

Abstract
The European Bioinformatics Institute (EMBL-EBI) provides access to a wide range of databases and analysis tools that are of key importance in bioinformatics. As well as providing Web interfaces to these resources, Web Services are available using SOAP and REST protocols that enable programmatic access to our resources and allow their integration into other applications and analytical workflows.
This unit describes the various options available to a typical researcher or bioinformatician who wishes to use our resources via Web interface or programmatically via a range of programming languages.
Keywords: Web Services, Programmatic access, SOAP, REST, analytical pipelines, workflows
INTRODUCTION
Since 2004 the European Bioinformatics Institute (EMBL-EBI) has provided access to a wide range of databases and analysis tools using Web Services technologies (McWilliam et al. 2013). This comprises services to search, retrieve and run analysis tools on the databases hosted at the institute and to explore the network of cross-references present in the data (e.g. EB-eye (Valentin et al. 2010)). In this protocol we introduce the reader to services used to retrieve entry data in various data formats and to access the data in specific fields (e.g. dbfetch), and analysis tool services, for example, sequence similarity search (e.g. FASTA (Pearson et al. 1988; UNIT 3.9) and NCBI BLAST (Altschul et al. 1997; UNIT 3.3)), multiple sequence alignment (e.g. Clustal Omega (Sievers et al. 2011; UNIT x.x)), pairwise sequence alignment and protein functional analysis (e.g. InterProScan (Jones et al. 2014; UNIT 2.7)). The REST/SOAP Web Services (http://www.ebi.ac.uk/Tools/webservices/) interfaces to these databases and tools allow their integration into other tools, applications, web portals, analysis pipeline processes and analytical workflows. To help get users started using the Web Services, sample clients and examples of usage are provided covering a range of popular bioinformatic programming languages.
STRATEGIC PLANNING
The most significant planning issues around the decision to use Web Services versions of EMBL-EBI services are detailed below.
Web Services have several potential uses over and above normal Web interface access to services, for example:
Offering our services behind or together with your service
Systematic access to resources
As a gateway to workflows
While these needs can also be served by local installation of individual tools and databases, doing so comes with additional technical support and skills burdens, for example the requirement of keeping local software and databases up to date, as well as a compute and storage burden. Web Services reduces these burdens by allowing a standardized interface to remotely managed servers (at EMBL-EBI in this instance) where the tools and database providers manage the software and database updating, as well as providing access to large compute resources and the management thereof.
Web Services still allows for programmatic access to services (for example using scripts), thus is suitable for mass/systematic analysis, or for using the services as part of a wider workflow or as the backend to another service.
There are some situations where Web Services are not suitable:
Where you want to perform analysis on a large volume of locally held data - carrying out operations remotely would require uploading a lot of data to the remote servers, which is time consuming and more vulnerable to connectivity quality.
Where the analysis is latency critical - the nature of remote services necessarily adds some latency to the process.
Where the data cannot leave the local computer/network for any reason - while Web Services use secure https protocols, license restrictions on datasets you own may prevent their transmission in any form over the internet.
Whilst using Web Services reduces the burden of maintaining software and data, it’s important to note that the user still needs to be familiar with programmatic concepts, although using a graphical workflow tool that interfaces with Web Services can alleviate some of the programming knowledge required.
BASIC PROTOCOL 1: RETRIEVING DATA FROM EMBL-EBI USING DBFETCH VIA THE WEB INTERFACE
In this protocol we introduce the reader to commonly used biological sequence databases and retrieving data from them using services at the EMBL-EBI.
A large number of databases exist that store biological data derived from experiments or computation. These aim to determine the order of nucleotides or amino acids; also known as the primary structure; and include methods such as Sanger sequencing (Sanger et al. 1975), NGS (Next Generation Sequencing (Pettersson et al., 2009) for whole genome and exom sequencing; peptide sequences from C and N-terminal analysis (Edman et al., 1950); Edman degradation (Roberts et al. 1976); enzyme digestion (Hernandez et al. 2006); mass spectrometry and use of x-ray crystallography of biomolecular structures (Franklin et al., 1956).
-
Nucleotide Sequences
The most commonly used nucleotide sequence database is the product of a tri-lateral agreement between the EMBL-EBI (European Molecular Biology Laboratory – European Bioinformatics Institute), the NCBI (National Center for Biological Information) and the DDBJ Centre (DNA Databank of Japan). These form the INSDC (International Nucleotide Sequence Database Collaboration). This collaborative database is known today as GenBank (Benson et al. 2008); European Nucleotide Archive (Cochrane et al., 2007) and DDBJ (Tateno et al., 2002). These three centers collect and share data on a daily basis forming perhaps the largest effort to exchange and share scientific data across the globe.
-
Genomes
Next Generation Sequencing (NGS) technology has evolved rapidly during the last 10 years. Traditional method sequencing speeds had been a delimiting factor for obtaining whole genomes. With NGS, it is possible today to sequence a human genome in a single day and at a fraction of the cost. This has led to an explosion in the number of genomes available for biomedical; agronomical; environmental and computational research today.
The largest collection of these genomes are spread in organism specific databases (e.g. FlyBase (Crosby et al., 2011); WormBase (Harris et al., 2012; UNIT 1.8) and SGD (Marsden et al., 2007; UNIT 1.20). ENSEMBL (Flicek et al., 2011; UNIT 1.15) and EnsemblGenomes (Kersey et al., 2011) is a recent effort to collect these and provide a single means to obtain and distribute these data. ENSEMBL is a joint project between the EMBL-EBI and the Wellcome Trust Sanger Insitute and is primarily focused on genomes from vertebrate and other eukaryotic organisms. EnsemblGenomes is based on the ENSEMBL and is divided into 5 web sites that focus on the genomes of bacteria, protists, fungi, plant and invertebrate metazoa.
-
Protein sequences
Amino acid sequences date back to the late 1940’s when Edman and Sanger developed methods for retrieving sequence from purified protein using a combination of biochemical methods. Just as with nucleotide sequences later, collecting and distributing these sequence became a task that would enable researchers to share and de-duplicate effort. The first such database was established in 1960’s by the NBRF (National Biochemical Research Foundation) and was known as the Atlas of Protein Sequence and Structure, published by Margaret Dayhoff. Her group pioneered methods for the comparison of protein sequence using computational methods. The NBRF established the PIR (Protein Information Resource) in 1984 to produce and distribute the PIR-PSD (PIR-Protein Sequence Database) (Wu et al. 2004), the first international database which grew out from Dayhoff’s Atlas of Protein Sequence and Structure. PIR, EMBL and the Swiss Institute of Bioinformatics joined efforts to produce a single and largest protein sequence database by unifying PIR-PSD, TrEMBL and SwissProt (Boeckmann et al., 2003) databases. This is known today as the UniProt Knowledgbase (Uniprot consortium., 2010). This service is one of the few in the world that can return sequences derived from structures in the PDB (Protein Data Bank). These include nucleotide and protein sequences as well as those from the Structural Genomics Initiative (SGD).
Retrieving sequences from EMBL-EBI using dbfetch Dbfetch (database fetch) (Lopez et al., 2003) is a system specifically designed to provide a single point of access for biological data spread across multiple resources. Dbfetch has been in operation since 2003 and provides today unified access to 40 databases (http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/dbfetch.databases). Dbfetch uses multiple data sources in order to provide a range of data formats wider than that available from a single source and mitigate the effect of a single data source being unavailable.
Necessary Resources
Hardware
Any Internet-connected computer
Software
A web browser, e.g. Google Chrome, Mozilla Firefox, Internet Explorer, Safari or Opera
General Procedure for using DbFetch over the web
-
Access the DbFetch web page.
Dbfetch can be access using a www browser using the following URL: http://www.ebi.ac.uk/Tools/dbfetch/dbfetch. It is worth noting at this time that the service can also be accessed using https (http+SSL) to provide encrypted data transfer where desired: https://www.ebi.ac.uk/Tools/dbfetch/dbfetch
The web interface of dbfetch is divided into two sections. The first section contains a pull-down menu where the user can select one of the available databases, followed by a text input box. Up to 200 valid database identifiers are used as input, each separated by a coma. These are then followed by two further pull down menus where the user selects the format and the style to download data. These are explained in detail in the following section. Figure 1 shows the web interface of dbfetch.
-
Select a database.
If you are using the first form to paste your database identifiers: choose a database name from this form; If you are using the second form to upload your database identifiers: the format required must be “database name”:”database identifier”
-
Enter database identifiers.
Maximum number of identifiers is 200. If you are using the first form: separate search items with a comma or space; if you are using the second form: separate search items with a new line.
-
Choose Format.
Here you can choose the simpler fasta format, or the databases’ default format for the chosen database.
Sequence database will distribute data in a variety of formats that are suitable for consumption in various systems and computing applications. Typically these formats can be complex documents that include annotations and sequences; often referred to as flat-files; or simple text documents that contain a one line header followed by a sequence; called fasta format. Other choices include annotations only; entrysize - useful for calculating and deciding on large sequence downloads; GFF3; seqxml; uniprotrdfxml and uniprotxml. For data orientated consumption there are special formats that ease importing these data into warehouses; relational databases; document indexers; etc. For example EMBL-XML, which can be used to import data into a relational system using the public EMBL/ENA schema.
-
Select Style.
You can get your results as text or html.
Styles may be HTML or RAW. HTML will contain hyperlinked ID and cross-reference information, suitable for consuming downloaded entries using a web browser. RAW, on the other hand, is just text, without hyperlinks.
Figures 2 and 3 show two screen shots of the dbfetch web interface with the format and style pull-down menu choices available for the UniProt Knowledgebase.
-
Bulk Processing.
For downloading sequences in bulk, use the second part of the form to upload a text file that contains lists of valid database identifiers. For example, to download data from ENA Archive, the database identifier must be on a line and in the format “database name”:”database identifier” E.g. embl:x56957. Figure 4 shows an example file containing identifiers for various entries containing erythroid ankyrin mRNA, CDS and protein from both ENA and UniProt.
-
Retrieve!
You are now ready to fetch your results, by pressing the Retrieve button.
Figure 1.
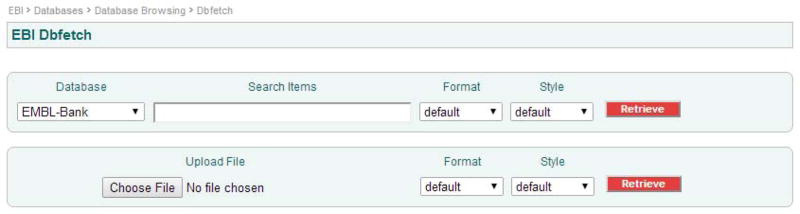
Web interface of dbfetch
Figure 2.
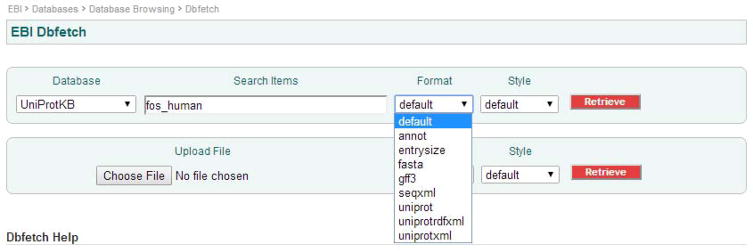
Dbfetch - format pull down menu choices for the UniProt Knowledgebase
Figure 3.
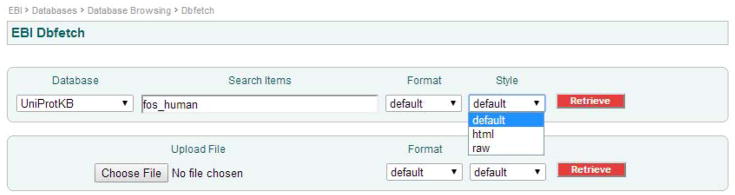
Dbfetch - style pull down menu choices for the UniProt Knowledgebase.
Figure 4.
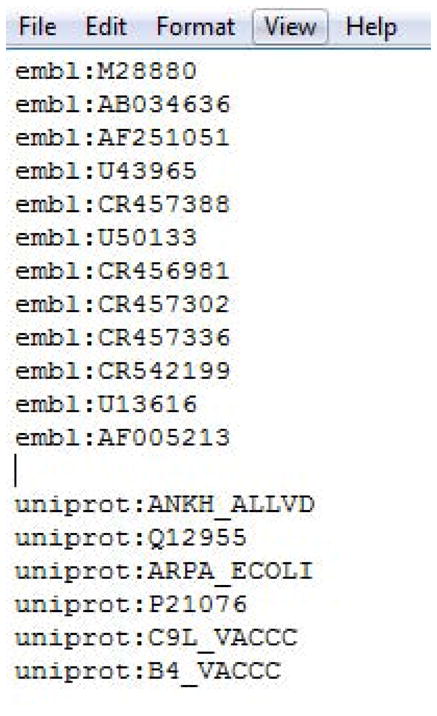
Examples of the format required to download data in bulk from dbfetch.
ALTERNATE PROTOCOL 1: RETRIEVING DATA FROM EMBL-EBI USING WSDBFETCH VIA REST INTERFACE
Dbfetch provides three modes of access to the user. As described above, one is using a web browser and the CGI interface. Two others exist that make use of data access standards called Web Services. Web Services consist of two protocols; SOAP (Simple Object Access Protocol) and REST (Representational State Transfer); that together complement each other and can be used to perform various data retrieval tasks. Like dbfetch, WSDbfetch (McWilliam et al., 2009) allows the user to retrieve entries. For the developer the advantage of these interfaces is that they allow the functionality of dbfetch to be integrated into their application, workflow or process pipeline. Since the Web Services technologies are language agnostic the developer can use the programming language of their choice. EMBL-EBI provides fully working example clients written in a variety of common programming languages, including Perl, PHP, Python, Ruby and VB.NET. These clients can be downloaded from http://www.ebi.ac.uk/Tools/webservices/services/dbfetch and give full access to the dbfetch service from the command line. The SOAP clients give ample examples of how to deal with processing errors and data resource outages. On the other hand, the REST clients provide an easier to use interface that lacks error reporting functionality apart from HTTP standard status codes (http://en.wikipedia.org/wiki/List_of_HTTP_status_codes). The REST interface can be consumed using a web browser or common web retrieval utilities such as wget, lynx and curl. In the following examples we will use URLs to demonstrate the WSDbfetch REST interface.
The fundamental syntax of the WSDbfetch REST interface is:
http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/{db}/{id}
Where {db} is the database name (e.g. “uniprotkb”) and {id} is the identifier (e.g. “WAP_RAT”). The following line shows how to fetch the mouse whey acidic protein precursor from UniProtKB using the RESTful interface:
http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/uniprotkb/WAP_MOUSE
As described earlier, dbfetch provides access to various format and styles to download data. WSDbfetch provides the same functionality. To download WAP_MOUSE in the UniProtKB XML format (“uniprotxml”) the URL would be:
http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/uniprotkb/WAP_MOUSE/uniprotxml
Likewise, to download WAP_MOUSE in UniProtKB flat-file format with HTML hyperlinks the following URL would be used:
http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/uniprotkb/WAP_RAT/uniprot?style=html
Dbfetch presently provides access to 40 databases. These are shown in Table 1 along with the acronym used in dbfetch and WSDbfetch as the database name.
Table 1.
Databases available to dbfetch and their names within dbfetch
| Database | dbfetch name |
|---|---|
| EDAM Ontology | edam |
| EMBL-Bank | embl |
| EMBLCDS | emblcds |
| EMBLCON | emblcon |
| EMBLCONEXP | emblconexp |
| EMBL-SVA | emblsva |
| Ensembl Gene | ensemblgene |
| Ensembl Genomes Gene | ensemblgenomesgene |
| EnsemblGenomes Transcript | ensemblgenomestranscript |
| Ensembl Transcript | ensembltranscript |
| European Patent Office Proteins | epo_prt |
| Human Genome Nomenclature Committee | hgnc |
| Human Mayor Histocompatibility Complex | imgthla |
| IMGT/LIGM-DB | imgtligm |
| InterPro | interpro |
| IPD-KIR Killer-cell Immunoglobulin-like Receptors | ipdkir |
| IPD-MHC Mayor Histocompatibility Complex | ipdmhc |
| IPRMC InterPro Matches | iprmc |
| IPRMC Uniparc | iprmcuniparc |
| Japanese Patent Office Proteins | jpo_prt |
| Korean Intelectual Property Off. Proteins | kipo_prt |
| MEDLINE | medline |
| Patent DNA Non Redundant L1 | nrnl1 |
| Patent DNA Non Redundant L2 | nrnl2 |
| Patent Protein Non Redundant RL1 | nrpl1 |
| Patent Protein Non Redundant L2 | nrpl2 |
| Patent Equivalents | patent_equivalents |
| PDB Structures and Sequences | pdb |
| RefSeq nucleotide | refseqn |
| RefSeq protein | refseqp |
| SGT | sgt |
| Taxonomy | taxonomy |
| ENA Trace Archive | tracearchive |
| UniParc | uniparc |
| UniProtKB | uniprotkb |
| UniRef100 | uniref100 |
| UniRef50 | uniref50 |
| UniRef90 | uniref90 |
| UniSave | unisave |
| USPTO Proteins | uspto_prt |
A listing of the available databases with a description of each database, details of the various available data formats and result styles and example entry identifiers can be found at: http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/dbfetch.databases
Hardware
An Internet-connected UNIX, Linux, Mac, or Windows workstation
Software
The “wget” utility. For MS Windows “wget” can be obtained in Cygwin (http://cygwin.com/) or from GnuWin (http://gnuwin32.sourceforge.net/). For OS X, Linux and UNIX systems “wget” is commonly installed by default. If “wget” is not installed it can be installed from the systems package manager or downloaded and installed from https://www.gnu.org/software/wget/.
Input
A database entry identifier in the format “database name”:”database identifier” supported by EMBL-EBI.
Steps for using WSDbfetch RESTful services from command line
-
Retrieve entry into a file.
Using the above URLs with a utility such as “wget” is quite simple and building this into a shell or batch language script should be straightforward. The following describe typical command lines using “wget” and the RESTful interface of WSDbftech:
Getting the nucleotide sequence of FFA1 (free fatty acid receptor- associated with diabetes type II) also known as Gene Protein Coupled Receptor 40 GPR40, and write this to a file you would use:
wget http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/embl/AF024687
A file called AF024687 will be present in the file system after wget finishes.
-
Retrieve entry into console or terminal.
Displaying the entry directly in the console (or terminal) is also possible. To do that use the wget –qO- flag:
wget –qO- http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/embl/AF024687
-
Retrieve entry annotation.
Retrieving the annotations section of the nucleotide sequence is done using:
wget -qO- http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/embl/CP000651/annot
-
Retrieve entry fasta format sequence.
Examining the above entry the user will notice that cross-references to Ensembl and UniProtKB are present in the annotation. The identifiers here can be used to obtain these entries. Suppose you want to obtain the protein sequence in fasta format. You would type:
wget -qO- http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/uniprot/O14842/fasta
-
Retrieve entry with cross-references and features.
Retrieving the Ensembl Gene is achieved by typing:
wget -qO- http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/ensemblgene/ENSG00000126266
The default for Ensembl Gene in dbfetch is to retrieve a sequence in fasta format. However, should you wish to retrieve annotations with cross-references and features in EMBL format you can use:
wget -qO- http://www.ebi.ac.uk/Tools/dbfetch/dbfetch/ensemblgene/ENSG00000126266/embl
ALTERNATE PROTOCOL 2: RETRIEVING DATA FROM EMBL-EBI USING WSDBFETCH VIA SOAP INTERFACE
The support team at EMBL-EBI has prepared example clients based on the SOAP interface. These provide the full functionality of the dbfetch service and demonstrate how a developer can integrate this service into his code. In this section we will limit the scope of use to the client themselves and not with the coding aspects of the services. Although they are only example clients, they may be suitable for a user’s needs without modification.
For a full description of the WSDbfetch SOAP Web Services, see http://www.ebi.ac.uk/Tools/webservices/services/dbfetch.
Necessary Resources
Hardware
An Internet-connected UNIX, Linux, Mac, or Windows workstation
Software
Perl (http://www.perl.org/) with the SOAP::Lite Perl module installed.
Download the Perl SOAP::Lite client from:
http://www.ebi.ac.uk/Tools/webservices/download_clients/perl/soaplite/wsdbfetch_soaplite.pl
See Support Protocol 1 for downloading and installing Perl SOAP Web Services Clients.
Steps to run WSDbfetch SOAP web services using Perl client. Downloading and installing the client is detailed in Support Protocol 1.
-
Display client usage
Run the script without specifying any parameters to print a brief help message (Figure 5).
The help message is divided into 3 sections. In the first methods which retrieve information about the available databases are detailed. The second section has instructions on how to retrieve a single entry using the “fetchData” method while the third provides information on how to retrieve entries in batch using the “fetchBatch” method.
-
Display a list of the databases supported by the service.
wsdbfetch_soaplite.pl getSupportedDBs
-
Display a list of the available databases with a comma separated list of supported formats:
wsdbfetch_soaplite.pl getSupportedFormats
-
Retrieve an entry.
To obtain the protein structure of the hepatocyte derived nuclear factor 4alpha from the PDB, which is described in the PDB entry 3CBB, enter the following command:
wsdbfetch_soaplite.pl fetchData pdb:3cbb
To get the sequences of all the chains in the structure, in fasta format, enter:
wsdbfetch_soaplite.pl fetchData pdb:3cbb fasta
This returns all four chains in the structure (Figure 6).
To get the sequence of a specific chain, instead of all the chains, the chain identifier is used as suffix for the entry identifier:
wsdbfetch_soaplite.pl fetchData pdb:3cbb_A fasta
Note: While PDB entry identifiers are not case sensitive, the PDB chain identifiers are. Thus “3cbb_a” and “3cbb_A” are not the same.
-
Retrieve a set of entries from a database.
Using the “fetchBatch” method a set of entries can be retrived. For example to fetch the sequences from the UniProtKB entries for the rat, mouse and pig whey acidic protein precursor, in fasta format, enter the following command:
wsdbfetch_soaplite.pl fetchBatch uniprotkb wap_rat,wap_mouse,wap_pig fasta raw
While the UniProtKB entry names are used in this command, these are not stable over time, so it is better whenever possible to use the UniProtKB entry accessions instead, for example:
wsdbfetch_soaplite.pl fetchBatch uniprotkb P01174,P01173,O46655 fasta raw
Figure 5.

Wsdbfetch soaplite client displaying help text
Figure 6.
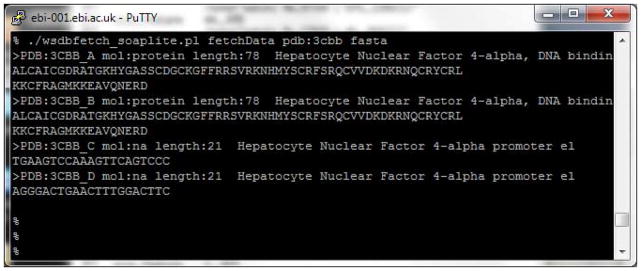
Wsdbfetch command line for retrieving amino acid sequences that correspond to chain identifiers from a 3D structure.
SUPPORT PROTOCOL 1: INSTALLING PERL SOAP WEB SERVICES CLIENTS
Perl is commonly used in bioinformatics and typically installed by default on UNIX and UNIX-like systems. Since many existing analytical pipelines are implemented in Perl, the Perl clients provide an option for integration of Web Services into existing pipelines.
Necessary Resources
Hardware
A MS Windows, Apple OS X, Linux or UNIX computer
Software
Perl (http://www.perl.org/).
A web browser, for example Google Chrome, Mozilla Firefox, MS Internet Explorer, Opera or Safari
-
Check that the SOAP::Lite Perl module has been installed.
On MS Windows open a Command Prompt. The procedure to do this varies according to different versions of Windows. On OS X, Linux or UNIX, open a terminal.
In the Command Prompt or terminal enter:
perl -MSOAP::Lite -e “use SOAP::Lite; print \$SOAP::Lite::VERSION;”
If a “Can’t locate SOAP/Lite.pm” error message is returned, the SOAP::Lite Perl module needs to be installed.
The SOAP::Lite Perl module can be installed via the operating system package manager on many Linux/UNIX systems. For example on Debain based Linux distributions (e.g. Bio-Linux, Linux Mint and Ubuntu) the “libsoap-lite-perl” package should be installed.
The SOAP::Lite Perl module can be installed from the Comprehensive Perl Archive Network (CPAN), see http://www.cpan.org/ for details.
-
Open a web browser and go to the EMBL-EBI Web Services pages at http://www.ebi.ac.uk/Tools/webservices/.
This page lists the available Web Service from EMBL-EBI. For each service a brief description and links to the service documentation are provided.
-
Click through to the service documentation pages, e.g. WSDbfetch (SOAP) (http://www.ebi.ac.uk/Tools/webservices/services/dbfetch/), This page displays information about the service., including links to sample Web Service clients.
Clients are provided in a number of programming languages and using a variety of Web Services tool-kits. For WSDbfetch (SOAP) this includes C#, Java, Perl, PHP, Python, Ruby, VB.NET clients. Dependencies and requirements for running each client are detailed on the right-hand side of the table on the web page.
Download the Perl SOAP::Lite script (e.g. wsdbfetch_soaplite.pl) by clicking on the link and using the “Save as” functionality in the web browser.
-
Test and run the client.
On MS Windows open a Command Prompt. The procedure to do this varies according to different versions of Windows. On OS X, Linux or UNIX, open a terminal.
Within the Command Prompt or terminal, change to the directory which contains the client program downloaded earlier.
To test the program (e.g. wsdbfetch_soaplite.pl), enter:
perl wsdbfetch_soaplite.pl
Help information will be displayed with further instructions on usage of the client.
BASIC PROTOCOL 2: SEQUENCE SIMILARITY SEARCH USING FASTA SEARCH VIA THE WEB INTERFACE
EMBL-EBI provides and maintains a comprehensive range of freely available analysis tools through web interfaces and web services (Goujon et al. 2010). The analysis services included 53 tools, divided in 12 categories. In this section, we aim at demonstrating how to run analysis tools and interpreting results through the web interface.
The Table 2 shows the analysis tools, the categories and the URLs of their web interfaces. The popular categories include Sequence Similarity Search (e.g. NCBI BLAST+ and FASTA), Multiple Sequence Alignment (e.g. Clustal Omega), Protein Functional Analysis (e.g. InterProScan, Phobius), etc.
Table 2.
Tools and categories of EMBL-EBI analysis tools
| Tool Category | Tools Included | Web Form URL |
|---|---|---|
| Sequence Similarity Search | NCBI BLAST+, WU-BLAST, FASTA, FASTM, PSI-BLAST, PSI-Search, ENA Sequence Search | www.ebi.ac.uk/Tools/sss/ |
| Multiple Sequence Alignment | Clustal Omega, ClustalW2, DbClustal, Kalign, MAFFT, MUSCLE, MView, T-Coffee, WebPRANK | www.ebi.ac.uk/Tools/msa/ |
| Protein Function Analysis | InterProScan, Phobius, CENSOR, FingerPRINTScan, Pratt, PROSITE Scan, RADAR | www.ebi.ac.uk/Tools/pfa/ |
| Sequence Format Conversion | Seqret, Readseq, MView | www.ebi.ac.uk/Tools/sfc/ |
| Phylogeny Analysis | ClustalW2 Phylogeny | www.ebi.ac.uk/Tools/phylogeny/ |
| Pairwise Sequence Alignment | Needle, Stretcher, Water, Matcher, LALIGN, Wise2DBA, GeneWise, PromoterWise | www.ebi.ac.uk/Tools/psa/ |
| RNA analysis | MapMi | www.ebi.ac.uk/Tools/rna/ |
| Sequence Operation | CENSOR, SeqCksum | www.ebi.ac.uk/Tools/so/ |
| Sequence Translation | Transeq, Sixpack, Backtranseq, Backtransmbig | www.ebi.ac.uk/Tools/st/ |
| Sequence Statistics | Pepinfo, Pepstats, Pepwindow, SAPS, Cpgplot, Newcpgreport, Isochore | www.ebi.ac.uk/Tools/seqstats/ |
| Structure | MACiE, PDBsum, PoreLogo, PoreWalker, ProFunc, SAS, Scorecons, PDBeFold, PDBeMotif, PDBePISA, MaxProut, DaliLite | www.ebi.ac.uk/Tools/structure/ |
| EMBOSS Tools | Needle, Stretcher, Water, Matcher, Transeq, Sixpack, Backtranseq, Backtransmbig, Pepinfo, Pepstats, Pepwindow, Cpgplot, Newcpgreport, Isochore, seqret | www.ebi.ac.uk/Tools/emboss/ |
In the following sections we will introduce the most commonly used sequence analysis tools using the Web interface and SOAP/REST Web Services client programs. EMBL-EBI provides freely available Web Services for analysis tools (http://www.ebi.ac.uk/Tools/webservices/) which mainly include Sequence Similarity Search (SSS), Multiple Sequence Alignment (MSA), Protein Functional Analysis (PFA), Phylogeny Analysis, Pairwise Sequence Alignment (PSA), RNA Analysis, Sequence Format Convert (SFC), Sequence Statistics, Sequence Translation and Structure Analysis. This section demonstrates examples using Web Servicesfor SSS, PFA and MSA.
Sequence similarity search (SSS) is a method of searching sequence databases by using alignment to a query sequence. By statistically assessing how well database and query sequences match one can infer homology and transfer information to the query sequence. The EMBL-EBI SSS Web Services contain analysis tools of NCBI BLAST+, WU-BLAST, FASTA, FASTM, PSI-BLAST and PSI-Search.
First we use the FASTA service web interface to run and interpret a FASTA search job. The FASTA package provides a comprehensive set of similarity/homology searching programs, similar to those provided by NCBI BLAST+, and some additional programs for searching with short peptides and oligonucleotides.
Necessary Resources
Hardware
Any internet-connected computer
Software
A web browser, e.g. Google Chrome, Mozilla Firefox, Internet Explorer, Safari or Opera
Files
A text file containing a sequence in one of the formats of FASTA, EMBL, GenBank, GCG, PIR, NBRF, PHYLIP and UniProtKB/Swiss-Prot. If the file is not available, the entry identifier in the format “database name”:”database identifier”, E.g. embl:x56957 can be used as input, or a sequence in one of the formats mentioned above can be pasted into the form.
This example uses the mouse protein “Glutathione S-transferase Mu 1” as input sequence from the UniProtKB database. The entry details can be found at http://www.uniprot.org/uniprot/P10649 and the FASTA-format sequence can be downloaded at http://www.uniprot.org/uniprot/P10649.fasta.
Steps to run FASTA search on the web interface
-
Point the browser to the SSS web page http://www.ebi.ac.uk/Tools/sss/.
The Sequence Similarity Search page (shown in figure 7) allows a user to select different tools to search against databases of proteins, nucleotides, genomes and vectors.
-
Click “Protein” search under the FASTA section or directly go to http://www.ebi.ac.uk/Tools/sss/fasta/.
Job submission on the input form (Figure 8) for FASTA is organized into four steps: Select Your Databases, Enter Your Input Sequence, Select Your Parameters and Submit Your job. If the user wants to see the documentation, click the “Help & Documentation” link on the top navigation bar.
-
Select the databases to search.
In the “Select your databases” step, click the database categories to expand or collapse the list for available databases. Check or uncheck the boxes of the databases to select the appropriate databases. Multiple databases can be chosen. In this example, we choose UniProKB/Swiss-Prot.
-
Enter the input sequence.
Browse and select the input sequence file. Alternatively copy the sequence and paste it into the sequence box. The user can also input the entry accession with the database identifier, e.g. UniProtKB:P10649. Select the correct input sequence type just above the input sequence box. In this example, we paste a protein sequence in FASTA-format and select the sequence type of PROTEIN.
-
Set the parameters.
Firstly, select the program to run. The programs available in this search include FASTA, FASTX, FASTY, SSEARCH, GLSEARCH and GGSEARCH. Secondly, click on the “More options” button to expand the section for the advanced parameters (Figure 9), which include matrix, gap penalties, ktup, e-values, output formats, etc. Change the settings of the parameters according to your need. For more detail on each parameter, click on the name of the parameter, then enter the “Help & Documentation” page. In this example, we choose FASTA program and leave other parameters as default.
-
Submit the job.
Two modes are available for job submission: Interactive Mode and Email Mode. The first allows the user get result as soon as the job is finished; while the later will notify the user via email when result is available.
For the interactive mode, just click on the “Submit” button. An intermediate page will show up to indicate the job is running until the result is ready.
-
For the email mode, click the check-box of “Be notified by email”, then type your email address and the title of job, finally click the “Submit” button to run the job. The next page will confirm your job has been submitted. When the user receives the email notification, click on the result link to view the result.
In this example, we submit the job through interactive mode. If the information provided in the submission is not correct, the page will show a Warning or Error message to offer clues to correction. Once the information is updated, the user can resubmit the job.
-
View job result summary.
The result pages provide multiple views: Summary Table, Tool Output, Visual Output, Functional Predictions and Submission Details. The default view is Summary Table. Click on the result tabs to switch between views.
The Summary Table (Figure 10) view lists information about the resulting top hits, including alignment numbers, database and identifier, length, bit score, percentages of identities and positives, E-value, description, and cross-references to other relevant databases. The user can click on the links of identifiers or cross-references to enter external resource pages.
The user can check or unchecked the boxes of alignments in the first column of the table, then click the left-side buttons in this view to show or hide annotations and alignments, and to download source data in different formats. They can also pass the selected sequences on to other tools for further analysis, for example a Multiple Sequence Alignment using Clustal Omega.
-
Display the tool raw output.
Click the “Tool Output” tab to display the raw output (Figure 11). This page also allows the user to download the raw output in text and XML formats, and to forward output to further tool analysis pages, e.g. MView.
-
Visualize the result.
Switch to the “Visual Output” view. The visualization image (Figure 12) lines up the query sequence and the subject matches with lengths and colors, showing the significance levels of the alignments. The user can switch the color scale between Fixed and Dynamic. To produce better quality images, you can download the SGV format image from this page.
-
Display functional predictions.
A protein search job result will contain the Functional Predictions view (Figure 13), which visualizes functional predictions using InterPro matches. Check or uncheck the boxes for the protein features to include features for the visualization. Visualization can be switched between query-based and subject-based, color scale can be changed between Fixed and Dynamic, and the image can be downloaded in SVG format.
-
Display your submission details.
The Submission Details view (Figure 14) shows information about the program and its version, database, job title, date and time for job launch, input and output files, command line executed and input parameter settings. The user can review these details to decide if the submission is correct and whether a re-submission is needed.
Figure 7.

Screenshot of SSS categories web page
Figure 8.

FASTA input form
Figure 9.

Advanced parameters for FASTA search
Figure 10.

FASTA Results Summary Table
Figure 11.

FASTA Tool Output tab
Figure 12.

Visual output from FASTA search.
Figure 13.

Functional Predictions tab from FASTA search
Figure 14.

Submission Details tab
BASIC PROTOCOL 3: SEQUENCE SIMILARITY SEARCH USING NCBI BLAST+ SOAP WEB SERVICES WITH PERL CLIENT
NCBI BLAST+ (Camacho et al. 2009) is one of the widest used and most useful applications for sequence analysis. This example uses a Perl client program to run NCBI BLAST+ search via the SOAP Web Service interface.
Necessary Resources
Hardware
A UNIX, Linux, Mac, or Windows workstations
Software
Download the client from: http://www.ebi.ac.uk/Tools/webservices/download_clients/perl/soaplite/ncbiblast_soaplite.pl
See Support Protocol 1 for downloading and installing Perl SOAP Web Services clients.
For the full description of the NCBI BLAST+ SOAP Web Services, see http://www.ebi.ac.uk/Tools/webservices/services/sss/ncbi_blast_soap.
Input
A text file containing a sequence in one of the formats of GCG, FASTA, EMBL, GenBank, PIR, NBRF, PHYLIP or UniProtKB/Swiss-Prot; or a database entry supported by EMBL-EBI in the format “database name”:”database identifier” E.g. embl:x56957.
Steps to run NCBI BLAST+ SOAP web services using Perl client
-
Display client usage
Switch to the directory containing the downloaded client program - ncbiblast_soaplite.pl. For details of how to use the client, run it without any arguments:
ncbiblast_soaplite.pl
The usage help will be shown on the screen. Alternatively, run it with argument “--help”:
ncbiblast_soaplite.pl –help
Table 3 provides the major options in command-line.
-
Display parameter details.
To display all parameters of the tool, run
ncbiblast_soaplite.pl --params
To see further details of the parameter, run with argument “--paramDetail <ParameterName>”. To see which BLAST programs are available, run
ncbiblast_soaplite.pl --paramDetail program
To see which BLAST databases are available, run
ncbiblast_soaplite.pl --paramDetail database
-
Run jobs in synchronous mode.
The jobs can be run in synchronous mode, to retrieve a result as soon as the job is finished or asynchronous mode, to retrieve a result later. Here we describe how to run synchronous jobs.
To run a BLAST search, decide which BLAST program to run, the database to search and the query sequence type. Either a full sequence file or just an entry identifier in the form “database name”:”database identifier” can be used as input. Also, specify an email address for communication in using the Web Services.
For example, run a BLASTP job against the UniProtKB database with a sequence input file:
ncbiblast_soaplite.pl --email < email@example.org> --program blastp --database uniprotkb --stype protein SeqFile.fasta
If you know the entry identifier of your query sequence, you can the search using this identifier as the input:
ncbiblast_soaplite.pl --email < email@example.org> --program blastp --database uniprotkb --stype protein <DB:Identifier>
The entry identifier should contain the database name and the entry accession, separated by colon, e.g. UniProtKB:GSTM1_MOUSE, the mouse protein entry GSTM1_MOUSE in UniProtKB.
In synchronous mode, the program will prompt out JobID and JobStatus (RUNNING/FINISHED) in stand output until result files are received. The results contain files of input sequence, output files in text, XML and SVG formats.
-
Run jobs in asynchronous mode.
If the user wants to retrieve a result later, run jobs in asynchronous mode using the argument “--async”:
ncbiblast_soaplite.pl --async --email < email@example.org> --program blastp --database uniprotkb --stype protein SeqFile.fasta
If the job submission is successful, the client will provide the job identifier (JobId) in STDOUT. The user has to use the JobId in the result retrieval. Please see the guidelines section for more information about the composition of the job identifier.
To check the job status before getting the results, run:
ncbiblast_soaplite.pl --status --jobid <JobId>
The client will tell if the job is FINISHED, RUNNING, ERROR, FAILURE OR FINISHED.
If the job status is FINISHED, get the result types:
ncbiblast_soaplite.pl --resultTypes –jobid <JobId>
The NCBI BLAST+ web services provide result types of plain output (out), plain input (sequence), alignment identifiers (ids), XML result (xml) and other visualization images in SVG and PNG formats.
If the user wants to retrieve the result of a specific result type, for example, the plain text output (out):
ncbiblast_soaplite.pl –polljob –outformat out -- jobid <JobId>
To retrieve all available results:
ncbiblast_soaplite.pl --polljob -- jobid <JobId>
If the job status is RUNNING, please check it again later. In the case of ERROR or FAILURE, please resubmit your job. If the user still experiences the same issue, please send us a support request via http://www.ebi.ac.uk/support/, making sure to include the JobId and the error message. In the case of NOT_FOUND, please check the JobId; if the JobId is correct, the job results might have expired, thus please resubmit the job.
Table 3.
Description of important command-line options for the NCBI BLAST+ client.
| Option | Type | Description |
|---|---|---|
| [Required] | ||
| -p, --program | : str : | BLAST program to use, see --paramDetail program |
| -D, --database | : str : | Database(s) to search, space separated. |
| --stype | : str : | Query sequence type, see --paramDetail stype |
| seqFile | : file : | Query sequence |
| [Optional] | ||
| -m, --matrix | : str : | Scoring matrix, see --paramDetail matrix |
| -e, --exp | : real : | 0<E<= 1000. Statistical significance threshold for reporting database sequence matches. |
| -f, --filter | filter the query sequence for low complexity regions, see --paramDetail filter | |
| -A, --align | : int : | Pairwise alignment format, see --paramDetail align |
| -s, --scores | : int : | Number of scores to be reported |
| -n, --alignments | : int : | Number of alignments to report |
| -u, --match | : int : | Match score (BLASTN only) |
| -v, --mismatch | : int : | Mismatch score (BLASTN only) |
| -o, --gapopen | : int : | Gap open penalty |
| -x, --gapext | : int : | Gap extension penalty |
| -d, --dropoff | : int : | Drop-off |
| -g, --gapalign | Optimize gapped alignments | |
| --compstats | : str : | Composition adjustment/statistics method, see--paramDetail compstats |
| --seqrange | : str : | Region within input to use as query |
| --multifasta | Treat input as a set of fasta formatted sequences | |
| [General] | ||
| --async | Forces to make an asynchronous query | |
| : str : | Email address | |
| --title | : str : | Title for job |
| --status | Get job status | |
| --resultTypes | Get available result types for job | |
| --polljob | Poll for the status of a job | |
| --jobid | : str : | Jobid that was returned when an asynchronous job was submitted. |
| --params | List input parameters | |
| --paramDetail | Display details for input parameter | |
BASIC PROTOCOL 4: ITERATIVE SEQUENCE SEARCH USING PSI-SEARCH REST WEB SERVICES WITH PERL CLIENT
PSI-Search (Li et al. 2012) is a highly accurate iterative motif-based similarity search tool for proteins. It combines an optimal Smith–Waterman local alignment sequence search, using SSEARCH (Pearson 1991), with the PSI-BLAST profile construction strategy. An optional sequence boundary-masking procedure, which prevents alignments from being extended after they are initially included, can reduce HOE errors (Gonzalez et al. 2010) in the PSSM profile. This example uses a Perl client program to run PSI-Search via the REST Web Service interface.
Necessary Resources
Hardware
An Internet-connected UNIX, Linux, Mac, or Windows workstation
Software
Download the client from: http://www.ebi.ac.uk/Tools/webservices/download_clients/perl/lwp/psisearch_lwp.pl
See Support Protocol 2 for downloading and installing Perl REST Web Services clients.
For the full description of the PSI-Search REST Web Services, see http://www.ebi.ac.uk/Tools/webservices/services/sss/psisearch_rest
Input
A text file containing a sequence in one of the formats of GCG, FASTA, EMBL, GenBank, PIR, NBRF, PHYLIP or UniProtKB/Swiss-Prot; or a database entry supported by EMBL-EBI.
Steps to run PSI-Search REST Web Services using the Perl client
-
Display client usage.
Switch to the directory containing the downloaded client program - psisearch_lwp.pl. For details of how to use the client, run it without any arguments:
psisearch_lwp.pl
Usage help will be shown on the screen. Alternatively, run it with the argument “--help”:
psisearch_lwp.pl –help
Table 4 provides the major options in command-line.
-
2. Display parameter details
To see parameter details, run the client with argument “--paramDetail <ParameterName>”. To see which PSI-Seach programs are available, run
psisearch_lwp.pl --paramDetail program
To see which databases are available, run
psisearch_lwp.pl --paramDetail database
-
3. Run jobs in synchronous mode
The user can run jobs in synchronous mode to retrieve result as soon as the job is finished, or asynchronous mode to retrieve result later. Here we describe how to run synchronous jobs.
The user can run a job with a sequence file or entry identifier as input. The user also needs to specify an email address for communication in using the Web Services.
For example, run a PSI-Seach job against the UniProtKB database with a sequence input file:
psisearch_lwp.pl --email < email@example.org> --database uniprotkb SeqFile.fasta
If you know the entry identifier of your query sequence, you can the search using this identifier as input:
psisearch_lwp.pl --email < email@example.org> --database uniprotkb <DB:Identifier>
The entry identifier should contain the database name and the identifier, separated by colon, e.g. UniProt:GSTM1_MOUSE, the mouse protein entry GSTM1_MOUSE in UniProtKB.
In synchronous mode, the program will prompt out JobID and JobStatus (RUNNING/FINISHED) in standard output until result files are received. The results contain files of input sequence, output files in text, XML and SVG formats.
To run the next iteration, please go to Step 5 underneath.
-
4. Run jobs in asynchronous mode.
If the user wants to retrieve a result later, please run jobs in asynchronous mode using the argument “--async”:
psisearch_lwp.pl --async --email < email@example.org> --database uniprotkb SeqFile.fasta
If the job submission is successful, the client will provide the job identifier (JobId) in STDOUT. The user has to use the JobId in the result retrieval.
To check the job status before getting the results, run:
psisearch_lwp.pl --status --jobid <JobId>
The client will say if the job is FINISHED, RUNNING, ERROR, FAILURE OR FINISHED.
If the job status is FINISHED, you can view the possible result types with the command: psisearch_lwp.pl --resultTypes –jobid <JobId>
The PSI-Search Web Service provides result types of plain text output (out), plain input (sequence), XML result (xml), alignment identifiers (ids), selected alignment identifiers (preselected_ids) for the next iteration, checkpoint file (asn) for the next iteration and other visualization images in SVG and PNG formats.
If the user wants to retrieve one specific result type, for example, the plain text output (out):
psisearch_lwp.pl –polljob –outformat out -- jobid <JobId>
To retrieve all available results:
psisearch_lwp.pl --polljob -- jobid <JobId>
If the job status is RUNNING, please check it again later. In the case of ERROR or FAILURE, please resubmit your job. If the user still experiences the same issue, please submit a support request to http://www.ebi.ac.uk/support/ including the JobId and the error message. In the case of NOT_FOUND, please checks the JobId; if the JobId is correct, the job results might have expired, thus please resubmit the job.
To run the next iteration, please go to Step 5 underneath.
-
5. Run further iterations.
Step 3 and Step 4 mention the first iteration search for PSI-Search. This step explains how to run second and further iterations.
tSupportedFormatsefine the profile (PSSM) used to perform the search after the first iteration the set of hits to be included in the generation of the PSSM needs to be specified in the next iteration. This can be either obtained from the previous iteration using the job identifier of the previous iteration, or be explicit specification of a file containing the list of identifiers.
Usage for running the second iteration:
psisearch_lwp.pl --email <email> [--selectedHits <selFile>] [options...]
For example, if the first iteration JobId is psisearch-R20140226-143924-0629-76338157-pg, make sure the selected-hits file psisearch-R20140226-143924-0629-76338157-pg.preselected_ids.txt is available. The user can modify the selected-hits file to add or removed the hit identifies. Then run the second iteration:
psisearch_lwp.pl --email email@example.org --database uniprotkb -- selectedHits psisearch-R20140226-143924-0629-76338157-pg.preselected_ids.txt SeqFile.fasta
Usage for running the third or further iteration:
psisearch_lwp.pl --email <email> --selectedHits <selFile> [options...]
For example, if the second iteration JobId is psisearch-R20140226-144011-0719-82303522-oy, run the third iteration:
psisearch_lwp.pl --email email@example.org --database uniprotkb --selectedHits psisearch-R20140226-144011-0719-82303522-oy.preselected_ids.txt SeqFile.fasta
Jobs run in synchronous mode will retrieve the results immediately after the job is finished. To retrieve results for jobs run in synchronous mode, follow the instructions mentioned in Step 4.
Table 4.
Description of important command-line options for the PSI-Search client.
| Option | Type | Description |
|---|---|---|
| [Required] | ||
| -D, --database | : str : | Database(s) to search, space separated. |
| seqFile | : file : | Query sequence |
| [Optional] | ||
| -M, --matrix | : str : | Scoring matrix, see --paramDetail matrix |
| -e, --expthr | : real : | 0<E<= 1000. Statistical significance threshold for reporting database sequence matches. |
| -h, --psithr | : real : | E-value limit for inclusion in PSSM |
| -v, --scores | : int : | Number of scores to be reported |
| -b, --alignments | : int : | Number of alignments to report |
| -G, --gapopen | : int : | Gap open penalty |
| -E, --gapext | : int : | Gap extension penalty |
| --hsps | : : | Enable multiple alignments per-hit |
| --nohsps | : : | Disable multiple alignments per-hit |
| --scoreformat | : str : | Score table format for FASTA output |
| --previousjobid | : str : | Job Id for last iteration |
| --selectedHits | : file : | Selected hits from last iteration for building search profile (PSSM) |
| -R, --cpfile | : file : | PSI-BLAST checkpoint from last iteration |
| --multifasta | : : | Treat input as a set of fasta formatted sequences |
| [General] | ||
| --async | Forces to make an asynchronous query | |
| : str : | Email address | |
| --title | : str : | Title for job |
| --status | Get job status | |
| --resultTypes | Get available result types for job | |
| --polljob | Poll for the status of a job | |
| --jobid | : str : | Jobid that was returned when an asynchronous job was submitted. |
| --params | List input parameters | |
| --paramDetail | Display details for input parameter | |
SUPPORT PROTOCOL 2: INSTALLING PERL REST WEB SERVICES CLIENTS
Perl is commonly used in bioinformatics and typically installed by default on UNIX and UNIX-like systems. Since many existing analytical pipelines are implemented in Perl, the Perl clients provide an option for integration of Web Services into existing pipelines.
Necessary Resources
Hardware
A MS Windows, Apple OS X, Linux or UNIX computer
Software
Perl (http://www.perl.org/).
A web browser, for example Google Chrome, Mozilla Firefox, MS Internet Explorer, Opera or Safari
-
Check that the required LWP and XML::Simple Perl modules have been installed.
On MS Windows open a Command Prompt. The procedure to do this varies according to different versions of Windows. On OS X, Linux or UNIX, open a terminal.
In the Command Prompt or terminal enter:
perl -MLWP -e “print \$LWP::VERSION;”
perl -MXML::Simple -e “print \$XML::Simple::VERSION;”
If a “Can’t locate LWP.pm” error message is returned, the LWP Perl module needs to be installed.
The LWP Perl module can be installed via the operating system package manager on many Linux/UNIX systems. For example on Debain based Linux distributions (e.g. Bio-Linux, Linux Mint and Ubuntu) the “libwww-perl” package should be installed.
The LWP Perl module can be installed from the Comprehensive Perl Archive Network (CPAN), see http://www.cpan.org/ for details.
If a “Can’t locate XML/Simple.pm” error message is returned, the XML::SImple Perl module needs to be installed.
The LWP Perl module can be installed via the operating system package manager on many Linux/UNIX systems. For example on Debain based Linux distributions (e.g. Bio-Linux, Linux Mint and Ubuntu) the “libxml-simple-perl” package should be installed.
The XML::Simple Perl module can be installed from the Comprehensive Perl Archive Network (CPAN), see http://www.cpan.org/ for details.
Locate the Web Services example client (see Support Protocol 1, steps 2 and 3).
Download the Perl LWP script (e.g. ncbiblast_lwp.pl) by clicking on the link and using the “Save as” functionality in the web browser.
-
Test and run the client.
On MS Windows open a Command Prompt. The procedure to do this varies according to different versions of Windows. On OS X, Linux or UNIX, open a terminal.
Within the Command Prompt or terminal, change to the directory which contains the client program downloaded earlier.
To test the program (e.g. ncbiblast_lwp.pl), enter:
perl ncbiblast_lwp.pl
Help information will be displayed with further instructions on usage of the client.
BASIC PROTOCOL 5: PROTEIN FUNCTIONAL ANALYSIS USING INTERPROSCAN 5 SOAP WEB SERVICES WITH JAVA CLIENT
InterProScan 5 (Jones et al. 2014) combines different protein signature recognition methods into one resource and allows user to scan sequences for matches against the InterPro collection of protein signature databases.
This example uses a Java client program to run InterProScan 5 search via the SOAP Web Service interface.
Necessary Resources
Hardware
A UNIX, Linux, Mac, or Windows workstations
Software
Download the client IPRScan5_Axis1.jar from: http://www.ebi.ac.uk/Tools/webservices/download_clients/java/jar/IPRScan5_Axis1.jar
Download required libraries: http://www.ebi.ac.uk/Tools/webservices/download_clients/java/jar/ebiws-lib.zip
See Support Protocol 3 for downloading and installing Java Web Services Clients.
For the full description of the InterProScan 5 SOAP Web Services, see http://www.ebi.ac.uk/Tools/webservices/services/pfa/iprscan5_soap.
Input
A text file containing a sequence in one of the formats of GCG, FASTA, EMBL, GenBank, PIR, NBRF, PHYLIP or UniProtKB/Swiss-Prot; or a database entry supported by EMBL-EBI.
Steps to run InterProScan 5 SOAP web services using Perl client
-
Display client usage.
Switch to the directory containing the downloaded client program - IPRScan5_Axis1.jar. Unzip the required libraries:
unzip ebiws-lib.zip
For details of how to use the client, run it without any arguments:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar
A usage help will be shown on the screen. Alternatively, run it with argument “--help”:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar –help
Table 5 provides the major options for the command-line.
-
Display parameter details.
To display all parameters of the tool, run
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --params
To see the details of a parameter, use with the argument “--paramDetail <ParameterName>”. To see which applications are available, run
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --paramDetail appl
-
Run jobs in synchronous mode.
The user can run jobs in synchronous mode to retrieve results as soon as the job is finished, or asynchronous mode to retrieve results at a later time. Here we describe how to run synchronous jobs.
To run an InterProScan 5 search, user has to decide the applications to run. The user can run a job with a sequence file or entry identifier as input. The user also needs to specify an email address for communication in using the Web Services.
For example, run an InterProScan 5 job using all InterPro applications with a sequence input file:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --email < email@example.org> SeqFile.fasta
If you know the entry identifier of your query sequence, you can the search using this identifier as input:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --email < email@example.org> <DB:Identifier>
The entry identifier should contain the database name and the entry identifier, separated by colon, e.g. UniProt:GSTM1_MOUSE, the mouse protein entry GSTM1_MOUSE in UniProtKB.
By default, all applications, GO terms and pathways are included in the analysis. To specify particular applications (e.g. PfamA, Gene3d and Phobius) without analysis of GO terms and pathways, run:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --email < email@example.org> --appl PfamA,Gene3d,Phobius --nogoterms –nopathways SeqFile.fasta
The application names are separated by comma in the command line above.
In synchronous mode, the program will prompt out JobID and JobStatus (RUNNING/FINISHED) in standard output until result files are received. The results contain files of input sequence, output files in text, XML and SVG formats.
-
Run jobs in asynchronous mode.
If the user wants to retrieve a result later, run jobs in asynchronous mode using the argument “--async”:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --async --email < email@example.org> -- SeqFile.fasta
If the job submission is successful, the client will provide the job identifier (JobId) in STDOUT. The user has to use the JobId in the result retrieval.
To check the job status before getting the results, run:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --status --jobid <JobId>
The client will tell if the job is FINISHED, RUNNING, ERROR, FAILURE OR FINISHED.
If the job status is FINISHED, get the result types:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --resultTypes –jobid <JobId>
The InterProScan 5 Web Services provide result types of plain output (out), plain input (sequence), XML result (xml), GFF output (gff), TSV table (tsv), a HTML tarball file (htmltarball) and the SVG image (svg).
If the user wants to retrieve a specific result type, for example, the plain text output (out):
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar –polljob –outformat out -- jobid <JobId>
To retrieve all available results:
java -Djava.ext.dirs=lib -jar IPRScan5_Axis1.jar --polljob -- jobid <JobId>
If the job status is RUNNING, please check it later. In the case of ERROR or FAILURE, please resubmit your job. If the user still experiences issues, please send us a support request via http://www.ebi.ac.uk/support/ including the JobId and the error message. In the case of NOT_FOUND, please check the JobId; if the JobId is correct, the job results might have expired, thus please resubmit the job.
Table 5.
Description of important command-line options for the InterProScan 5 client.
| Option | Type | Description |
|---|---|---|
| [Required] | ||
| seqFile | : file : | Query sequence |
| [Optional] | ||
| --appl | : str : | Comma separated list of signature methods to run |
| --goterms | : : | Enable retrieval of GO terms |
| --nogoterms | : : | Disable retrieval of GO terms |
| --pathways | : : | Disable retrieval of pathway terms |
| --nopathways | : : | Disable retrieval of pathway terms |
| --multifasta | : : | Treat input as a set of fasta formatted sequences |
| [General] | ||
| --params | : : | List tool parameters |
| --paramDetail | : str : | Information about a parameter |
| : str : | Email address, required to submit job | |
| --title | : str : | Title for the job |
| --async | : : | Perform an asynchronous submission |
| --jobid | : str : | Job identifier |
| --status | : : | Get status of a job |
| --resultTypes | : : | Get list of result formats for a job |
| --polljob | : : | Get results for a job |
SUPPORT PROTOCOL 3: INSTALLING JAVA WEB SERVICES CLIENTS
Commonly installed Java provides a platform independent option for developing and deploying software.
Necessary Resources
Hardware
A MS Windows, Apple OS X, Linux or UNIX computer
Software
A Java runtime environment, see http://www.java.com/.
A web browser, for example Google Chrome, Mozilla Firefox, MS Internet Explorer, Opera or Safari
Locate the Web Services example client (see Support Protocol 1, steps 2 and 3).
Download the Java executable jar (e.g. NCBIBlast_Axis1.jar) by clicking on the link.
Download the dependencies archive: “ebiws-lib.zip”
Extract the files in the dependencies archive. For example on MS Windows this can be down using Explorer to open the archive and copy the “lib” directory to the desired location.
-
Test and run the client.
On MS Windows open a Command Prompt. The procedure to do this varies according to different versions of Windows. On OS X, Linux or UNIX, open a terminal.
Within the Command Prompt or terminal, change to the directory which contains the client program downloaded earlier.
To test the program (e.g. NCBIBlast_Axis1.jar), enter:
java –Djava.ext.dirs=lib -jar NCBIBlast_Axis1.jar
Where “lib” is the location of the “lib” directory created by extracting the dependencies archive.
Help information will be displayed with further instructions on usage of the client.
If “java” is not found, but has been installed it may need to be added to the current PATH, see step 6.
-
Adding Java to the PATH.
-
For MS Windows check the location used to install Java using Explorer. This will usually be something like “C:\Program Files (x86)\Java\jre7”. In the Command Prompt add the location of the Java “bin” directory to the PATH, by entering:
set PATH=%PATH%;C:\Program Files (x86)\Java\jre7\bin
The “java” command should now be found.
-
On Linux, OS X and UNIX systems the method to add a directory to the PATH depends on the shell being used. First locate the Java installation, and then add the Java “bin” directory to the PATH. For example for a Java installation in “/usr/lib/jvm/java-7-openjdk-amd64/”
-
For sh or bash shells:
export PATH=${PATH}:/usr/lib/jvm/java-7-openjdk-amd64/bin
-
For csh or tcsh shells:
setenv PATH ${PATH}:/usr/lib/jvm/java-7-openjdk-amd64/bin
-
-
BASIC PROTOCOL 6: MULTIPLE SEQUENCE ALIGNMENT USING CLUSTAL OMEGA VIA WEB INTERFACE
Multiple Sequence Alignment (MSA) is generally the alignment of three or more biological sequences. From the output, homology can be inferred and the evolutionary relationships between the sequences studied.
Clustal Omega (Sievers et al. 2011) is a fast, large-scale multiple sequence alignment program that uses seeded guide trees and HMM profile-profile techniques to generate alignments.
Hardware
Any internet-connected computer
Software
A web browser, e.g. Google Chrome, Mozilla Firefox, Internet Explorer, Safari or Opera
Files
A text file containing three or more sequences in one of the following formats: FASTA, EMBL, GCG, PIR, NBRF, PHYLIP and UniProtKB/Swiss-Prot.
This example uses a FASTA format multiple sequence file containing a collection of Myosin sequences. The example file can be downloaded from http://www.ebi.ac.uk/Tools/examples/protein/sequence12.txt
Steps to run Clustal Omega via the web interface
-
(Optional) To view the range of MSA tools available at EMBL-EBI, point the browser to the MSA web page http://www.ebi.ac.uk/Tools/msa/
The Multiple Sequence Alignment page (shown in Figure 15) allows a user to select between different MSA tools.
-
Click “Launch Clustal Omega” under the Clustal Omega section, or directly go to http://www.ebi.ac.uk/Tools/msa/clustalo/.
Job submission in via this page (Figure 16) is organized into three steps: Enter Your Input Sequences, Set Your Parameters and Submit Your job. If the user wants to see the Help documentation, click the “Help & Documentation” link on the top navigation bar.
-
Enter the input sequences.
Browse and select the input sequences file. Alternatively copy the sequences and paste them into the sequence box. Select the correct input sequence type just above the input sequence box. In this example, we paste a set of protein sequences in FASTA-format and select the sequence type of PROTEIN.
-
Set the parameters.
Firstly, select the output format. To examine further options, click on the “More options” button to expand the section for the advanced parameters, which for Clustal Omega include options to de-align input sequences, the number of iterations for the guide tree and HMM stages etc. Change the settings of the parameters according to your need. For more detail on each parameter, click on the name of the parameter, or visit the “Help & Documentation” page. In this example, we leave the parameters at their default settings.
-
Submit the job.
Two modes are available for job submission: Interactive Mode and Email Mode. The first allows the user get result as soon as the job is finished; while the later will notify the user via email when result is available.
For the interactive mode, just click on the “Submit” button. An intermediate page will show up to indicate the job is running until the result is ready.
-
For the email mode, click the check-box of “Be notified by email”, then enter your email address and the title of job, finally click the “Submit” button to run the job. The next page will confirm your job has been submitted. When the user receives the email notification, click on the result link in the email to view the result.
In this example, we submit the job through interactive mode. If the information provided in the submission is not correct, the page will show a warning or error message and offer clues to correct this. Once the information is updated, the user can re-submit the job.
-
View results.
The result pages provide multiple views: Alignments, Result Summary, Phylogenetic Tree and Submission Details. The default view is Alignments. Click on the result tabs to switch between views.
The “Alignments” tab (Figure 17) shows the alignment produced by Clustal Omega. There are buttons to download the alignment, send the alignment to a Phylogenetic program, and for protein alignments, to color the alignment by physico-chemical property.
-
View all output files.
Click the “Result Summary” tab to display the list of all outputs from the program, including the Percent Identity Matrix (Figure 18). This page also allows users to launch a Jalview (Waterhouse et al. 2009) applet with the alignment, which provides further visualization options.
-
View the phylogenetic tree.
Switch to the “Phylogenetic Tree” view. This page shows a simple (by default: Neighbour-joining) phylogenetic tree calculated from your alignment. The first part of the page (Figure 19) contains the full tree data, which can be downloaded for use in third-party tree viewer programs. The second part of the page (Figure 20) contains a visualization of the tree data with options to display fixed or scaled branch lengths.
-
Display your submission details.
The Submission Details view (Figure 21) shows information about the program and its version, job title, date and time for job launch, input and output files, command line executed and input parameter settings. The user can review these details to decide if the submission is correct and if a re-submission is needed.
Figure 15.

Multiple Sequence Alignment tools page
Figure 16.

Clustal Omega input form
Figure 17.

The Alignments tab from Clustal Omega results.
Figure 18.
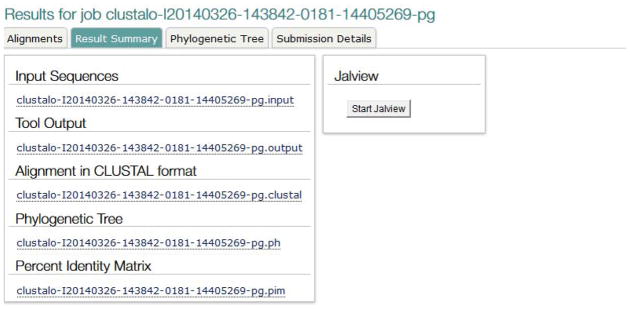
Result Summary tab from Clustal Omega
Figure 19.

Phylogenetic tree data.
Figure 20.
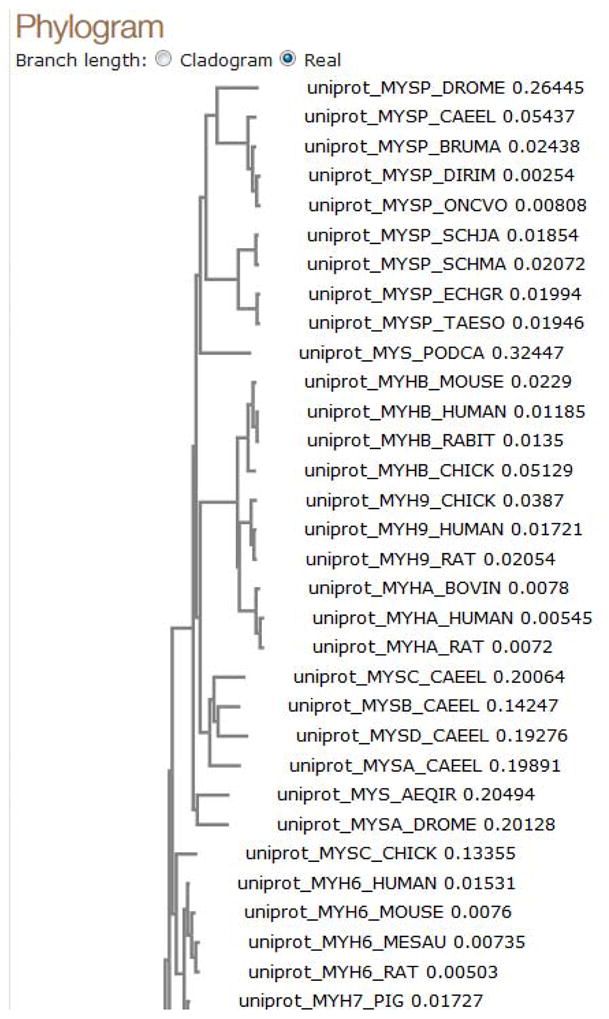
Phyogenetic tree vizualisation
Figure 21.

Submission Details tab for Clustal Omega
ALTERNATE PROTOCOL 3: MULTIPLE SEQUENCE ALIGNMENT USING CLUSTAL OMEGA VIA C# .NET CLIENT
Multiple Sequence Alignment (MSA) is generally the alignment of three or more biological sequences. From the output, homology can be inferred and the evolutionary relationships between the sequences studied.
Clustal Omega (Sievers et al. 2011) is a fast, large-scale multiple sequence alignment program that uses seeded guide trees and HMM profile-profile techniques to generate alignments.
This protocol demonstrates a Clustal Omega multiple sequence alignment via Web Services using a C# .Net client.
Necessary Resources
Hardware
A UNIX, Linux, Mac, or Windows workstations
Software
Download the example client from: http://www.ebi.ac.uk/Tools/webservices/download_clients/csharp/bin/ClustalOCliClient.exe
See Support Protocol 4 for downloading and installing C#.NET Web Services Clients.
For the full description of the Clustal Omega SOAP Web Services, see http://www.ebi.ac.uk/Tools/webservices/services/msa/clustalo_soap
Input
A text file containing three or more sequences in one of the following formats: FASTA, EMBL, GCG, PIR, NBRF, PHYLIP and UniProtKB/Swiss-Prot.
Steps to run Clustal Omega SOAP web services using the example .NET client
-
Display client usage.
Switch to the directory containing the downloaded client program - ClustalOCliClient.exe. For details of how to use the client, run it without any arguments:
ClustalOCliClient
A usage help will be shown on the screen. Alternatively, run it with argument “--help”:
ClustalOCliClient –help
Table 6 shows the main options for the command-line.
-
Display parameter details.
To display all parameters of the tool, run
ClustalOCliClient --params
To see further details of the parameter, run with argument “--paramDetail <ParameterName>”. For example to see what input types are available run
ClustalOCliClient --paramDetail stype
-
Run jobs in synchronous mode.
The user can run jobs in synchronous mode to retrieve the results as soon as the job is finished, or asynchronous mode to retrieve results at a later time.
To run a Clustal Omega alignment the user has to supply a minimum of an input file containing three or more sequences in the correct format and their email address.
For example:
ClustalOCliClient --email < email@example.org> sequence12.txt
In synchronous mode, the program will output the JobID and JobStatus (RUNNING/FINISHED) to standard output until result files are received. The results contain files of input sequence, output files in text, and XML formats.
-
Run jobs in asynchronous mode.
If the user wants to retrieve result at a later time, please run jobs in asynchronous mode using the argument “--async”:
ClustalOCliClient--async --email < email@example.org> -- sequence12.txt
If the job submission is successful, the client will provide the job identifier (JobId) in STDOUT. The user has to use the JobId to retrieve the result.
To check the job status before getting the results, run:
ClustalOCliClient --status --jobid <JobId>
The client will say if the job status is FINISHED, RUNNING, ERROR, FAILURE OR FINISHED.
If the job status is FINISHED, you can view the available result types with:
ClustalOCliClient --resultTypes –jobid <JobId>
With default options the Clustal Omega Web Services provides result types of plain output (out), plain input (sequence), alignment (aln-clustal), phylogenetic tree data (phylotree) and the Percent Identity Matrix (pim)
If the user wants to retrieve a specific result type, for example, the plain text output (out):
ClustalOCliClient –polljob –outformat out -- jobid <JobId>
To retrieve all available results:
ClustalOCliClient --polljob -- jobid <JobId>
If the job status is RUNNING, please check it again later. In the case of ERROR or FAILURE, please resubmit your job. If the user still experiences issues, please contact us via http://www.ebi.ac.uk/support/ including the JobId and the error message. In the case of NOT_FOUND, check the JobId; if the JobId is correct, the job results might have expired (7 days after submission), so you will need to resubmit the job.
Table 6.
Description of important command-line options for the Clustal Omega .NET client.
| Option | Type | Description |
|---|---|---|
| [Required] | ||
| seqFile | : file : | sequences to align (“-” for STDIN) |
| [Optional] | ||
| --stype | : str : | input sequence type, see --paramDetail stype. |
| --guidetreeout | enable output of guide tree. | |
| --noguidetreeout | disable output of guide tree. | |
| --dismatout | enable output of distance matrix. | |
| --nodismatout | disable output of distance matrix. | |
| --dealign | enable de-alignment of input sequences. | |
| --nodealign | disable de-alignment of input sequences. | |
| --mbed | enable mbed-like clustering guide-tree. | |
| --nombed | disable mbed-like clustering guide-tree. | |
| --mbediteration | enable mbed-like clustering iteration. | |
| --nombediteration | disable mbed-like clustering iteration. | |
| --iterations | : int: | number of iterations, see --paramDetail iterations. |
| --gtiterations | : int : | maximum guide tree iterations, see --paramDetail gtiterations. |
| --hmmiterations | : int : | maximum HMM iterations, see --paramDetail hmmiterations. |
| --outfmt | : str : | output alignment format, see --paramDetail outfmt. |
| [General] | ||
| -h, --help | prints this help text | |
| --async | forces to make an asynchronous query | |
| : str : | e-mail address | |
| --title | : str : | title for job |
| get job status | ||
| --resultTypes | get available result types for job | |
| --polljob | poll for the status of a job | |
| --jobid | : str : | jobid that was returned when an asynchronous job was submitted. |
| --outfile | : str : | file name for results (default is jobid; “-” for STDOUT) |
| --outformat | : str : | result format to retrieve |
| --params | list input parameters | |
| --paramDetail | : str : | display details for input parameter |
| --quiet | decrease output | |
| --verbose | increase output | |
| --trace | show SOAP messages being interchanged | |
SUPPORT PROTOCOL 4: INSTALLING C# .NET WEB SERVICES CLIENTS
.NET is a platform and programming language independent environment allowing .NET programs to be written in various programming languages and run on various platforms. C# is the primary programing language for .NET. Commonly used on MS Windows platforms, and installed by default on recent desktop MS Windows versions, the .NET environment is also available for Linux and UNIX-like platforms through the work of the Mono Project.
Necessary Resources
Hardware
A MS Windows, Apple OS X, Linux or UNIX computer
Software
A .NET runtime environment. This can be the Microsoft .NET environment included with recent versions of MS Windows (i.e. Vista, 7, 8 or 8.1), a Microsoft .NET version obtained via Windows Update or as an Internet download (see http://www.microsoft.com/net), or on Apple OS X, Linux or UNIX systems the alternative .NET implementation from the Mono Project (http://www.mono-project.com/).
A web browser, for example Google Chrome, Mozilla Firefox, MS Internet Explorer, Opera or Safari
-
Open a web browser and go to the EMBL-EBI Web Services pages at http://www.ebi.ac.uk/Tools/webservices/.
This page lists the available Web Service from EMBL-EBI. For each service a brief description and links to the service documentation are provided. For example in the “Sequence Similarity Search (SSS)” section the Web Services providing sequence search functionality are listed (Figure 22).
-
Clicking through to the service documentation pages, e.g. for NCBI BLAST (SOAP) (http://www.ebi.ac.uk/Tools/webservices/services/sss/ncbi_blast_soap), displays information about the service., including links to sample Web Service clients.
Clients are provided in a number of programming languages and using a variety of Web Services tool-kits. For NCBI BLAST (SOAP) this includes C#, Java, Perl, PHP, Python, Ruby, VB.NET clients (Figure 23). Dependencies and requirements for running each client are detailed on the right-hand side of the table.
Download the C# .NET executable (e.g. NcbiBlastCliClient.exe) by clicking on the link.
-
Test and run the client:
-
On MS Windows open a Command Prompt. The procedure to do this varies according to different versions of Windows.
Within the Command Prompt, change to the directory which contains the client program downloaded earlier.
To test the program (e.g. NcbiBlastCliClient.exe), enter:
NcbiBlastCliClient.exe
Help information will be displayed with further instructions on usage of the client.
-
On OS X, Linux or UNIX, open a terminal.
Within the terminal, change to the directory which contains the client program downloaded earlier.
To test the program (e.g. NcbiBlastCliClient.exe), enter:
mono NcbiBlastCliClient.exe
Help information will be displayed with further instructions on usage of the client.
-
Figure 22.

Sequence Similarity Search section of Web Services page
Figure 23.

Available NCBI BLAST SOAP clients
BASIC PROTOCOL 7: PUTTING SERVICES TOGETHER IN A WORKFLOW
One of the advantages of using Web Services is the ease with which workflows can be constructed, enabling multiple operations and analyses to be carried out automatically.
The simple workflow below demonstrates a brief sequence investigation: Beginning with an unknown sequence file, a search is carried out against the UniProtKB/Swiss-Prot database to find whether the sequence is already in the database and whether there are any homologous sequences. The top hits are then retrieved and a multiple sequence alignment is carried out to identify conserved regions. This alignment is retrieved together with a rough phylogenetic tree.
Necessary Resources
Hardware
A UNIX, Linux, Mac, or Windows workstations with Internet access
Software
-
Download the example clients from:
http://www.ebi.ac.uk/Tools/webservices/download_clients/perl/soaplite/fasta_soaplite.pl
http://www.ebi.ac.uk/Tools/webservices/download_clients/perl/soaplite/wsdbfetch_soaplite.pl
http://www.ebi.ac.uk/Tools/webservices/download_clients/perl/soaplite/clustalo_soaplite.pl
See Support Protocol 1 for downloading and installing the Web Services Perl clients.
Input
A sequence file in any format supported by FASTA. In this example a file called input.fasta containing a sequence in FASTA format shown in Figure 24.
Figure 24.

The file input.fasta, used as an example query.
Steps to run workflow
Open a shell and navigate to the local directory on your computer containing the client and input files.
-
Construct a suitable command line for running a search against UniProtKB/Swiss-Prot for our input sequence.
SSEARCH is an accurate similarity search tool, and is part of the fasta package defined by the --program option. A suitable command would be:
fasta_soaplite.pl --email email@example.org --program ssearch --database uniprotkb_swissprot --stype protein --scores 10 --sequence input.fasta --outformat ids --quiet --outfile ids.txt
-
Construct a command line for retrieving sequences from a list of identifiers in the DB:Identifier format supplied via a file.
We can use wsdbfetch for this, and it’s handy to specify retrieving the sequences in FASTA format. A suitable command would be:
wsdbfetch_soaplite.pl fetchData @ids.txt fasta > seqs.fasta
-
Construct a command line to align these sequences.
Clustal Omega can be used for this:
clustalo_soaplite.pl --email email@example.org seqs.fasta
-
Chain these commands together.
Chaining these commands can be achieved in a number of ways via a batch or shell script. One way is to pipe them together, for example:
fasta_soaplite.pl --email email@example.org --program ssearch --database uniprotkb_swissprot --stype protein --scores 10 --sequence input.fasta --outformat ids --quiet --outfile ids.txt | wsdbfetch_soaplite.pl fetchData@ids.txt fasta > seqs.fasta | clustalo_soaplite.pl --email email@example.org seqs.fasta
When piping there is no need to save the intermediary files (though they might be useful anyway) so you can use the ‘-‘ character to direct the output to STDOUT and use STDIN as input, i.e.:
fasta_soaplite.pl --email email@example.org --program ssearch --database uniprotkb_swissprot --stype protein --scores 10 --sequence input.fasta --outformat ids --quiet --outfile - | wsdbfetch_soaplite.pl fetchData @- fasta | clustalo_soaplite.pl --email email@example.org -
-
Examine the output.
The result of this workflow is a list of files from the final alignment in Clustal Omega. The names of the files starts with the jobId and the different results are contained in files with different suffixes. The alignment is in the .aln-clustal.clustal file, the phylogenetic tree data is in the .phylotree.ph file.
GUIDELINES FOR UNDERSTANDING RESULTS
The interpretation of the scientific results from the wide variety of tools that are available through the EMBL-EBI web interface and Web Services is beyond the scope of this unit, however in this section we present some of the common outcomes from successful or unsuccessful uses of the service.
When a job is submitted through the web interface (Basic Protocol 2), a quick check on the input is carried out and only once the data passes this validation check is it submitted to the compute clusters where the actual request/analysis is executed. This check allows us to reduce the number of invalid submissions to the clusters and allows the user to quickly correct a simple error. If the input check is not passed an error box appears on the webpage with some detail about the error and what action the user can take to correct it (Figure 25). If the check is passed, a temporary running page will be displayed with the job identifier until the results are ready to be viewed (Figure 26). The unique job identifier currently consists of the name of the tool; the method of submission (I, E, R or S - representing: Interactive, Email, REST or SOAP); date and time of submission; and finally an identifier which is helpful to us internally relating to the running of jobs on our compute clusters.
Figure 25.
Clustal Omega input page showing error message from failed input validation.
Figure 26.
Clustal Omega successful submission/job running page.
Causes of failing the validation check are usually simple user mistakes, such as failing to select a database to search against in the case of FASTA, or accidently hitting the Submit button before a set of sequences has been uploaded or entered into the input box for Clustal Omega. Errors are also returned when the data input is too large. On popular tools there are FAQs in the Help & Documentation pages that address common causes of validation check failure.
Unfortunately passing the quick input validation check does not guarantee that the job completes successfully as there can be situations in which the underlying tool produces an error once it is run. An example is where a user has accidentally truncated the input for a multiple sequence alignment such that sequence file header text now appears in the middle of the sequence data for a different entry (Figure 27). In this case the validation check is too simple to catch the input error, but a tool such as MUSCLE may give an error when it encounters this non-sequence data. When we detect that a tool has failed to provide the expected results (or has produced an error) we highlight this to the user in place of the normal results pages, and present links to the user that contains as much information possible to help them determine the cause of the error (Figure 28). This information includes the data that was submitted to the tool, and any errors that the tool outputs. When encountering this page users should read any error messages (Figure 29) from the tool and check their input carefully for errors. If they still need help then the job identifier should be sent to our helpdesk using the Feedback link at the top of the page or via http://www.ebi.ac.uk/support/.
Figure 27.

Example input mistake for a multiple sequence alignment. Note how the first two sequences have been merged so that the header information for the second sequence (UniRef50_A4IL81) appears as part of the sequence data for the first sequence.
Figure 28.

MUSCLE results page following input mistake.
Figure 29.
Tool Error Details page.
Attempting to view the results of a job a long time after it was submitted may not succeed as results are not kept indefinitely - currently they are deleted after seven days. Doing so generates a job not available page as seen in Figure 30. To generate the results again the user will need to carry out a new job submission.
Figure 30.
Job not available page for an expired job.
The situation when using Web Services is similar. Incorrect usage of a command-line client, for example supplying an incorrect parameter, returns an error such as ‘Unknown option:’. The user should run the client without any parameters to display correct usage and available parameters. Omission of data required for a job (for example, failing to select a database or supplying an input file for multiple sequence alignment that only contains one sequence) results an error being passed to the user in exactly the same terms as when the validation check fails on the website - behind the scenes it is in fact the same check as for the web interface.
Successful Web Service requests result in a job status of ‘FINISHED’ - this is analogous to the results page being displayed for web interface submissions. Problems with the running of the job (for example due to server failure) result in a status of ‘ERROR’ or ‘FAILURE’. Requests for an invalid Job ID, either because the ID is incorrect, or because the result has expired, returns a status of ‘NOT FOUND’ (Figure 31).
Figure 31.

Error message returned when attempting to retrieve an invalid job ID via Web Services.
If there is a problem and the tool generates an error then error files are produced, together with your input and any standard output from the tool (Figure 32). Error files can be identified by their suffix of “.error” and contain information about the error. These error files are of particular value when requesting assistance from our helpdesk. Common causes of errors include: incorrect or missing parameters; using input that is incorrectly formatted or unsuitable for the tool; and attempted retrieval of results beyond the period which they are available.
Figure 32.

Files created from MUSCLE Web Services job using the bad input file from figure 27. The .error.txt file contains the tool error details. The .out.txt file contains the standard output from the tool. The .sequence.txt file contains the input that was submitted for the job.
Note that there are situations when an incorrect analysis has been requested yet the tool appears to run fine - for example when a search is carried out against a protein database using DNA input. Correct usage would be to use a tool such as FASTX to translate the DNA input, however if the user incorrectly uses FASTA the tool will still run and produce a result of sorts. This is because there are amino acids corresponding to the same single letter characters used for DNA bases, so the program does not prevent the search. Another example might be the use of a multiple sequence alignment tool, such as Clustal Omega, for situations which it is not designed for, for example for pairwise alignment or to align short primers to a longer sequence. In general, if the standalone tool allows an analysis to be carried out then we attempt to allow it at EMBL-EBI as well - it is up to the user to decide what uses they put the tools to and they should examine the results for the unexpected. We do offer documentation and training courses (http://www.ebi.ac.uk/training/) to educate users on correct usage of the tools and our helpdesk is available for further assistance at http://www.ebi.ac.uk/support/.
Acknowledgments
Continued development and support for the EMBL-EBI services mentioned in this protocol is provided centrally by the EMBL.
LITERATURE CITED
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2008;36:D25–D30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane G, Akhtar R, Aldebert P, Althorpe N, Baldwin A, Bates K, Bhattacharyya S, Bonfield J, Bower L. Priorities for nucleotide trace, sequence and annotation data capture at the Ensembl Trace Archive and the EMBL Nucleotide Sequence Database. Nucleic Acids Res. 2007;36:D5–D12. doi: 10.1093/nar/gkm1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby MA, Goodman JL, Strelets VB, Zhang P, Gelbart WM FlyBase Consortium. FlyBase: genomes by the dozen. Nucl Acids Res. 2007;35:D486–D491. doi: 10.1093/nar/gkl827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edman P, Högfeldt E, Sillén LG, Kinell P. Method for determination of the amino acid sequence in peptides. Acta Chem Scand. 1950;4:283–293. [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, Gordon L, Hendrix M, Hourlier T, Johnson N, Kähäri A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Kulesha E, Larsson P, Longden I, McLaren W, Overduin B, Pritchard B, Riat HS, Rios D, Ritchie GR, Ruffier M, Schuster M, Sobral D, Spudich G, Tang YA, Trevanion S, Vandrovcova J, Vilella AJ, White S, Wilder SP, Zadissa A, Zamora J, Aken BL, Birney E, Cunningham F, Dunham I, Durbin R, Fernández-Suarez XM, Herrero J, Hubbard TJ, Parker A, Proctor G, Vogel J, Searle SM. Ensembl 2011. Nucleic Acids Res. 2011;39:D800–D806. doi: 10.1093/nar/gkq1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RE. Structure of Tobacco Mosaic Virus: Location of the Ribonucleic Acid in the Tobacco Mosaic Virus Particle. Nature. 1956;177:928–30. [Google Scholar]
- Gonzalez MW, Pearson WR. Homologous over-extension: a challenge for iterative similarity searches. Nucleic Acids Res. 2010;38:2177–2189. doi: 10.1093/nar/gkp1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Antoshechkin I, Bieri T, Blasiar D, Chan J, Chen WJ, De La Cruz N, Davis P, Duesbury M, Fang R, Fernandes J, Han M, Kishore R, Lee R, Müller HM, Nakamura C, Ozersky P, Petcherski A, Rangarajan A, Rogers A, Schindelman G, Schwarz EM, Tuli MA, Van Auken K, Wang D, Wang X, Williams G, Yook K, Durbin R, Stein LD, Spieth J, Sternberg PW. WormBase: a comprehensive resource for nematode research. Nucleic Acids Res. 2009;38:D463–7. doi: 10.1093/nar/gkp952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez P, Müller M, Appel RD. Automated protein identification by tandem mass spectrometry: Issues and strategies. Mass Spectrometry Reviews. 2006;25:235–254. doi: 10.1002/mas.20068. [DOI] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu031. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey PJ, Staines DM, Lawson D, Kulesha E, Derwent P, Humphrey JC, Hughes DST, Keenan S, Kerhornou A, Koscielny G, Langridge N, McDowall MD, Megy K, Maheswari U, Nuhn M, Paulini M, Pedro H, Toneva I, Wilson D, Yates A, Birney E. Ensembl Genomes: An integrative resource for genome-scale data from non-vertebrate species. Nucleic Acids Res. 2011;40:D91–D97. doi: 10.1093/nar/gkr895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, McWilliam H, Goujon M, Cowley A, Lopez R, Pearson WR. PSI-Search: iterative HOE-reduced profile SSEARCH searching. Bioinformatics. 2012;28:1650–1651. doi: 10.1093/bioinformatics/bts240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R, Duggan K, Harte N, Kibria A. Public services from the European Bioinformatics Institute. Brief Bioinform. 2003;4:332–340. doi: 10.1093/bib/4.4.332. [DOI] [PubMed] [Google Scholar]
- Marsden RL, Lewis TA, Orengo CA. Towards a comprehensive structural coverage of completed genomes: a structural genomics viewpoint. BMC Bioinformatics. 2007;8:86. doi: 10.1186/1471-2105-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam H, Li W, Uludagi M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam H, Valentin F, Goujon M, Li W, Narayanasamy M, Martin J, Miyar T, Lopez R. Web services at the European Bioinformatics Institute-2009. Nucleic Acids Res. 2009;37:W6–W10. doi: 10.1093/nar/gkp302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR. Searching protein sequence libraries: Comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics. 1991;11:635–650. doi: 10.1016/0888-7543(91)90071-l. [DOI] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A. 1988;85:2444–8. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson E, Lundeberg J, Ahmadian A. Generations of sequencing technologies. Genomics. 2009;93:105–11. doi: 10.1016/j.ygeno.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Roberts RJ. Restriction endonucleases. CRC Crit Rev Biochem. 1976;4:123–64. doi: 10.3109/10409237609105456. [DOI] [PubMed] [Google Scholar]
- Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94:441–8. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno Y, Imanishi T, Miyazaki S, Fukami-Kobayashi K, Saitou N, Sugawara H, Gojobori T. DNA Data Bank of Japan (DDBJ) for genome scale research in life science. Nucleic Acids Res. 2002;30:27–30. doi: 10.1093/nar/30.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2010;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin F, Squizzato S, Goujon M, McWilliam H, Paern J, Lopez R. Fast and efficient searching of biological data resources--using EB-eye. Brief Bioinform. 2010;11:375–84. doi: 10.1093/bib/bbp065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2 - a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Nebert DW. Update on genome completion and annotations: Protein Information Resource. Human genomics. 2004;1:229–233. doi: 10.1186/1479-7364-1-3-229. [DOI] [PMC free article] [PubMed] [Google Scholar]