domain size, or number of layers. The element-specific mechanistic insights obtained here are highly relevant as a framework for the understanding and further optimization of a range of compound 2D materials and more complex CVD-grown heterostructures. After CVD, we find that exposure to ambient air leads to intercalation of oxygen under h-BN on Cu. The stability of h-BN following this air exposure is strongly dependent on the catalyst interaction and accumulated oxygen dose, where high oxygen levels lead to drastic catalytically mediated h-BN etching and h-BN disintegration on Cu upon processing at elevated temperatures. In turn, however, these etching effects could potentially open new routes to control the growth morphology of h-BN when oxygen is pre-dosed or co-fed under controlled conditions during h-BN CVD. Large-scale processing schemes for h-BN integration and processes involving growth of in-plane/out-of-plane h-BN/2D material heterostructures4−11 will have to carefully consider these findings in terms of allowable levels of oxygen exposure during (re-)processing and suggested use of h-BN as high-temperature passivation layers.40
Methods
CVD of h-BN using borazine ((HBNH)3, Fluorochem, UK) as precursor was performed in customized in situ compatible cold-wall CVD reactors12,14 on cold-rolled polycrystalline Cu foils (Alfa Aesar, 25 μm thick, 99.999% purity) at ∼950–1000 °C and at Pborazine ≈ 1 × 10–4–5 × 10–3 mbar for 5–30 min. Typical base pressures were ∼10–6 mbar for all ex situ and in situ chambers. Borazine vapor was introduced into the reaction chambers via leak valves from liquid borazine reservoirs. The CVD process commonly consisted of a H2 pre-treatment step (∼0.2 mbar), followed by pump-down to base pressure, and then the precursor was introduced to initiate h-BN growth. After a pre-defined growth time the borazine precursor was removed, followed by pump-down to base pressure and cooling in vacuum. Figure 1 summaries the salient steps during the h-BN CVD process. For experiments with air pre-dosing, the dosing was performed after H2 annealing, i.e., between steps 2 and 3 in Figure 1.
The ISISS end station of FHI MPG at the BESSY II synchrotron was used for in situ XPS measurements. Further details on the experimental setup/measurements can be found elsewhere.12,98
In situ XRD (θ–2θ geometry) was performed at the BM20 beamline (Rossendorf beamline) of the European Synchrotron Radiation Facility (ESRF) in a cold-wall stainless steel reactor mounted on a high-precision six-circle goniometer.12 The Kapton windows of the reaction chamber allow transmission of X-rays at different scattering geometries. A monochromatic X-ray beam of 11.5 keV (corresponding wavelength of 1.078 Å) was used, and the diffracted X-rays were measured with a 1D line detector (Mythen). Cu powder (Alfa Aesar, <5 μm, 99.9% purity) pressed into a thick granular film onto a sapphire wafer was used as the model system since the high degree of texture in cold rolled Cu foils prevented reliable measurement of the powder diffraction geometry.12 A boron nitride-coated graphite resistive heating element (Boralectric) was used to heat the sample inside the chamber.12 The sample was clamped down with alumina spacers, and the temperature was measured with a thermocouple in contact with the sapphire substrate.12 Gases were fed via computer-controlled mass flow controllers and the borazine via a manual leak valve. Quantitative lattice parameters were derived by Rietveld refinement using X’Pert Plus and file 64699 (fcc Cu) from the Inorganic Crystal Structure Database (ICSD).
Scanning electron microscopy (SEM, Carl Zeiss SIGMA VP, 1–2 kV) and Raman spectroscopy (Renishaw in-Via 532 nm laser) after transfer, using a PMMA support layer as described in detail elsewhere,14,99,100 were used to characterize h-BN growth. We note that the presented SEM images are representative of measurements across several macroscopically separated points on the samples. An FEI Titan 80-300 ETEM equipped with a monochromator at the electron gun and a spherical aberration (Cs)-corrector for the objective lens was used for acquiring TEM images. Samples are heated for 1 h at 350 °C in a vacuum prior to imaging. Images were acquired at 80 kV, energy spread <0.3 eV, and corrector alignment to minimize Cs to obtain a resolution better than 0.12 nm.
Acknowledgments
P.R.K. acknowledges funding from the Cambridge Commonwealth Trust and the Lindemann Trust Fellowship. R.S.W. acknowledges a research fellowship from St. John’s College, Cambridge. S.H. acknowledges funding from ERC grant InsituNANO (no. 279342), EPSRC under grant GRAPHTED (project reference EP/K016636/1), Grant EP/H047565/1, and EU FP7 Work Programme under grant GRAFOL (project reference 285275). The European Synchrotron Radiation Facility (ESRF) is acknowledged for provision of synchrotron radiation and assistance in using beamline BM20/ROBL. We acknowledge Helmholtz-Zentrum-Berlin Electron storage ring BESSY II for synchrotron radiation at the ISISS beamline and continuous support of our experiments.
Supporting Information Available
Figures S1–S4, showing additional SEM and AFM images, and further XPS measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# P.R.K.: Department of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
The authors declare no competing financial interest.
Supplementary Material
References
- Butler S. Z.; Hollen S. M.; Cao L.; Cui Y.; Gupta J. A.; Gutiérrez H. R.; Heinz T. F.; Hong S. S.; Huang J.; Ismach A. F.; Johnston-Halperin E.; Kuno M.; Plashnitsa V. V.; Robinson R. D.; Ruoff R. S.; Salahuddin S.; Shan J.; Shi L.; Spencer M. G.; Terrones M.; Windl W.; Goldberger J. E. ACS Nano 2013, 7, 2898–2926. [DOI] [PubMed] [Google Scholar]
- Xu M.; Liang T.; Shi M.; Chen H. Chem. Rev. 2013, 113, 3766–3798. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Connell J. W. Nanoscale 2012, 4, 6908–6939. [DOI] [PubMed] [Google Scholar]
- Haigh S. J.; Gholinia A.; Jalil R.; Romani S.; Britnell L.; Elias D. C.; Novoselov K. S.; Ponomarenko L. A.; Geim A. K.; Gorbachev R. Nat. Mater. 2012, 11, 764–767. [DOI] [PubMed] [Google Scholar]
- Roth S.; Matsui F.; Greber T.; Osterwalder J. Nano Lett. 2013, 13, 2668–2675. [DOI] [PubMed] [Google Scholar]
- Wang M.; Jang S. K.; Jang W.-J.; Kim M.; Park S.-Y.; Kim S.-W.; Kahng S.-J.; Choi J.-Y.; Ruoff R. S.; Song Y. J.; Lee S. Adv. Mater. 2013, 25, 2746–2752. [DOI] [PubMed] [Google Scholar]
- Miyata Y.; Maeda E.; Kamon K.; Kitaura R.; Sasaki Y.; Suzuki S.; Shinohara H. Appl. Phys. Express 2012, 5, 085102. [Google Scholar]
- Levendorf M. P.; Kim C.-J.; Brown L.; Huang P. Y.; Havener R. W.; Muller D. A.; Park J. Nature 2012, 488, 627–632. [DOI] [PubMed] [Google Scholar]
- Sutter P.; Cortes R.; Lahiri J.; Sutter E. Nano Lett. 2012, 12, 4869–4874. [DOI] [PubMed] [Google Scholar]
- Liu L.; Park J.; Siegel D. A.; McCarty K. F.; Clark K. W.; Deng W.; Basile L.; Idrobo J. C.; Li A.-P.; Gu G. Science 2014, 343, 163–167. [DOI] [PubMed] [Google Scholar]
- Han G. H.; Rodríguez-Manzo J. A.; Lee C.-W.; Kybert N. J.; Lerner M. B.; Qi Z. J.; Dattoli E. N.; Rappe A. M.; Drndic M.; Johnson A. T. C. ACS Nano 2013, 7, 10129–10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidambi P. R.; Bayer B. C.; Blume R.; Wang Z.-J.; Baehtz C.; Weatherup R. S.; Willinger M.-G.; Schloegl R.; Hofmann S. Nano Lett. 2013, 13, 4769–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidambi P. R.; Bayer B. C.; Weatherup R. S.; Ochs R.; Ducati C.; Szabó D. V.; Hofmann S. Phys. Status Solidi—Rapid Res. Lett. 2011, 5, 341–343. [Google Scholar]
- Kidambi P. R.; Ducati C.; Dlubak B.; Gardiner D.; Weatherup R. S.; Martin M.-B.; Seneor P.; Coles H.; Hofmann S. J. Phys. Chem. C 2012, 116, 22492–22501. [Google Scholar]
- Weatherup R. S.; Bayer B. C.; Blume R.; Baehtz C.; Kidambi P. R.; Fouquet M.; Wirth C. T.; Schlögl R.; Hofmann S. ChemPhysChem 2012, 13, 2544–2549. [DOI] [PubMed] [Google Scholar]
- Weatherup R. S.; Baehtz C.; Dlubak B.; Bayer B. C.; Kidambi P. R.; Blume R.; Schloegl R.; Hofmann S. Nano Lett. 2013, 13, 4624–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.; Hsu A.; Jia X.; Kim S.; Shi Y. ACS Nano 2012, 6, 8583–8590. [DOI] [PubMed] [Google Scholar]
- Dean C. R.; Young A. F.; Meric I.; Lee C.; Wang L.; Sorgenfrei S.; Watanabe K.; Taniguchi T.; Kim P.; Shepard K. L.; Hone J. Nat. Nanotechnol. 2010, 5, 722–726. [DOI] [PubMed] [Google Scholar]
- Kubota Y.; Watanabe K.; Tsuda O.; Taniguchi T. Science 2007, 317, 932–934. [DOI] [PubMed] [Google Scholar]
- Guo N.; Wei J.; Fan L.; Jia Y.; Liang D.; Zhu H.; Wang K.; Wu D. Nanotechnology 2012, 23, 415605. [DOI] [PubMed] [Google Scholar]
- Lee K. H.; Shin H.-J.; Lee J.; Lee I.-Y.; Kim G.-H.; Choi J.-Y.; Kim S.-W. Nano Lett. 2012, 12, 714–718. [DOI] [PubMed] [Google Scholar]
- Kim K. K.; Hsu A.; Jia X.; Kim S. M.; Shi Y.; Hofmann M.; Nezich D.; Rodriguez-Nieva J. F.; Dresselhaus M.; Palacios T.; Kong J. Nano Lett. 2012, 12, 161–166. [DOI] [PubMed] [Google Scholar]
- Preobrajenski A. B.; Vinogradov A. S.; Mårtensson N. Surf. Sci. 2005, 582, 21–30. [Google Scholar]
- Song L.; Ci L.; Lu H.; Sorokin P. B.; Jin C.; Ni J.; Kvashnin A. G.; Kvashnin D. G.; Lou J.; Yakobson B. I.; Ajayan P. M. Nano Lett. 2010, 10, 3209–3215. [DOI] [PubMed] [Google Scholar]
- Ismach A.; Chou H.; Ferrer D. a; Wu Y.; McDonnell S.; Floresca H. C.; Covacevich A.; Pope C.; Piner R.; Kim M. J.; Wallace R. M.; Colombo L.; Ruoff R. S. ACS Nano 2012, 6, 6378–6385. [DOI] [PubMed] [Google Scholar]
- Chatterjee S.; Luo Z.; Acerce M.; Yates D. M.; Johnson A. T. C.; Sneddon L. G. Chem. Mater. 2011, 23, 4414–4416. [Google Scholar]
- Shi Y.; Hamsen C.; Jia X.; Kim K. K.; Reina A.; Hofmann M.; Hsu A. L.; Zhang K.; Li H.; Juang Z.-Y.; Dresselhaus M. S.; Li L.-J.; Kong J. Nano Lett. 2010, 10, 4134–4139. [DOI] [PubMed] [Google Scholar]
- Suzuki S.; Pallares R. M.; Hibino H. J. Phys. D. Appl. Phys. 2012, 45, 385304. [Google Scholar]
- Orofeo C. M.; Suzuki S.; Kageshima H.; Hibino H. Nano Res. 2013, 6, 335–347. [Google Scholar]
- Kim G.; Jang A.-R.; Jeong H. Y.; Lee Z.; Kang D. J.; Shin H. S. Nano Lett. 2013, 13, 1834–1839. [DOI] [PubMed] [Google Scholar]
- Vinogradov N. A.; Zakharov A. A.; Ng M. L.; Mikkelsen A.; Lundgren E.; Mårtensson N.; Preobrajenski A. B. Langmuir 2012, 28, 1775–1781. [DOI] [PubMed] [Google Scholar]
- Xu M.; Fujita D.; Chen H.; Hanagata N. Nanoscale 2011, 3, 2854–2858. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Fu L.; Zhao S.; Zhou Y.; Peng H.; Liu Z. Adv. Mater. 2014, 26, 1776–1781. [DOI] [PubMed] [Google Scholar]
- Morscher M.; Corso M.; Greber T.; Osterwalder J. Surf. Sci. 2006, 600, 3280–3284. [Google Scholar]
- Ćavar E.; Westerström R.; Mikkelsen A.; Lundgren E.; Vinogradov A. S.; Ng M. L.; Preobrajenski A. B.; Zakharov A. A.; Mårtensson N. Surf. Sci. 2008, 602, 1722–1726. [Google Scholar]
- Sutter P.; Lahiri J.; Albrecht P.; Sutter E. ACS Nano 2011, 5, 7303–7309. [DOI] [PubMed] [Google Scholar]
- Sutter P.; Lahiri J.; Zahl P.; Wang B.; Sutter E. Nano Lett. 2013, 13, 276–281. [DOI] [PubMed] [Google Scholar]
- Lu J.; Yeo P. S. E.; Zheng Y.; Xu H.; Gan C. K.; Sullivan M. B.; Castro Neto A. H.; Loh K. P. J. Am. Chem. Soc. 2013, 135, 2368–2373. [DOI] [PubMed] [Google Scholar]
- Park J.-H.; Park J. C.; Yun S. J.; Kim H.; Luong D. H.; Kim S. M.; Choi S. H.; Yang W.; Kong J.; Kim K. K.; Lee Y. H. ACS Nano 2014, 8, 8520–8528. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Gong Y.; Zhou W.; Ma L.; Yu J.; Idrobo J. C.; Jung J.; MacDonald A. H.; Vajtai R.; Lou J.; Ajayan P. M. Nat. Commun. 2013, 4, 2541. [DOI] [PubMed] [Google Scholar]
- Li X.; Cai W.; An J.; Kim S.; Nah J.; Yang D.; Piner R.; Velamakanni A.; Jung I.; Tutuc E.; Banerjee S. K.; Colombo L.; Ruoff R. S. Science 2009, 324, 1312–1314. [DOI] [PubMed] [Google Scholar]
- Predel B.Cu-N (Copper-Nitrogen). SpringerMaterials: The Landolt-Börnstein Database, http://www.springermaterials.com/docs/info/10086090_1091.html (accessed May 26, 2014).
- Rogl P.; Schuster J. C.. Boron-Copper-Nitrogen Ternary Alloy Phase Diagram. ASM International, http://www.asminternational.org/materials-resources/-/journal_content/56/10192/APD-B-CU-N-1100491 (accessed May 26, 2014).
- Gamalski A. D.; Perea D. E.; Yoo J.; Li N.; Olszta M. J.; Colby R.; Schreiber D. K.; Ducati C.; Picraux S. T.; Hofmann S. ACS Nano 2013, 7, 7689–7697. [DOI] [PubMed] [Google Scholar]
- Björk M. T.; Ohlsson B. J.; Sass T.; Persson A. I.; Thelander C.; Magnusson M. H.; Deppert K.; Wallenberg L. R.; Samuelson L. Appl. Phys. Lett. 2002, 80, 1058. [Google Scholar]
- Li X.; Yin J.; Zhou J.; Guo W. Nanotechnology 2014, 25, 105701. [DOI] [PubMed] [Google Scholar]
- Corso M.; Auwärter W.; Muntwiler M.; Tamai A.; Greber T.; Osterwalder J. Science 2004, 303, 217–220. [DOI] [PubMed] [Google Scholar]
- Gorbachev R. V.; Riaz I.; Nair R. R.; Jalil R.; Britnell L.; Belle B. D.; Hill E. W.; Novoselov K. S.; Watanabe K.; Taniguchi T.; Geim A. K.; Blake P. Small 2011, 7, 465–468. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Hamsen C.; Jia X.; Kim K. K.; Reina A.; Hofmann M.; Hsu A. L.; Zhang K.; Li H.; Juang Z.-Y.; Dresselhaus M. S.; Li L.-J.; Kong J. Nano Lett. 2010, 10, 4134–4139. [DOI] [PubMed] [Google Scholar]
- Reich S.; Ferrari A.; Arenal R.; Loiseau A.; Bello I.; Robertson J. Phys. Rev. B 2005, 71, 205201. [Google Scholar]
- Widmayer P.; Boyen H.-G.; Ziemann P.; Reinke P.; Oelhafen P. Phys. Rev. B 1999, 59, 5233–5241. [Google Scholar]
- Kim K. K.; Hsu A.; Jia X.; Kim S. M.; Shi Y.; Hofmann M.; Nezich D.; Rodriguez-Nieva J. F.; Dresselhaus M.; Palacios T.; Kong J. Nano Lett. 2012, 12, 161–166. [DOI] [PubMed] [Google Scholar]
- Trehan R. J. Vac. Sci. Technol. A: Vac., Surf., Film 1990, 8, 4026. [Google Scholar]
- Deng J.; Chen G.; Song X. Int. J. Mod. Phys. B 2002, 16, 4339–4342. [Google Scholar]
- Park K. S. J. Vac. Sci. Technol. A: Vac., Surf., Film 1997, 15, 1041. [Google Scholar]
- Joshi S.; Ecija D.; Koitz R.; Iannuzzi M.; Seitsonen A. P.; Hutter J.; Sachdev H.; Vijayaraghavan S.; Bischoff F.; Seufert K.; Barth J. V.; Auwärter W. Nano Lett. 2012, 12, 5821–5828. [DOI] [PubMed] [Google Scholar]
- Koitz R.; Seitsonen A. P.; Iannuzzi M.; Hutter J. Nanoscale 2013, 5, 5589–5595. [DOI] [PubMed] [Google Scholar]
- Feng X.; Salmeron M. Appl. Phys. Lett. 2013, 102, 053116. [Google Scholar]
- Trehan R. J. Vac. Sci. Technol. A: Vac., Surf., Film 1990, 8, 4026. [Google Scholar]
- Orlando F.; Larciprete R.; Lacovig P.; Boscarato I.; Baraldi A.; Lizzit S. J. Phys. Chem. C 2012, 116, 157–164. [Google Scholar]
- Guimon C.; Gonbeau D.; Pfister-Guillouzo G.; Dugne O.; Guette A.; Naslain R.; Lahaye M. Surf. Interface Anal. 1990, 16, 440–445. [Google Scholar]
- Müller F.; Hüfner S.; Sachdev H.; Gsell S.; Schreck M. Phys. Rev. B 2010, 82, 075405. [Google Scholar]
- Rybakov S. Y.; Sharapov V. M.; Gavrilov L. E. J. Phys. IV 1995, 05, C5-921––C5-926. [Google Scholar]
- Baiker A.; Monti D. Ber. Bunsenges. Phys. Chem. 1983, 87, 602–605. [Google Scholar]
- Biemolt W.; Jansen A. P. J.; Neurock M.; van de Kerkhof G. J. C. S.; van Santen R. A. Surf. Sci. 1993, 287–288, 183–187. [Google Scholar]
- Chakrabarti D. J.; Laughlin D. E. Bull. Alloy Phase Diagrams 1982, 3, 45–48. [Google Scholar]
- Simonov K. A.; Vinogradov N. A.; Ng M. L.; Vinogradov A. S.; Mårtensson N.; Preobrajenski A. B. Surf. Sci. 2012, 606, 564–570. [Google Scholar]
- Larciprete R.; Ulstrup S.; Lacovig P.; Dalmiglio M.; Bianchi M.; Mazzola F.; Hornekær L.; Orlando F.; Baraldi A.; Hofmann P.; Lizzit S. ACS Nano 2012, 6, 9551–9558. [DOI] [PubMed] [Google Scholar]
- Grånäs E.; Knudsen J.; Schröder U. A.; Gerber T.; Busse C.; Arman M. A.; Schulte K.; Andersen J. N.; Michely T. ACS Nano 2012, 6, 9951–9963. [DOI] [PubMed] [Google Scholar]
- Mu R.; Fu Q.; Jin L.; Yu L.; Fang G.; Tan D.; Bao X. Angew. Chem., Int. Ed. 2012, 51, 4856–4859. [DOI] [PubMed] [Google Scholar]
- Sutter P.; Sadowski J. T.; Sutter E. A. J. Am. Chem. Soc. 2010, 132, 8175–8179. [DOI] [PubMed] [Google Scholar]
- Goriachko A.; He Y. B.; Over H. J. Phys. Chem. C 2008, 112, 8147–8152. [Google Scholar]
- Usachov D.; Adamchuk V. K.; Haberer D.; Grüneis A.; Sachdev H.; Preobrajenski A. B.; Laubschat C.; Vyalikh D. V. Phys. Rev. B 2010, 82, 075415. [Google Scholar]
- Brugger T.; Ma H.; Iannuzzi M.; Berner S.; Winkler A.; Hutter J.; Osterwalder J.; Greber T. Angew. Chem., Int. Ed. 2010, 49, 6120–6124. [DOI] [PubMed] [Google Scholar]
- Preobrajenski A. B.; Nesterov M. A.; Ng M. L.; Vinogradov A. S.; Mårtensson N. Chem. Phys. Lett. 2007, 446, 119–123. [Google Scholar]
- Preobrajenski A.; Vinogradov A.; Ng M.; Ćavar E.; Westerström R.; Mikkelsen A.; Lundgren E.; Mårtensson N. Phys. Rev. B 2007, 75, 245412. [Google Scholar]
- Vinogradov N. A.; Schulte K.; Ng M. L.; Mikkelsen A.; Lundgren E.; Mårtensson N.; Preobrajenski A. B. J. Phys. Chem. C 2011, 115, 9568–9577. [Google Scholar]
- Goriachko A.; Zakharov A. A.; Over H. J. Phys. Chem. C 2008, 112, 10423–10427. [Google Scholar]
- Hao Y.; Bharathi M. S.; Wang L.; Liu Y.; Chen H.; Nie S.; Wang X.; Chou H.; Tan C.; Fallahazad B.; Ramanarayan H.; Magnuson C. W.; Tutuc E.; Yakobson B. I.; McCarty K. F.; Zhang Y.-W.; Kim P.; Hone J.; Colombo L.; Ruoff R. S. Science 2013, 342, 720–723. [DOI] [PubMed] [Google Scholar]
- Wang L.; Wu B.; Chen J.; Liu H.; Hu P.; Liu Y. Adv. Mater. 2014, 26, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Tay R. Y.; Griep M. H.; Mallick G.; Tsang S. H.; Singh R. S.; Tumlin T.; Teo E. H. T.; Karna S. P. Nano Lett. 2014, 14, 839–846. [DOI] [PubMed] [Google Scholar]
- Weatherup R. S.; Amara H.; Blume R.; Dlubak B.; Bayer B. C.; Diarra M.; Bahri M.; Cabrero-Vilatela A.; Caneva S.; Kidambi P. R.; Martin M.-B.; Deranlot C.; Seneor P.; Schlögl R.; Ducastelle F.; Bichara C.; Hofmann S. J. Am. Chem. Soc. 2014, 136, 13698–13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachov D.; Fedorov A.; Vilkov O.; Adamchuk V. K.; Yashina L. V.; Bondarenko L.; Saranin A. A.; Grüneis A.; Vyalikh D. V. Phys. Rev. B 2012, 86, 155151. [Google Scholar]
- Hubacek M.; Sato T. J. Mater. Sci. 1997, 2, 3293–3297. [Google Scholar]
- Lee K.; Kim Y.; Tosa M. Appl. Surf. Sci. 2001, 170, 420–424. [Google Scholar]
- Nie S.; Wu W.; Xing S.; Yu Q.; Bao J.; Pei S.; McCarty K. F. New J. Phys. 2012, 14, 093028. [Google Scholar]
- Suenaga K.; Kobayashi H.; Koshino M. Phys. Rev. Lett. 2012, 108, 075501. [DOI] [PubMed] [Google Scholar]
- Caretti I.; Jiménez I. J. Appl. Phys. 2011, 110, 023511. [Google Scholar]
- Gibb A. L.; Alem N.; Chen J.; Erickson K. J.; Ciston J.; Gautam A.; Linck M.; Zettl A. J. Am. Chem. Soc. 2013, 135, 6758–6761. [DOI] [PubMed] [Google Scholar]
- Meyer J. C.; Chuvilin A.; Algara-Siller G.; Biskupek J.; Kaiser U. Nano Lett. 2009, 9, 2683–2689. [DOI] [PubMed] [Google Scholar]
- Kotakoski J.; Jin C. H.; Lehtinen O.; Suenaga K.; Krasheninnikov A. V. Phys. Rev. B 2010, 82, 113404. [Google Scholar]
- Cun H.; Iannuzzi M.; Hemmi A.; Osterwalder J.; Greber T. ACS Nano 2014, 8, 7423–7431. [DOI] [PubMed] [Google Scholar]
- Slotman G. J.; Fasolino A. J. Phys.: Condens. Matter 2013, 25, 045009. [DOI] [PubMed] [Google Scholar]
- Boutilier M. S. H.; Sun C.; O’Hern S. C.; Au H.; Hadjiconstantinou N. G.; Karnik R. ACS Nano 2014, 8, 841–849. [DOI] [PubMed] [Google Scholar]
- Li L. H.; Cervenka J.; Watanabe K.; Taniguchi T.; Chen Y. ACS Nano 2014, 8, 1457–1462. [DOI] [PubMed] [Google Scholar]
- Xi K.; Kidambi P. R.; Chen R.; Gao C.; Peng X.; Ducati C.; Hofmann S.; Kumar R. V. Nanoscale 2014, 6, 5746–5753. [DOI] [PubMed] [Google Scholar]
- Dlubak B.; Martin M.-B.; Weatherup R. S.; Yang H.; Deranlot C.; Blume R.; Schloegl R.; Fert A.; Anane A.; Hofmann S.; Seneor P.; Robertson J. ACS Nano 2012, 6, 10930–10934. [DOI] [PubMed] [Google Scholar]
- Bluhm H.; Hävecker M.; Knop-gericke A.; Kiskinova M.; Schlögl R.; Salmeron M. MRS Bull. 2007, 32, 1022–1030. [Google Scholar]
- Meyer J.; Kidambi P. R.; Bayer B. C.; Weijtens C.; Kuhn A.; Centeno A.; Pesquera A.; Zurutuza A.; Robertson J.; Hofmann S. Sci. Rep. 2014, 4, 5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvila A.; Kidambi P. R.; Kling J.; Wagner J. B.; Robertson J.; Hofmann S.; Meyer J. J. Mat. Chem. C 2014, 2, 6940–6945. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.