

Abstract
The trinuclear platinum complexes ([{Pt(NH3)3}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}]6+, TriplatinNC‐A; [{trans-Pt(NH3)2(NH2(CH2)6NH3+)}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}]8+, TriplatinNC) belong to a class of biologically active agents that bind to DNA via nonbonding noncovalent (hydrogen bonding, electrostatic) interactions. Charge delocalization (6+ to 8+) in these linear trinuclear platinum complexes results in a high cellular uptake and promising cytotoxic activity in several carcinoma cell lines. We show in the present work with the aid of the methods of biophysical chemistry that in particular TriplatinNC condenses DNA with unprecedented potency which is much higher than that of conventional DNA condensing agents. In addition, in contrast to other DNA condensing agents, both platinum complexes induce aggregation of small transfer RNA molecules. We also demonstrate for the first time that TriplatinNC-A and TriplatinNC in particular completely inhibit DNA transcriptional activity at markedly lower concentration than naturally occurring spermine. Notably, the topoisomerase I-mediated relaxation of supercoiled DNA was inhibited by TriplatinNC-A and TriplatinNC at ~60-fold and ~250-fold lower concentration than that of spermine, respectively. We suggest that the general mechanisms of biological activity of TriplatinNC-A and TriplatinNC may be associated with their unique ability to condense/aggregate nucleic acids with consequent inhibitory effect on crucial enzymatic activities.
Keywords: DNA condensation, transfer RNA condensation, platinum complexes, DNA transcription, topoisomerase I
Polynuclear platinum complexes represent a discrete class of anticancer drugs structurally distinct from the mononuclear compounds. The trinuclear TriplatinNC-A ([{Pt(NH3)3}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}]6+ and TriplatinNC [{trans-Pt(NH3)2(NH2(CH2)6NH3+)}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}]8+,) (Chart 1B,C) bind to DNA via noncovalent interactions. The basis of these interactions is the phosphate clamp, a third mode of ligand binding to DNA, discrete from the canonical intercalation and minor-groove binding.[1, 2]
Chart 1.
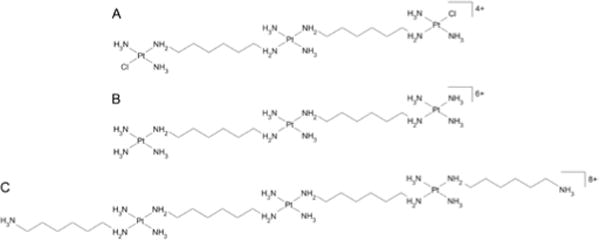
Schematic representation of the trinuclear linear Pt compounds. A. BBR3464, [{trans-PtCl-(NH3)2}-μ-trans-Pt(NH3)2{H2N(CH2)6NH2}2]4+; B. TriplatinNC-A, [{Pt(NH3)3}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}]6+; C. TriplatinNC, [{trans-Pt(NH3)2(NH2(CH2)6NH3+)}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}]8+.
Polynuclear platinum compounds and TriplatinNC in particular, have interesting biological properties including in vitro toxicity in several ovarian carcinoma cell lines,[3, 4] high cellular accumulation and transformed cell selectivity.[5]
The modular nature of the polynuclear Pt complex binding results in high DNA binding affinity with subsequent stabilization and induction of B→Z and B→A transitions in susceptible sequences at concentrations lower than those required by cobalt hexammine[6] and displacement of ethidium bromide from DNA.
The molecular details of phosphate clamp binding to DNA has analogies with the arginine fork mode of recognition on nucleic acids.[1,2] The structure, especially of the non-covalent compounds, also bears analogy with the naturally occurring polyamines such as spermidine and spermine. In a previous paper,[7] we reported on the DNA binding affinity, condensation properties and sequence selectivity of the dinuclear polyamine(spermidine/spermine)-linked complexes and the trinuclear TriplatinNC-A. All polynuclear platinum compounds induce DNA condensation at more than 1 order of magnitude lower concentrations than conventional condensing agent, such as the naturally occurring polyamine spermine4+ itself (H2N(CH2)3NH(CH2)4NH(CH2)3NH2). TriplatinNC-A binds to DNA in a sequence-dependent manner protecting DNA from enzymatic cleavage by endonuclease deoxyribonuclease I (DNase I).[7] This result is consistent with the structurally demonstrated minor groove spanning of TriplatinNC-A,[2] its protection of the minor groove toward alkylation,[8] and the increase of DNA-binding affinity of Hoechst dye.[9]
The mechanism of biological activities of polynuclear platinum complexes, which bind to DNA via noncovalent interactions, is not fully understood. However our results suggest that it may be associated with the unique ability of these compounds to condense DNA along with their sequence-specific DNA binding and possibly on the competition with naturally occurring polyamines for intracellular binding sites, but with altered function. Herein we compare their nucleic acid binding effects by biophysical methods and report on the condensation and aggregation of the DNA and tRNA induced by TriplatinNC and TriplatinNC-A in cell-free media. Furthermore, because DNA-binding ligands in general may impair crucial enzymatic processes acting directly on DNA or RNA, we have investigated their effects on the DNA transcriptional activity and activity of eukaryotic DNA topoisomerase I (topoI).
We employed total intensity light scattering[10] to determine the efficacy of TriplatinNC and TriplatinNC-A to condense/aggregate calf thymus (CT) DNA and tRNA. This method is based on the measurement of the intensity of light scattered by the diluted DNA or RNA solution at 90°. The scattered light intensity is low in the absence or low concentrations of condensing agent, however a marked increase in the intensity of the scattered light appears at a critical concentration of added agent due to the formation of condensed DNA or RNA particles. The increase in the intensity of the scattered light is concentration dependent up to a certain concentration of the condensing agent and then it levels off. The efficacy of various condensing agents to induce DNA or RNA condensation can be quantified by determining the EC50 value, which is the concentration of a condensing agent at the midpoint of the condensation. As it can be seen in Table 1, TriplatinNC having a higher charge of 8+ was more efficient in inducing CT DNA condensation than TriplatinNC-A with 6+ charge. The EC50 value of TriplatinNC (0.15 ± 0.01 μM) is ~27-fold lower than that of spermine obtained at the same conditions.[7]
Table 1.
EC50 Values (μM) of spermine and Pt compoundsa
| CT DNA | tRNA | |
|---|---|---|
| spermine | 4.1 ±0.5b | >50 |
| TriplatinNC-A | 0.20±0.01b | 0.27±0.01 |
|
|
||
| TriplatinNC | 0.15±0.01 | 0.24±0.01 |
All measurements were conducted in 10 mM sodium cacodylate buffer, pH 7.2 at 298 K. EC50 values were determined by plotting the scattered light intensity against concentration of condensing agents. Data are mean ± SD of three separate measurements.
Data previously reported.[7]
The molecules of CT DNA having average length of several kilobase pairs can be compacted by either monomolecular condensation with distinguishable morphologies[11,12] or multimolecular aggregation with irregular morphology. The tRNA molecules, on the other hand, are typically 60–95 nucleotides in length and are too short to be individually condensed. The only way tRNA particles can be formed is by multimolecular aggregation.[13,14]
The results summarized in Table 1 show that TriplatinNC and TriplatinNC-A were also efficient in tRNA aggregation. Notably, the conventional DNA condensing agent spermine was not capable of inducing tRNA aggregation at the concentrations up to even ~200-fold higher. Thus, TriplatinNC-A and TriplatinNC, in particular, represent unique agents capable of condensing tRNA. The outstanding and unprecedented efficiency to condense/aggregate nucleic acids was confirmed by the UV/Vis spectrophotometry, gel retardation assay and by investigating the effect of TriplatinNC on the morphology of DNA or tRNA condensates by atomic force microscopy (Figures 1 and S1–S8).
Figure 1.
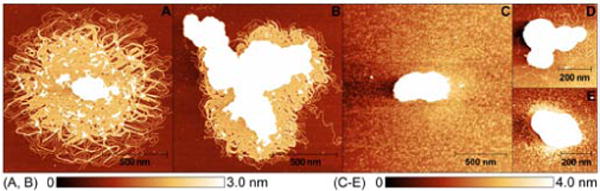
Representative AFM images of linearized plasmid pSP73 (A, B) and tRNA (C–E) in the presence of TriplatinNC at 6.25 μM concentration. For other details, see Figures S7 and S8.
It has been previously demonstrated[15] that DNA transcriptional activity is a function of spermine concentration. When the concentration of spermine was kept below ~1 mM the DNA transcriptional activity is enhanced while increasing the concentrations of spermine above ~1 mM results in an inhibition of the DNA transcription. We employed the same procedure described by Luckel et al.[15] to examine the transcriptional activity of the circular form of pBR322 DNA in the presence of TriplatinNC-A and TriplatinNC. The DNA transcription was monitored by measuring the fluorescence intensity resulting from the separation of the AmNS group from UTP-gamma-AmNS during RNA polymerization. The actual amounts of transcription products in the presence of various concentrations of spermine, TriplatinNC-A and TriplatinNC as a function of time are shown in Figure 2A. It can be seen that the DNA transcriptional activity was inhibited in the presence of 2 μM and 8 μM of TriplatinNC and TriplatinNC-A, respectively, while the concentration of spermine so high as 800 μM still had even slightly enhancing effect. The plots of relative transcriptional activity (Figure 2B), defined as the ratio of RNA products at given TriplatinNC and TriplatinNC-A concentrations to the amount of RNA products in the absence of any complex, show that the addition of ~4 μM TriplatinNC and ~20 μM TriplatinNC-A completely inhibited transcription of the DNA. These concentrations are approximately 350-fold and 70-fold lower than the concentration of spermine necessary to achieve the same effect (>1400 μM).[15]
Figure 2.
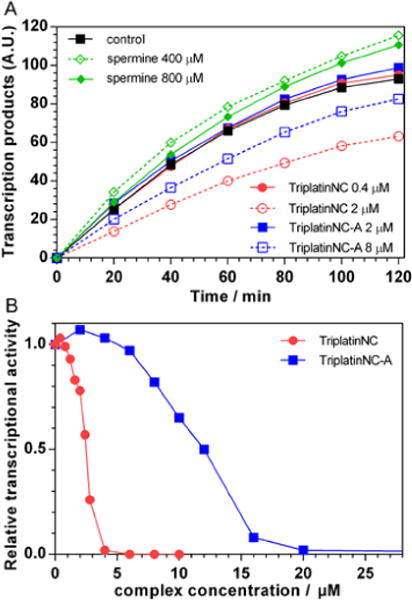
A. Transcription kinetics of circular pBR322 plasmid DNA in the absence and in the presence of various concentrations of spermine, TriplatinNC-A and TriplatinNC. B. Relative transcriptional activity of pBR322 plasmid DNA as a function of TriplatinNC (●) and TriplatinNC-A (■) concentration.
The results show that TriplatinNC and TriplatinNC-A are ~27-fold and ~20-fold more potent inducers of mammalian CT DNA condensation than spermine and that, unlike spermine, both complexes cause aggregation of relatively small tRNA molecules (typically 60–95 nucleotides in length). The high resistance of DNA and tRNA condensates/aggregates formed by TriplatinNC-A and in particular TriplatinNC against the treatment by high concentrations of NaCl (Figures S2B, S3 and S6) indicates strong interaction of both complexes with nucleic acids. Therefore, we suggest that the high affinity of TriplatinNC-A and TriplatinNC for molecules of nucleic acids and high efficiency of these polynuclear platinum complexes in condensing DNA preclude reading DNA sequences by RNA polymerases when they produce a complementary, antiparallel RNA strand (a primary transcript). Notably, DNA compaction is expected to prevent the binding/sliding of RNA polymerase to DNA.[16–18] RNA polymerases function like a molecular motor that can convert chemical energy into the work of translocation along the DNA during transcription.[19] Hence, it is also possible that this DNA translocation mechanism is insufficiently powerful to displace TriplatinNC and TriplatinNC-A tightly bound to condensed template DNA. Since TriplatinNC and TriplatinNC-A are markedly more effective in condensing DNA and inhibiting the transcriptional activity of DNA compared to conventional spermine, our findings suggest an enhanced regulatory role of TriplatinNC and TriplatinNC-A at the level of genome transcription in comparison with spermine. The activity of platinum drugs against cancer is mediated by a combination of processes including cell entry, drug activation, DNA-binding, and last but not least transcription inhibition.[20] Our results reveal a correlation between the high affinity of TriplatinNC and TriplatinNC-A for molecules of nucleic acids and high efficiency of these polynuclear platinum complexes to condense/aggregate DNA on one hand and the capability of these complexes to inhibit DNA transcription on the other hand.
Topoisomerases are ubiquitous and vital enzymes because of their role in the control of the topological state of DNA. Hence, topoisomerases participate in nearly all events related to DNA metabolism including replication, transcription and recombination. Topoisomerases are now viewed as important therapeutic targets and in particular, topoisomerase inhibitors are considered promising anticancer agents.[21–23]
We utilized a DNA relaxation assay to examine whether activity of eukaryotic topoI is affected in the presence of TriplatinNC and TriplatinNC-A. This assay is based on the monitoring of the conversion of naturally negatively supercoiled pSP73KB plasmid preincubated with increasing concentrations of TriplatinNC, TriplatinNC-A and spermine into relaxed covalently closed circular DNA. As can be seen in Figure 3, TriplatinNC and TriplatinNC-A completely inhibited topoI-catalyzed DNA relaxation at concentrations as low ~16 μM and ~64 μM, respectively. These values are approximately ~250- and ~60-fold lower than the concentration of natural polyamine spermine (~4 mM) resulting in the same effect (Figure S9).
Figure 3.
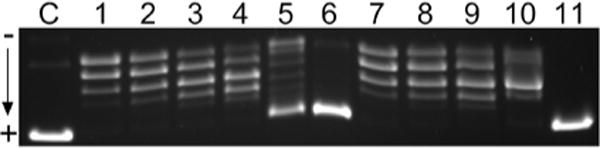
Inhibition of topoI-mediated relaxation of negatively supercoiled pSP73KB plasmid in the presence of increasing concentrations of TriplatinNC-A and Triplatin NC analyzed by agarose gel electrophoresis. Lane C: nonmodified plasmid DNA in the absence of topoI; lane 1: nonmodified plasmid DNA in the presence of topoI; lanes 2–6: plasmid DNA in the presence of topoI and 4, 8, 16, 32, and 64 μM of TriplatinNC-A, respectively; lanes 7–11: plasmid DNA in the presence of topoI and 1, 2, 4, 8, and 16 μM of TriplatinNC, respectively. The DNA concentration in the samples was 200 μM (0.5 μg).
The inhibitory effect of TriplatinNC-A on topoI-mediated plasmid DNA relaxation is comparable to that of some synthetic hexamines examined previously,[24] whereas TriplatinNC is ~4-fold more potent. Interestingly, both trinuclear platinum complexes induce DNA topoI inhibition at concentrations that are similar or even lower than those of several conventional topoI inhibitors, such as camptothecin derivatives, used in the treatment of cancer, inducing a similar inhibition effect.[25]
It was reported previously[24] that the types of structures of the DNA aggregates formed by different polyamines might play a key role in the topoI catalysis. For instance, DNA aggregation by the natural polyamine spermine yields a highly fluid DNA aggregate. By contrast, the polyamines with the longest central chains produce very different non-fluid aggregates with a high packing density of DNA molecules. Polyamines yielding aggregates of the second type were markedly more efficient inhibitors of DNA relaxation by topoI than considerably shorter natural polyamines (spermine) yielding the fluid aggregates.[7,24]
Hence, trinuclear platinum complexes tested in the present work, in particular TriplatinNC, can be grouped with longer polyamines yielding the non-fluid aggregates (Figure S7) responsible for inhibition of DNA relaxation by topoI.[24] The observation that TriplatinNC-A is a somewhat less efficient inhibitor of the DNA topoI activity than TriplatinNC (Figure 2) is consistent with the fact that the DNA condensates induced by TriplatinNC-A are less compact and that at higher concentrations of TriplatinNC DNA particles are predominantly formed instead of rather flat and single-layered compact DNA patterns typically observed for TriplatinNC-A (cf. Figures 5 and 7 in Ref.[7]). Moreover, it was proposed previously[24] that, in contrast to spermine, the longer polyamines such as hexamines, could make more cross-links (cross-links through electrostatic interaction and hydrogen bonding bridging the groove of the helices or helix[26–28]) between different DNA molecules or different parts of the same DNA molecule leading to the formation of highly condensed non-fluid aggregates that are responsible for the inhibition of the topoI activity. Thus, it is reasonable to assume that the capability of the longer trinuclear Pt complexes tested in the present work to form cross-links similar to those formed by hexamines might contribute to the formation of highly condensed non-fluid aggregates and consequently to the inhibition of the topoI activity by these agents. The details of the mechanism of the inhibition of the topoI activity by the condensed non-fluid aggregates formed by long polyamines, such as TriplatinNC-A, TriplatinNC or hexamines, are unclear. A plausible suggestion, which needs experimental confirmation, might be that formation of the highly condensed non-fluid aggregates prevents topoI to access its specific recognition sites, or if the enzyme binds DNA it is unable to cleave and transfer the densely packed DNA strand.[24]
Both trinuclear platinum complexes, TriplatinNC and TriplatinNC-A demonstrate high efficiency in inducing the condensation and aggregation of nucleic acids. The higher overall charge (8+) and greater length of TriplatinNC are probably responsible for its higher potency to induce DNA condensation and tRNA aggregation in comparison with the (6+) TriplatinNC-A. The presence of dangling amines on the terminal Pt(II) atoms of TriplatinNC, besides increasing the charge, also extends the length of the molecule and might enable cross-links by condensing counterions between the different DNA or RNA molecules or between the different domains of the same DNA or RNA molecule in the process of condensation or aggregation through electrostatic interaction and hydrogen bonding bridging the groove of the helices.[26–28] AFM studies of DNA condensation in the presence of TriplatinNC have revealed (Figures S7, S8) that the morphologies of DNA condensates become more compact with increasing concentration of the platinum complex. The higher concentrations of TriplatinNC lead to the formation of DNA particles instead of rather flat and single-layered compact DNA patterns typically observed for TriplatinNC-A. The results from AFM experiments (Figures 1, S7 and S8) correlate well with the results of other experiments and confirm that TriplatinNC and TriplatinNC-A condense DNA with much higher potency than spermine and, in contrast to spermine, both platinum complexes induce aggregation of short tRNA molecules. These properties also distinguish the phosphate clamp DNA binding mode of polynuclear platinum complexes (with their high positive charge) from typical minor-groove binders, despite the affinity for A-T-rich sequences.[7] Thus, the chemotype is distinct from both the polyamine class and minor-groove binders.
The high affinity and DNA conformational changes induced by the complexes also have consequences for protein-DNA interactions. The enzymatic studies have shown that TriplatinNC and TriplatinNC-A inhibit DNA transcriptional activity and topoI-mediated relaxation of supercoiled DNA at markedly lower concentrations than naturally occurring spermine. These results, combined with the demonstration of nucleolus targeting of TriplatinNC[29] suggest new approaches to eventual inhibition of cellular proliferation, distinct from the covalent Pt-DNA formation approach.[30]
Within the concept of a non-covalently binding polynuclear Pt agent, this paper and previous studies have summarized DNA affinity and conformational changes by systematic changes of the modular nature of the chemotype: {Pt(tetraamine} versus polyamine central linker and the use of the ‘dangling’ amine versus NH3 as ligand to add further overall charge. There is overall similarity in consequences of DNA binding for all compounds independent of specific structure but yet, the complexes differ in their biological consequences. Especially TriplatinNC is cytotoxic at low micromolar concentrations whereas TriplatinNC-A consistently is 5–10 times less potent. Both compounds are significantly more cytotoxic than the dinuclear polyamine-linked analogs.[31,32] For the two compounds studied here, cytotoxicity also correlates with cellular accumulation mediated by heparan sulfate binding on the cell surface.[33,34] Thus, while nucleic acid condensation effects may contribute overall to the general cellular effects of these compounds, differentiation of cytotoxic effects may reflect differences in cellular accumulation.[5,9] This distinction may allow for consideration of these compounds as dual-function agents where ‘traditional’ DNA-binding effects may be complemented by effects on the cell surface.[30] In addition, these trinuclear platinum complexes may also have the potential for gene delivery because the transfection of DNA in gene therapy largely depends on the possibility of obtaining its condensation by simple artificial molecules.[35–37]
Supplementary Material
Footnotes
This work was supported by the Czech Science Foundation [grant number 13-08273S] and the Ministry of Education of the CR [grant number LH13096]. NF acknowledges support from NIH [grant number RO1 CA-78754].
Supporting information for this article is available on the WWW under http://dx.doi.org/xxxxxxxxxx.
Contributor Information
Jaroslav Malina, Institute of Biophysics, Academy of Sciences of the Czech Republic, v.v.i., Kralovopolska 135, CZ-61265 Brno (Czech Republic).
Nicholas P. Farrell, Department of Chemistry, Virginia Commonwealth University, Richmond, VA 23284-2006, USA
Viktor Brabec, Institute of Biophysics, Academy of Sciences of the Czech Republic, v.v.i., Kralovopolska 135, CZ-61265 Brno (Czech Republic).
References
- 1.Komeda S, Moulaei T, Woods KK, Chikuma M, Farrell NP, Williams LD. J Am Chem Soc. 2006;128:16092–16103. doi: 10.1021/ja062851y. [DOI] [PubMed] [Google Scholar]
- 2.Komeda S, Moulaei T, Chikuma M, Odani A, Kipping R, Farrell NP, Williams LD. Nucleic Acids Res. 2011;39:325–336. doi: 10.1093/nar/gkq723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farrell N. In: Platinum-based Drugs in Cancer Therapy. Kelland LR, Farrell NP, editors. Humana Press Inc; Totowa/NJ: 2000. pp. 321–338. [Google Scholar]
- 4.Harris AL, Yang X, Hegmans A, Povirk L, Ryan JJ, Kelland L, Farrell NP. Inorg Chem. 2005;44:9598–9600. doi: 10.1021/ic051390z. [DOI] [PubMed] [Google Scholar]
- 5.Harris AL, Ryan JJ, Farrell N. Mol Pharmacol. 2006;69:666–672. doi: 10.1124/mol.105.018762. [DOI] [PubMed] [Google Scholar]
- 6.Qu Y, Harris A, Hegmans A, Petz A, Kabolizadeh P, Penazova H, Farrell N. J Inorg Biochem. 2004;98:1591–1598. doi: 10.1016/j.jinorgbio.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 7.Malina J, Farrell NP, Brabec V. Inorg Chem. 2014;53:1662–1671. doi: 10.1021/ic402796k. [DOI] [PubMed] [Google Scholar]
- 8.Qu Y, Moniodis JJ, Harris AL, Yang X, Hegmans A, Povirk LF, Berners-Price SJ, Farrell NP. In: Polyamine Drug Discovery. Woster PM, Casero R, editors. The Royal Society of Chemistry; 2012. pp. 191–204. [Google Scholar]
- 9.Harris A, Qu Y, Farrell N. Inorg Chem. 2005;44:1196–1198. doi: 10.1021/ic048356p. [DOI] [PubMed] [Google Scholar]
- 10.Vijayanathan V, Thomas T, Shirahata A, Thomas TJ. Biochemistry. 2001;40:13644–13651. doi: 10.1021/bi010993t. [DOI] [PubMed] [Google Scholar]
- 11.He SQ, Arscott PG, Bloomfield VA. Biopolymers. 2000;53:329–341. doi: 10.1002/(SICI)1097-0282(20000405)53:4<329::AID-BIP5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 12.Noguchi H, Saito S, Kidoaki S, Yoshikawa K. Chem Phys Lett. 1996;261:527–533. [Google Scholar]
- 13.Widom J, Baldwin RL. J Mol Biol. 1980;144:431–453. doi: 10.1016/0022-2836(80)90330-7. [DOI] [PubMed] [Google Scholar]
- 14.Post CB, Zimm BH. Biopolymers. 1982;21:2123–2137. doi: 10.1002/bip.360211104. [DOI] [PubMed] [Google Scholar]
- 15.Luckel F, Kubo K, Tsumoto K, Yoshikawa K. FEBS Lett. 2005;579:5119–5122. doi: 10.1016/j.febslet.2005.07.095. [DOI] [PubMed] [Google Scholar]
- 16.Tsumoto K, Luckel F, Yoshikawa K. Biophys Chem. 2003;106:23–29. doi: 10.1016/s0301-4622(03)00138-8. [DOI] [PubMed] [Google Scholar]
- 17.Yamada A, Kubo K, Nakai T, Yoshikawa K, Tsumoto K. Appl Phys Lett. 2005;86:223901. [Google Scholar]
- 18.Kabata H, Kurosawa O, Arai I, Washizu M, Margarson SA, Glass RE, Shimamoto N. Science. 1993;262:1561–1563. doi: 10.1126/science.8248804. [DOI] [PubMed] [Google Scholar]
- 19.Yin YW, Steitz TA. Cell. 2004;116:393–404. doi: 10.1016/s0092-8674(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 20.Ang WH, Myint M, Lippard SJ. J Am Chem Soc. 2010;132:7429–7435. doi: 10.1021/ja101495v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman LW, Rohr LR, Bronstein IB, Holden JA. Human Pathol. 2002;33:599–607. doi: 10.1053/hupa.2002.124911. [DOI] [PubMed] [Google Scholar]
- 22.Palumbo M, Gatto B, Moro S, Sissi C, Zagotto G. Biochim Biophys Acta. 2002;1587:145–154. doi: 10.1016/s0925-4439(02)00077-7. [DOI] [PubMed] [Google Scholar]
- 23.Ulukan H, Swaan PW. Drugs. 2002;62:2039–2057. doi: 10.2165/00003495-200262140-00004. [DOI] [PubMed] [Google Scholar]
- 24.Sukhanova A, Devy J, Pluot M, Bradley JC, Vigneron JP, Jardillier JC, Lehn JM, Nabiev I. Bioorg Med Chem. 2001;9:1255–1268. doi: 10.1016/s0968-0896(01)00009-8. [DOI] [PubMed] [Google Scholar]
- 25.Yang CHJ, Horton JK, Cowan KH, Schneider E. Cancer Res. 1995;55:4004–4009. [PubMed] [Google Scholar]
- 26.Liquori AM, Costantino L, Crescenzi V, Elia V, Giglio E, Puliti R, De Santis Savino M, Vitagliano V. J Mol Biol. 1967;24:113–122. [Google Scholar]
- 27.Pelta J, Livolant F, Sikorav JL. J Biol Chem. 1996;271:5656–5662. doi: 10.1074/jbc.271.10.5656. [DOI] [PubMed] [Google Scholar]
- 28.Kabir A, Kumar GS. PLoS ONE. 2013;8:e70510. doi: 10.1371/journal.pone.0070510. doi:70510.71371/journal.pone.0070510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wedlock LE, Kilburn MR, Liu R, Shaw JA, Berners-Price SJ, Farrell NP. J Chem Soc Chem Comm Chem Comm. 2013;49:6944–6946. doi: 10.1039/c3cc42098a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mangrum JB, Engelmann BJ, Peterson EJ, Ryan JJ, Berners-Price SJ, Farrell NP. J Chem Soc Chem Comm Chem Comm. 2014;50:4056–4058. doi: 10.1039/c3cc49695c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrell N. In: Metal Ions in Biological Systems. Sigel A, Sigel H, editors. Vol. 42. Marcel Dekker, Inc; New York, Basel: 2004. pp. 251–296. [PubMed] [Google Scholar]
- 32.Mangrum JB, Farrell NP. Chem Commun. 2010;46:6640–6650. doi: 10.1039/c0cc01254h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benedetti BT, Peterson EJ, Kabolizadeh P, Martinez A, Kipping R, Farrell NP. Mol Pharmaceutics. 2011;8:940–948. doi: 10.1021/mp2000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva H, Frézard F, Peterson EJ, Kabolizadeh P, Ryan JJ, Farrell NP. Mol Pharmaceutics. 2012;9:1795–1802. doi: 10.1021/mp300098t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volcke C, Pirotton S, Grandfils C, Humbert C, Thiry PA, Ydens I, Dubois P, Raes M. J Biotechnol. 2006;125:11–21. doi: 10.1016/j.jbiotec.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 36.Sansone F, Dudic M, Donofrio G, Rivetti C, Baldini L, Casnati A, Cellai S, Ungaro R. J Am Chem Soc. 2006;128:14528–14536. doi: 10.1021/ja0634425. [DOI] [PubMed] [Google Scholar]
- 37.Farrell N, Kloster MGB. 6,310,047 US Patent.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.