

Abstract
We examined regional changes in brain volume in healthy adults (N = 167, age 19-79 years at baseline; N = 90 at follow-up) over approximately two years. With latent change score models, we evaluated mean change and individual differences in rates of change in 10 anatomically-defined and manually-traced regions of interest (ROIs): lateral prefrontal cortex (LPFC), orbital frontal cortex (OF), prefrontal white matter (PFw), hippocampus (HC), parahippocampal gyrus (PhG), caudate nucleus (Cd), putamen (Pt), insula (In), cerebellar hemispheres (CbH), and primary visual cortex (VC). Significant mean shrinkage was observed in the HC, CbH, In, OF, and the PhG, and individual differences in change were noted in all regions, except the OF. Pro-inflammatory genetic variants mediated shrinkage in PhG and CbH. Carriers of two T alleles of interleukin-1β (IL-1βC-511T, rs16944) and a T allele of methylenetetrahydrofolate reductase (MTHFRC677T, rs1801133) polymorphisms showed increased PhG shrinkage. No effects of a pro-inflammatory polymorphism for C-reactive protein (CRP-286C>A>T, rs3091244) or apolipoprotein (APOE) ε4 allele were noted. These results replicate the pattern of brain shrinkage observed in previous studies, with a notable exception of the LPFC thus casting doubt on the unique importance of prefrontal cortex in aging. Larger baseline volumes of CbH and In were associated with increased shrinkage, in conflict with the brain reserve hypothesis. Contrary to previous reports, we observed no significant linear effects of age and hypertension on regional brain shrinkage. Our findings warrant further investigation of the effects of neuroinflammation on structural brain change throughout the lifespan.
Keywords: aging, MRI, inflammation, longitudinal, parahippocampal gyrus, cerebellum
1. Introduction
Brain volume declines with age and significant shrinkage occurs in multiple gray and white matter regions within a relatively short time (Driscoll et al., 2009; Fjell et al., 2009, 2013; Pfefferbaum et al., 1998; Raz et al., 2005, 2010, 2013; Resnick et al., 2003; Scahill et al., 2003). Notably, the magnitude and rate of volume reduction vary across brain regions. Shrinkage is especially pronounced in the cerebellum, tertiary association cortices, medial temporal lobe and neostriatum (Fjell et. al., 2009; Raz et al., 2005; Tamnes et al., 2013), whereas primary visual cortex shows minimal age-related decline (Fjell et al., 2009; Raz et al., 2005, 2010). The rates of change vary, not only across brain regions, but also across individuals. Understanding individual differences in brain aging and elucidating their mechanisms has been hampered by the daunting number of proposed antecedents, and the cumulative record of the relevant studies remains sparse. Among multiple potential modifiers of the rate and extent of brain aging, vascular risk factors (Jagust, 2013; Raz & Rodrigue, 2006) and neuroinflammation (Finch, 2010; Godbout & Johnson, 2006) have attracted significant attention.
To date, several studies have attempted to gauge the influence of vascular risk (VR) and inflammation on regional brain volumes. Hypertension, a major and probably the most common VR, has been associated with shrinkage and smaller volume of age-sensitive brain regions, such as the hippocampus (Burns et al., 2012; Korf et al., 2004; Raz et al., 2005, 2007a, 2007b), and tertiary association cortices (Raz et al., 2003a, 2005, 2007a, 2007b).
Several biomarkers of inflammation have also been linked to individual differences in brain structure. Elevated circulating levels of pro-inflammatory cytokines (e.g., interleukin (IL)-6, and tumor necrosis factor α), C-reactive protein (CRP), and homocysteine (Hcy) correlate with reduced volumes of the hippocampus, cerebral cortex, and cerebral white matter, as well as increase in the volume of white matter hyperintensities (Bettcher et al., 2012; Choe et al., 2014; den Heijer et al., 2003; Jefferson et al., 2007; Marsland et al., 2008; Raz et al., 2012; Satizabal et al., 2012; Seshadri et al., 2008; Shimomura et al., 2011; Taki et al., 2013; van Dijk et al., 2005; Williams et al., 2002, but see Feng et al., 2013; Morra et al., 2009; Scott et al., 2004).
In exploring the effects of pro-inflammatory biomarkers on the brain in humans, one must contend with two problems: the inability to manipulate the levels of biomarkers directly through induced inflammation, and the uncertain relationship between blood levels and brain content (Banks et al., 1995). For ethical reasons, blood levels of pro-inflammatory biomarkers cannot be safely manipulated in healthy humans, and there are doubts as to whether human IL-1β can cross the blood-brain barrier (Banks, 2005; Banks et al., 2001), although in rodents, chronic inflammation increases IL-1β gene expression in the hippocampus (Bardou et al., 2013). These challenges can be overcome, at least to some extent, by Mendelian randomization (Katan, 1986), which takes advantage of known genetic variants (single nucleotide polymorphisms; SNPs) that predispose individuals to various levels of the pro-inflammatory biomarkers in the brain and peripheral circulation. Thus, the study of persons with genetic polymorphisms that are associated with variable levels of a given risk factor may shed light on neuroinflammation in correlational studies. To date, several genetic variants associated with vascular health and inflammation have been linked to age-related and individual differences in brain volume and white matter integrity.
Several SNPs associated with various levels of proinflammatory response have been identified. Methylenetetrahydrofolate reductase (MTHFR C677T), a polymorphism of a gene that controls production of the enzyme necessary for metabolizing Hcy, modulates plasma concentration of Hcy (Bathum et al., 2007; de Lau et al., 2010). Variants in the IL-1β gene (e.g., IL-1β C-511T) are associated with release of the eponymous cytokine in response to infection (Yarlagadda et al., 2009). Several SNPs that promote inflammation have been associated with structural brain differences. For instance, MTHFR C677T has been linked to smaller white matter volumes and accelerated shrinkage in periventricular fronto-parietal and parieto-occipital regions (Rajagopalan et al., 2011, 2012). G homozygotes of the polymorphism of the IL-6 gene (IL-6A-174G, rs1800795) have greater hippocampal grey matter volumes than heterozygotes and A homozygotes (Baune et al., 2012). In healthy adult carriers (Raz et al., 2012) but not in cognitively impaired individuals (Rajagopalan et al., 2012) the variant T allele of IL-1β C-511T and CRP-286 C>A>T polymorphisms are associated with increased burden of white matter hyperintensities (WMH). Notably, none of the extant studies of pro-inflammatory SNPs investigated their effect on rate of change.
Several other polymorphisms have been linked to individual differences in brain volumes and in the rate of age-related declines. The most prominent among these is a well-established genetic risk factor for late-onset Alzheimer’s disease (AD; Roses, 1996), and the only consistently reported single-gene marker for longevity (Brooks-Wilson, 2013): the ε4 allele of the APOE gene. The APOE ε4 allele controls availability of the apolipoprotein E (APOE), a major factor in lipid transport. APOE ε4 has also been linked to increased risk of cardiovascular disease (Mahley & Rall, 2000) and hyperlipidemia (Davignon et al., 1988). However, the extant literature concerning the effect of APOE ε4 on brain aging and cognition is contradictory (Reinvang et al., 2013) and some suggest that positive findings may reflect inclusion of participants with incipient pathology (Cherbuin et al., 2007).
Reliance on cross-sectional comparison studies significantly impedes understanding of age-related change (see Raz & Kennedy, 2009 for a review). Although it has been made abundantly clear that cross-sectional studies cannot capture true dynamics of age-related change and the role of various mediators in shaping the age trajectories (e.g., Hofer & Sliwinski, 2001; Lindenberger & Pötter, 1998; Lindenberger et al., 2011; Maxwell & Cole, 2007), longitudinal studies remain relatively scarce. Moreover, a significant share of the extant literature on age-related brain shrinkage is focused solely on the mean change across individuals, without considering individual differences in change. This focus on a measure of central tendency, commonly expressed in a simple autoregressive or a residual regression model, precludes the understanding of differential aging. Whereas development of latent longitudinal methods, such as the latent change score model (LCSM; McArdle, 2008, 2009; McArdle & Nesselroade, 1994), enables quantification of individual differences in age-related change, application of these methods is still relatively rare. With a few exceptions (McArdle et al., 2004; Raz et al., 2005, 2010, 2013), most of the extant longitudinal studies (e.g., Fjell et al., 2009; Pfefferbaum et al, 1998; Resnick et al., 2003) are limited to evaluation of mean change.
In this study, our aim was to address the limitations outlined above. First, we aimed to document regional brain changes occurring over two years in healthy adults and replicate previously reported findings. We expected, based on the extant literature, to observe mean shrinkage of the cerebellum, hippocampus, striatum, and prefrontal cortices, but no significant mean change in the volume of the primary visual cortex. Second, we examined individual variation in age-related change in regional brain volumes. Based on previous studies, we hypothesized to find significant variance (i.e., individual differences) in change across all regions of interest (ROIs). Third, we gauged the influence of putative modulators of brain aging trajectories on the rate of change in regional brain volumes. Specifically, we tested the effects of VR factors and biomarkers, such as arterial hypertension, and genetic variants associated with increased pro-inflammatory response, IL-1β C-511T, CRP -286 C>A>T, and MTHFR C677T. In addition, we evaluated the effect of a common genetic risk factor for AD (APOE ε4) on rates of change in regional brain volumes.
2. Methods
2.1. Participants
The data for this study were collected in a major metropolitan area in the USA. The participants were volunteers recruited through media advertisement and flyers. Persons who reported a history of cardiovascular, neurological or psychiatric disease, head trauma with loss of consciousness in excess of five minutes, thyroid dysfunction, diabetes mellitus, or history of treatment for drug and alcohol abuse or a habit of consuming three or more alcoholic drinks per day were excluded from the study. Participants who were taking anti-seizure medication, anxiolytics or antidepressants were additionally excluded, and persons suffering from claustrophobia were advised not to participate in the study. Persons with a reported diagnosis of hypertension were included. These participants were taking anti-hypertensive medications, such as beta-blockers, calcium channel blockers, angiotensin converting enzyme inhibitors or potassium-sparing diuretics.
All participants were screened for dementia and depression using Mini-Mental State Examination (MMSE; Folstein et al., 1975), with a cut-off of 26 (87%) correct responses, and Center for Epidemiologic Studies Depression Scale (CES-D; Radloff, 1977) with a cut-off of 15. All participants were right-handed, as indicated by a score above 75% on the Edinburgh Handedness Questionnaire (Oldfield, 1971). An on-call neuroradiologist examined the magnetic resonance (MR) scans for suspected space-occupying lesions and signs of significant pathology.
Of 1055 persons who replied to advertisements, 360 were eligible after the telephone screening for right-handedness and history of cardiovascular, psychiatric or neurological disease. Of the eligible participants, 43 were excluded due to MRI contraindications (e.g., metal in body, claustrophobia, or pregnancy at baseline), 99 discontinued after starting the study because of various logistic and motivational problems as well as exclusionary findings (e.g., psychiatric illness, excessive alcohol consumption, head trauma) that were not reported at the initial screening. Five participants had incidental MRI findings (e.g., aneurysm, multiple sclerosis plaques, cerebellar infarct). Sixteen scans were acquired with sequences that differed significantly from the coronal sequence described below and were not included in the analyses. Of the remaining 197 participants who were eligible for the longitudinal follow-up, 167 were scanned at baseline on a 4 Tesla (4T) scanner and one on a 3 Tesla (3T) magnet that replaced the 4T scanner during the study at the same site; an additional 29 participants were scanned on a 3T magnet at follow-up. The data acquired on the 3T scanner were not included in the analyses presented here. Thus, the final baseline sample consisted of 167 participants (85% of the eligible cohort). Women composed 65% of the sample. Participants with controlled hypertension were included in the study (N = 33, 20% of the sample). The process of recruitment, selection and attrition is depicted in a flow chart in Figure 1.
Figure 1.

A flow chart illustrating the process of recruitment, selection and attrition of participants. A. Recruitment – the number of persons, who answered the ads, were contacted by telephone, interviewed and selected to receive health questionnaires. B. Selection – number of participants, who passed the health screening, subsequent screening for MRI eligibility, were scanned on the 4T scanner with the same coronal MPRAGE sequence, did not have incidental findings, and did not acquire MRI contraindications during the follow-up period. C. Attrition – Participants who were unable to start the baseline measurements in spite of full eligibility and those who had baseline measurements but failed to appear at follow-up. The full details of exclusionary criteria and description of the screening instruments can be found in the Method section.
Of the 167 participants who completed baseline scanning on the 4T magnet, 90 (54%) had the follow-up on the same scanner. The mean ± SD follow-up interval was 24.99 ± 2.19 months, with a range of 20-33 months. Attrition was unrelated to sex (b =.24, p = .40), level of education (b = −.02, p = .83), race (b = 0.30, p = .55), CES-D score (b =.31, p =.24), or diagnosis of hypertension (b =.99, p = .08). Participants with advanced age (b = −.04, p = .002) and higher MMSE scores (b = −11.66, p = .02) were less likely to drop out of the study. The demographic descriptors of the sample are presented in Table 1.
Table 1.
Descriptive Sample Statistics at Baseline.
| Variables | N | Minimum | Maximum | Mean | SD |
|---|---|---|---|---|---|
| Age | 167 | 19 | 79 | 52.80 | 15.72 |
| Education | 167 | 12 | 20 | 15.41 | 2.30 |
| SBP mmHg | 166 | 75.47 | 156.67 | 122.47 | 12.91 |
| DBP mmHg | 166 | 60.00 | 106.67 | 75.47 | 7.42 |
Note. N = sample size, SD = standard deviation, SBP = Systolic blood pressure, DBP = Diastolic blood pressure, mmHg = millimeter of mercury. Education measured as years of formal schooling.
2.2. Covariates
2.2.1. Blood pressure measures
Blood pressure was measured on three separate days with a mercury sphygmomanometer (BMS 12-S25) using a standard blood pressure cuff (Omron Professional) on the left arm while participants were seated and their forearm was positioned on the table. Measurements were conducted by trained laboratory technicians. The systolic and diastolic measures were averaged across measurement occasions and for each individual, the mean values of systolic and diastolic blood pressure were used.
2.2.2. Genotyping
We used methods and assays described in previous publications (e.g., Raz et al. 2012). DNA was isolated from buccal samples obtained in mouthwash in 145 participants (N = 88 at follow-up) who signed a separate informed consent for genotyping. Isolation was performed using a Gentra Autopure LS with the standard buccal cell protocol. For genotyping quality control, 37% direct repeats and DNA sequencing for verification were performed. Both control DNA and no-template controls were used. First run success rate of genotyping was 96.25%. However, after direct repeat quality control runs with at least four successful identifications required for a call, only one sample showed allele dropout (99.38%). After direct repeat analyses were performed seven times, the sample was genotyped with an estimated error of .33%. Thus, the final genotyping success rate for this study was 100%.
2.2.2.1. IL-1β C-511T (rs16944)
The IL-1β C-511T polymorphism was interrogated with the 5′-nuclease assay using a Taqman SNP Genotyping assay (Applied Biosystems, Foster City, CA, USA). The allelic distribution of the IL-1β-C-511 polymorphism met the Hardy–Weinberg equilibrium (HWE; χ2 = .08, p = .78), with 34% of participants being C homozygotes (N = 49), 50% C/T heterozygotes (N = 72), whereas 24 participants had the homozygous TT genotype (16%). Variant allele frequency was 0.59. Two groups, TT versus C/T and CC, were used in the analyses: IL-1β-511 T homozygotes (N = 24) vs. C allele carriers (N = 121). The two groups did not differ in age (t = −.47, p = .64), education level (t = 1.77, p = .08), or MMSE scores (Mann-Whitney U = 1274.50, Z = −.99, p = .32).
2.2.2.2. MTHFR C677T (rs1801133)
The MTHFR C677T polymorphism was interrogated with the 5′-nuclease assay using a TaqMan SNP Genotyping Assay (Applied Biosystems, Foster City, CA, USA). The assays were run using an Applied Biosystems 7900. Genotyping revealed that 57 (39%) participants were heterozygous (CT), 14 (10%) were homozygous for the T allele (TT) and 74 (51%) were homozygous for the C allele (CC). The distribution of the alleles was consistent with the HWE: χ2 = .38, p = .54. Because of few TT homozygotes in the sample, we conducted analyses comparing carriers of the T allele (N = 71) and CC homozygotes (N = 74). The carriers of the T allele did not differ from MTHFR C677T homozygotes in age (t = −1.35, p = .18.), education (t = −.08, p = .93), or MMSE (Mann-Whitney U = 2402.00, Z = −.93, p = .35).
2.2.2.3. CRP-286C>A>T (rs3091244)
The CRP polymorphism was PCR amplified using forward 5′ AGGTGTCAGAGCTTCGGGAAGAG-3′ and reverse primers 5′-GCTCTGGGAGGAGCATGTTTGTTT-3′ in a 25 μl reaction containing 2.5 mmol/l MgCl2, 0.5 μmol/l of the primers, 1.25 U AmpliTaq Gold polymerase, and 200 μmol/l dNTPs. The mixture was denatured at 95 °C for 5 min and amplification achieved by 15 cycles of 94 °C for 30 s, 66 °C for 30 s, and 72 °C for 1 min, followed by a final extension at 72 °C for 10 min. The amplicons were purified using Sephadex and the purified products were analyzed on an ABI PRISM 3700 DNA Analyzer using a 50 cm capillary array.
One sample was mishandled, and 144 samples were available for genotyping. The sample comprised of 10 homozygote A carriers (7%), 51 A/C carriers (35%), 13 A/T carriers (9%), 54 homozygote C carriers (38%), 11 C/T carriers (8%), and 5 homozygote carriers of the T allele (3%). The distribution of C>A, met the HWE requirements (χ2 = 0.18, p = .68), but not carriers of the C>T alleles (χ2 = 10.31, p = .001). There were no age differences between the allelic variants (t = .65, p = .52). Neither did they differ in years of education (t = −.42, p =.68) or MMSE (Mann-Whitney U = 1449.50, Z = −1.14, p = .26). As there were only 5 TT homozygotes, in subsequent analyses, we compared T allele carriers (N = 29) to non-carriers of that allele, i.e. AA, A/C and CC genotypes (N = 115).
2.2.2.4. APOE variants (rs429358 and rs7412)
APOE polymorphisms (rs429358 and rs7412) were pre-amplified with forward 5′-CAATGCTACCGAGTTTTCTTCC-3′ and reverse primers 5′-TTCAGATTCTTCACAGATGCGTA-3′ in a 25 μl reaction containing 2.5 mmol/l MgCl2, 0.5 μmol/l of the primers, 1.25 U AmpliTaq Gold polymerase, and 200 μmol/l dTTPs. The mixture was denatured at 95°C for 10 minutes and amplification achieved by 15 cycles of 94°C for 30 seconds, 58°C for 30 seconds, and 72°C for 1 minute, followed by a final extension at 72°C for 10 minutes. One μl of this reaction was subsequently used for rs429358 and rs7412 5′-nuclease assays under standard conditions. The primers and probes for the rs429358 assay were 5′-GCGGGCACGGCTGT-3′, 5′-GCTTGCGCAGGTGGGA-3′, VICCATGGAGGACGTGTGC-NFQ and FAM-ATGGAGGACGTGCGC-NFQ. The primers and probes for the rs7412 assay were 5′-TCCGCGATGCCGATGAC-3′, 5′-CCCCGGCCTGGTACAC-3′, VIC-CAGGCGCTTCTGC-NFQ and FAM-CAGGCACTTCGCNFQ. APOE genotyping yielded 18 APOE ε2/3, 5 ε2/4, 81 ε3/ε3, 38 ε3/ε4, 3 ε4/ε4, and no ε2/ε2 genotypes. Because of the small number of APOE ε4 homozygotes (N = 3), carriers of one or two copies of the APOE ε4 (N = 46) were compared with 99 persons who had no ε4 allele. Allelic frequency met the assumption of HWE (χ2 = .02, p =.90). Frequency of APOE ε4 did not differ between Caucasian and African-American participants: χ2 = 3.75, p =.15. There were no age differences between the allelic variants (t = −.86, p = .39). Neither did they differ in years of education (t = −.08, p =.93), or MMSE (Mann-Whitney U = 2402.00, Z = −.93, p = .35).
2.3. MRI Protocol
Images were acquired on a 4T MRI system (Bruker Biospin, Ettlingen, Germany) with an 8-channel radio frequency coil. For volume measurements, we acquired magnetization-prepared rapid gradient echo (MPRAGE) T1-weighted images in the coronal plane. Acquisition parameters were as follows: echo time (TE) = 4.38 ms, repetition time (TR) =1600 ms, inversion time (TI) = 800 ms, field of view (FOV) = 256×256 mm2, resolution = .67×.67×1.34 mm3, matrix size = 384×384, and flip angle (FA) = 8°.
The MPRAGE was acquired as part of a longer protocol that included several other sequences, including a set of 50 contiguous axial slices of fluid-attenuated inversion recovery (FLAIR) images, which was acquired with the following parameters: TR = 8440 ms, TE = 112 ms, TI = 2200 ms, FA = 150°, FOV = 256×256 mm2, in plane resolution= 1×1 mm2, slice thickness = 2 mm, matrix size = 256×256. All MPRAGE and FLAIR images were inspected for potential pathology and possible incidental findings were reviewed by an experienced radiologist.
2.4. MRI processing
In post-processing we followed procedures described in our previous studies (e.g., Raz et al., 2004). All image manipulations and measurements were conducted with Analyze 10.0 software (Biomedical Imaging Resource, Mayo Clinic College of Medicine). To correct for asymmetrical positioning of the head in the scanner, images were rotated to adjust for variation in head pitch (forward - backward), tilt (left - right), and rotation (right - left). All images were processed by the same group of expert operators who attained reliability of at least .90 as measured by the intraclass correlation formula ICC (2) that assumes random raters (Shrout & Fleiss, 1979). The tracers were blind to the time of the image acquisition, and the demographic characteristics of the participants.
The volumes were computed from measured areas of the ROIs on consecutive slices multiplied by the slice thickness. To control for sex differences in body size and for possible changes in unknown variables that could have affected the volumes between baseline and follow-up measurements, all regional volumes were adjusted for the volume of the intracranial vault through analyses of covariance (ANCOVA), described in previous publications (Jack et al., 1989; Raz et al., 2004). For all ROIs, the assumptions of ANCOVA were tested, and if, as in the case of VC, the slopes of ROI regression on ICV varied between the sexes, separate adjustments were performed for men and women.
2.5. Manual Morphometry
2.5.1. Intracranial volume (ICV)
The ICV was estimated in the axial plane and covered all contents within the cranial vault, including the orbits and nasal cavity. Tracing began at the occipital pole (i.e., the last slice on which brain tissue was visible) and proceeded anterior, for a total of 10 slices traced at an increment of 15 (10 mm inter-slice distance). Previous work indicates that there is minimal reduction in accuracy and reliability at this sampling density compared to tracing every slice (Eritaia et al., 2000).
2.5.2. Lateral prefrontal cortex (LPFC)
The lateral prefrontal cortex was measured on every other slice for a total of 10–14 coronal slices located within the posterior 40% of the distance between the tip of the frontal pole to the genu of the corpus callosum The upper boundary was the most dorsomedial point of the white matter of the superior frontal gyrus, and included all grey matter ventrolaterally from that point to the lateral orbital sulcus. Unattached white matter or areas containing cerebrospinal fluid (CSF) were manually removed from the measurement.
2.5.3. Orbito-frontal cortex (OF)
Orbito-frontal cortex was measured on the same coronal slices as the lateral prefrontal cortex. Tracing proceeds from the lateral boundary of the LPFC (i.e., the lateral orbital sulcus) to the olfactory sulcus and includes all gray matter in between these sulci. As with the LPFC, unattached white matter or areas containing CSF were manually removed from the measurement.
2.5.4. Prefrontal white matter (PFw)
The range is identical to lateral prefrontal and orbital frontal cortices and includes subcortical white matter associated with both cortical regions, manually excluding the ventricles and other CSF spaces.
2.5.5. Hippocampus (Hc)
The hippocampal slices were aligned perpendicular to its long axis in the right hemisphere, and the region was traced on every other slice starting from the level of the mammillary bodies to the slice showing the columns of fornix rising from the fimbria, a total of 17 to 21 slices per brain. The hippocampus included sectors Cornu Ammonis 1–4, the dentate gyrus, and the subiculum as a single volume.
2.5.6. Parahippocampal gyrus (PhG)
Included in the measure of parahippocampal gyrus were the entorhinal cortex and the ambient gyrus, but not the subiculum. The anterior boundary was the most anterior slice on which the temporal lobe white matter (the temporal stem) became continuous with the rest of the cortex. The posterior boundary was the most posterior slice on which the pulvinar was still visible, yet connected to the cortical white matter (i.e., not separated by the lateral ventricle). The lateral border of PhG was a horizontal line drawn from the furthest medial point of the cortex of the PhG to the white matter (note: this is the line used as the inferior border of the hippocampal formation). The medial border of the PhG is the collateral sulcus. The entire cortex between these two landmarks was included in the measure. It was traced on an increment of 2 for a total of 18 to 22 slices.
2.5.7. Insula (In)
The right and left insula cortices were traced on the coronal plane at an increment of 2 for approximately 10 to 15 slices beginning at the optic chiasm (the first slice on which the optic tracts become continuous) until the appearance of the posterior commissure. The insula cortex lies inside the lateral fissure, and was demarcated dorsally and ventrally by the circular sulcus. The white matter served as the medial boundary, whereas the lateral boundary was the CSF space outside of the cortex.
2.5.8. Caudate nucleus (Cd)
The volumes of the head and body of the caudate were traced for a total of 15-21 coronal slices. The most rostral slice was the one on which the caudate was first visualized. The caudate was traced on every fourth slice with an inter-slice distance of 2.67 mm until the caudate body was no longer visualized. The protocol excluded the tail of the Cd. The medial boundary was the lateral ventricle and the subcortical white matter laterally and dorsally. On rostral sections, the stria terminalis defines the ventral boundary, and on caudal sections, the septal nucleus.
2.5.9. Putamen (Pt)
The volume of the putamen was measured from 13-16 coronal slices (every fourth slice, inter-slice distance of 2.67 mm). The most rostral slice was the one on which the putamen was first visualized and continued to the last caudal slice on which it was visible. The external capsule was the lateral boundary throughout the range and the dorsal boundary was the white matter. The internal capsule was the medial border until the anterior commissure, after which the globus pallidus became the medial border. The limen insula, optic radiations, amygdala, temporal horn, and anterior commissure served as the ventral boundary of the putamen on several slices.
2.5.10. Cerebellar hemispheres (CbH)
The cerebellar hemispheres were traced on every other coronal slice on which the structure was visualized. The volume included the hemispheric gray and white matter, corpus medullare, and cerebellar tonsils. The vermis, the superior medullary velum, and the fourth ventricle were excluded. The cerebellar peduncles were included in the cerebellar white matter on the slices that preceded the appearance of the anterior vermis. Caudal to that slice, the peduncles were easily distinguishable from the cerebellar gray matter and were reliably excluded. The CbH were traced on approximately 30 slices.
2.5.11. Primary visual cortex (VC)
The range of the VC covered the posterior 50% of the coronal slices between the mid-vermis slice and the occipital pole. The region was defined as the cortical ribbon lining the calcarine sulcus. The inferior and superior boundaries of this ROI were the point of sulcus opening. Only cortex within the calcarine sulcus was included, confining this ROI to a part of area 17 and excluding other areas. The VC was traced at an increment of 2 for 13-20 slices.
3. Statistical Analyses
Latent change score models were fitted to data from two measurement occasions separated by an average of 25 months. The LCSM simultaneously estimates cross-sectional individual differences, and individual differences in change (individual-level). On a conceptual level, latent change scores are analogous to traditional change scores estimated as the difference between follow-up and initial measurements. However, unlike raw change scores, latent change score estimates are free of measurement error. Consequently, unlike traditional change scores (e.g., Cronbach & Furby, 1970), latent change scores do not suffer from contamination by measurement error and unreliability (McArdle, 2001).
The LCSM combines the properties of autoregressive cross-lagged models and latent curve models (Grimm et al., 2012; see Figure 2 for an illustration of a latent change score model). Construction of LCSM starts with creating separate latent variables from manifest variables at each measurement occasion (measurement models). Hence, the volume of each ROI is represented by a latent variable identified by measurements of right and left hemisphere volumes (VLT1 and VRT1; VLT2 and VRT2), with equal factor loadings on their common latent variable. Subsequently, change was modeled as a second order latent variable (ΔV). The latent change score was by definition free of measurement error, because measurement error was estimated from the two manifest variables that together represent common ROI volume. The properties of the latent change score are implied by the structure imposed between the two latent variables at the two measurement occasions. The factor at follow-up (VT2) is defined as the sum of the factor at baseline (VT1) and the latent change score (ΔV). Thus, if VT2 = VT1 + ΔV, then ΔV = VT2 – VT1. Each LCSM estimated the following parameters: mean volume at baseline (β), variance in volumes at baseline (α), mean volume change (δ), variance in change (γ), and covariance of the initial brain volume with change in volume across the measurement occasions (ε). For all ROIs, except for CbH and VC, we specified equal residual variance across the two hemispheres and across time (b and c). For all ROIs, we further constrained equality of the factor loading of the right hemisphere (d), and of the auto-correlated residuals of both hemispheres across time (e).
Figure 2.

A latent change score model for the assessment of two-occasion changes in regional brain volume. Squares represent observed variables, circles are latent variables. The triangle indicates that the model contains means. Free parameters are marked by an asterisk. Parameters with equal sign and the same subscript are constrained to be equal to each other. T1 is baseline; T2 - follow-up; VL is a regional volume of the left hemisphere; VR - a regional volume of the right hemisphere; β - mean volume at baseline; α - variance in volumes at baseline; δ - mean volume change, γ - variance in change; ε –covariance between individual differences in regional brain volume at baseline and individual differences in regional brain volume changes between baseline and follow-up. The model contains four observed variables and 9 free parameters, with 5 degrees of freedom.
As specified in the participant section, the sample underwent attrition over time. To make full use of all available data, we assumed that the data were missing at random (MAR; Rubin, 1976; Schafer & Graham, 2002). The MAR assumption allows the probability of missingness to depend on observed data, but not on missing data. Parameter estimates were derived using full information maximum likelihood (FIML) estimation (e.g. Little, 1995; McArdle & Nesselroade, 1994). The likelihood function is calculated for each individual based on the available data. Hence, no data were deleted nor imputed. To improve the performance of FIML, by reducing its estimation bias, it is possible to include auxiliary variables related to the missingness mechanism (Collins et al, 2001; Schafer & Graham, 2002). To identify such auxiliary variables, we tested the effects of covariates assessed at baseline that are known to lead to attrition (Lindenberger et al., 2002), such as age, level of education, MMSE scores, and diagnosis of hypertension. We found that age was an important predictor of attrition. Hence, to reduce estimation bias, we included age in the final longitudinal models.
With respect to estimation quality and statistical power, the FIML approach with auxiliary variables has repeatedly been shown to be superior to more traditional ad-hoc procedures, such as list-wise deletion, which limits all analyses to the complete cases. That is, had we limited our analyses to only include participants with complete longitudinal data (i.e., 90 rather than 167 participants), we would have obtained a selective and biased set of estimates (Allison, 2001; McArdle & Nesselroade, 1994; Schafer & Graham, 2002; West, 2001).
In a step-up fashion (e.g., Raudenbush & Bryk, 2002), we first estimated a series of LCSMs without covariates for each ROI. This preliminary step allows modeling each ROI with the most appropriate specification of the LCSM. In particular, we were concerned with the statistical significance of the average change score (δ) and of the variance in change (γ). Subsequently, we added age at baseline, genetic polymorphisms and diagnosis of hypertension as covariates with the intent to explain individual differences in baseline volume (α) and in change in volume (γ). Chronological age was centered at the grand mean. Time-invariant covariates measured at baseline were first added to all models. We also performed a set of follow-up analyses on a subsample of participants who were normotensive at baseline and follow-up T2 (N = 131). The nominal critical alpha level was set to .05, with the multiple comparisons alpha adjusted using Bonferroni correction (Dunn, 1961).
4. Results
4.1. Descriptive statistics
Descriptive statistics are presented in Tables 1 and 2. Prior to the analyses, we identified the potential outliers by Tukey upper and lower fences, multiplied by a factor of 2.2 (3Q (3rd quartile) + 2.2 × IQR (interquartile range), and 1Q − 2.2 × IQR; Hoaglin & Iglewicz, 1987). Three univariate outliers in the volumes of PFw, Hc, and Cd (one per ROI) were found. Upon exclusion of these observations (1.8% of the sample), all models were re-evaluated to assess the influence of the outliers on the results. Results with and without outliers were virtually identical.
Table 2.
Descriptive statistics of regional brain volumes at two measurement occasion
| T1 (baseline) | T2 (follow-up) | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Mean | SD | CV | Mean | SD | CV | |
| LPFC | 19.80 | 2.91 | .15 | 19.62 | 2.68 | .14 |
| OF | 8.93 | 1.36 | .15 | 8.69 | 1.33 | .15 |
| PFw | 37.64 | 5.81 | .15 | 36.80 | 5.97 | .16 |
| Hc | 5.69 | .68 | .11 | 5.62 | .59 | .10 |
| PhG | 3.67 | .50 | .13 | 3.62 | .47 | .13 |
| CbH | 53.74 | 6.44 | .11 | 51.42 | 6.44 | .12 |
| Cd | 7.88 | 1.02 | .13 | 7.77 | 1.02 | .13 |
| Pt | 8.90 | 1.02 | .11 | 8.75 | .89 | .10 |
| In | 7.20 | .89 | .12 | 6.97 | .77 | .11 |
| VC | 5.17 | 1.06 | .20 | 5.17 | 1.09 | .21 |
Note. Volumes, averaged across the hemispheres, are in cm3. LPFC = Lateral Prefrontal Cortex; OF = Orbito-Frontal Cortex; PFw = Prefrontal white matter; Hc = Hippocampus; PhG = Parahippocampal Gyrus; CbH = Cerebellar hemispheres; Cd = Caudate; Pt = Putamen; In = Insula; VC =Visual Cortex; SD = standard deviation; CV = SD/Mean, coefficient of variation.
All ROI volumes evidenced baseline-to-follow-up stability, with correlations ranging from r = .90 for the PhG to r = .98 for the OF. Longitudinal plots in Figures 3 and 4 show the individual trajectories of ROI volume as a function of age and illustrate individual differences in change.
Figure 3.

Longitudinal plots of cortical volumes as a function of age at the time of the MRI scan. Volumes are adjusted for the intracranial volume (see text for details). Unconnected observations are the data points acquired only at baseline. The plot for the hippocampal volume does not show an outlier that was removed without any effect on the results as specified in the Results section, thus reducing the number of plotted baseline observations to N = 166 but keeping follow-up observations at N = 90.
Figure 4.

Longitudinal plots of changes in the volumes of the basal ganglia, prefrontal white matter, and cerebellar hemispheres between the baseline and the follow-up as a function of age at the time of the MRI scan. Volumes are adjusted for the intracranial volume (see text for details). Unconnected observations are the data points acquired only at baseline. The plots do not show two region-specific outliers: one for the caudate nucleus and one for the prefrontal white matter. Thus, the number of plotted observations for the caudate is N = 166 at baseline, and N =89, at follow-up, and for the Prefrontal white matter - N = 166 at baseline and N = 90 at follow-up. As specified in the Results, removal of the outliers did not affect the outcome of the analyses.
4.2. Latent Change Score Models
4.2.1. Testing for individual differences in brain shrinkage
The results of the LCSMs for each ROI volume are summarized in Table 3. Standardized factor loadings for both hemispheres ranged from .86 for PhG to .99 for CbH and were all significant. Hemispheric loadings within an ROI were forced to be equal across waves, thus establishing metric invariance.
Table 3.
Unstandardized Parameter Estimates from Latent Change Score Models for each Region of Interest’s Volumes.
| Time 1 | Change | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| ROI | Mean (β) | Variance (α) | Mean (δ) | Variance (γ) | rT1-ΔT (ε) | Res. Var. | d T1-T2 | Change Prop. |
Prop. Var |
| LPFC | 9. 704 (.029)*** | 1.918 (.223) *** | .029 (.032) | .046(.015)** | −.202(.156) | .233*** | .021 | .298 | .024 |
| OF | 4.542(.056)*** | .433 (.053) *** | −.049 (.013)*** | .002(.003) | .052(.337) | 091*** | −.074 | 1.078 | .005 |
| PFw | 18.685(.228)*** | 7.897(.908) *** | −.014 (.065) | .252(.058)*** | .100(131) | .783*** | −.005 | .075 | .032 |
| Hc | 2.805(.028)*** | .097(.012) *** | −.037(.009)*** | .006(.001) *** | −.214(.146) | .033*** | −.119 | 1.319 | .062 |
| PhG | 1.849 (.021)*** | .055(.007) *** | −.029(.011)** | .007(.002) *** | −.230(.143) | .019*** | −.124 | 1.568 | .127 |
| CbH | 5.404(.050)*** | .412(.048)*** | −.140(.031)*** | .085(.013) *** | −.299(.093)*** | .009*** | −.218 | 2.591 | .206 |
| Cd | 3.888(.040)*** | .248(.028) *** | −.017(011) | .005(.002)** | .091 (.172) | .015*** | −.034 | .437 | .020 |
| Pt | 4.609(.042)*** | .262(.031) *** | −.022(.016) | .011(.004) *** | −.154(.182) | .031*** | −.043 | .477 | .042 |
| In | 3.601 (.036)*** | .184 (.022)*** | −.080 (.023)*** | .046(.008)*** | −.513(.096)*** | .029*** | −.187 | 2.221 | .250 |
| VC | 2.207(.110)*** | .201(.028) *** | .006(.017) | .024(.004) *** | −.119(113) | .061*** | .013 | .272 | .119 |
Note. LPFC = Lateral Prefrontal Cortex; OF = Orbito-Frontal Cortex; PFw = Prefrontal white matter; Hc = Hippocampus; PhG = Parahippocampal Gyrus; CbH = Cerebellar hemispheres Cd = Caudate; Pt = Putamen; In = Insula, VC = Visual Cortex; rT1, ΔT= Correlation between Time 1 volume and Change; Res. Var. = Residual Variance; All volumes are in cm3. The volumes of the right and left CbH were divided by 10 prior to analyses because of the large variance of the variable. dT1-T2 was calculated as δ/√α. Proportional change was calculated as ∣δ / β∣× 100. Proportion of Change Variance = γ / α.
p< .000
p< .01
p< .05
The separate models for all ROIs, except VC, showed acceptable to very good fit, according to conventional criteria: Comparative Fit Index (CFI) > .95, Standardized Root Mean Square Residual (SRMR) < .06, and Root-Mean-Square Errors of Approximation (RMSEA) < .05 (Browne & Cudeck, 1993; Hu & Bentler 1998, 1999). More specifically, CFI ranged from .99 for OF, to 1.00 for LPFC, Hc, Cd and CbH, RMSEA from .000 (LPFC, Hc, CbH and Cd) to .084 (OF), with all 90% confidence intervals including 0 (except for VC). The model for the VC fit less well: CFI = .99, SRMR = .15, RMSEA = .11, (CI 90 % .047-.175), χ2 (5) = 14.75, p = .011. Complete information on model fit indices is available in the supplementary material.
At baseline, mean and variance for all ROI volumes significantly differed from zero (see Table 3). Two-year mean changes were observed in volumes of OF, Hc, PhG, In, and CbH, but not in LPFC, PFw, Cd, Pt, or VC volumes. To compare the magnitude of change among the regions, we computed effect size as the ratios of the mean latent change scores and the standard deviations of the baseline latent volumes. We also computed an estimate of the proportion of change, as the ratio of the mean change in volume over the mean volume at baseline (expressed in percentage). Both estimates of effect size allow comparing the magnitude of change across ROIs. According to the effect size comparison (see Table 3, columns 8-9), the greatest relative negative mean change (shrinkage) occurred in CbH, followed by In, PhG, Hc, and OF; the smallest (essentially zero) effects were observed in LPFC, PFw, VC, Cd, and Pt.
As indicated in Table 3, individual differences in change were the largest in the In followed by CbH, PhG and VC. Smaller and more moderate variance in change was observed in HC, Pt, PFw, LPFC and Cd. Only negligible variance of change was evident in OF. Further, in two regions, CbH and In, larger baseline volumes were associated with greater shrinkage over time (see Table 3, column 6). Estimating the models without the outliers produced virtually identical results.
4.2.2. Explaining Individual Differences in Brain shrinkage
To explain individual differences in change observed in all ROIs except OF, we expanded the LCSM to include theoretically relevant covariates. We adjusted all α levels for multiple comparisons by Bonferroni correction to α’, depending on the number of comparisons. As we found no effects of education, MMSE, or length of retest interval across any of the ROIs, we eliminated these covariates from further analyses in the interest of parsimony.
4.2.2.1. Covariates’ effects on baseline volumes
The results of the LCSM with respect to effects of covariates on baseline volumes are presented in Table 4. Advanced age was strongly associated with smaller baseline brain volumes across all ROIs. However, the magnitude of association varied across the regions, with the greatest age differences found in the Pt and the smallest in the VC. Hypertension (HBP) was unrelated to regional brain volumes at baseline. Although possession of at least one copy of the APOE ε4 allele was linked to smaller baseline In volume, the association was rendered non-significant by Bonferroni correction for six comparisons (p =.046, Bonferroni α’ = .01). No associations between regional volumes and any examined SNP were noted. All models were re-estimated after exclusion of the outliers in three regions (PFw, Hc, Cd) and all results remained essentially unchanged. To gauge the magnitude of the covariates’ effects, the last column in Table 4 presents the percentage of variance in baseline volume predicted by the covariates. The size of these effects varied from low (11% for VC) to moderately large (45% for Pt).
Table 4.
Standardized Estimates of Covariates Effects on Regional Brain Volumes at Baseline.
| ROI | Age | HBP | IL1-βT | CRP T | MTHFRT | APOE ε4 | R2 |
|---|---|---|---|---|---|---|---|
| LPFC | −.619 (.059) | −.096 (.072) | .033 (.067) | .018 (.067) | −.033 (.069) | −.030 (.067) | .440 (.065) |
| PFw | −.470 (.074) | .030 (.083) | .082 (.077) | −.041 (.078) | −.038 (.079) | .044 (.077) | .225 (.063) |
| Hc | −.395 (.084 | .038 (.090) | .003 (.085) | .054 (.085) | .141 (.086) | .052 (.084) | .161 (.061) |
| PhG | −.552 (.071) | −.043 (.083) | −.057 (.078) | .027 (.078) | −.021 (.079) | −.092 (.077) | .334 (.072) |
| CbH | −.439 (.073) | −.069 (.081) | .023 (.076) | .015 (.076) | .000 (.078) | .044 (.076) | .226 (.062) |
| Cd | −.605 (.062) | .070 (.075) | .050 (.070) | .075 (.070) | −.024 (.072) | −.080 (.070) | .359 (.065) |
| Pt | −.620 (.058) | −.049 (071) | .009 (.066) | .063 (.066) | −0.117 (.068) | .030 (.066) | .454 (.064) |
| In | −.488 (.072) | −.125 (.080) | −.032 (.075) | .028 (.075) | .084 (.077) | −.149 (.074) | .305 (.068) |
| VC | −.297 (.088) | −.034 (.092) | .013 (.086) | −.063 (.086 | −.074 (.088) | .041 (.086) | .111 (.053) |
Note. The standard errors of the parameter estimates are presented within the parentheses. LPFC = Lateral Prefrontal Cortex; PFw = Prefrontal white matter; Hc = Hippocampus; PhG = Parahippocampal Gyrus; CbH = Cerebellar hemispheres; Cd = Caudate; Pt = Putamen; In = Insula; VC = Visual Cortex; Age = age at baseline; HBP = Diagnosis of hypertension at baseline (0 = no, 1 = yes); IL1-βT = presence of the T allele of IL1-βC-511T ; CRPT = presence of the T allele of CRP −286 C>A>T; MTHFRT = presence of the T allele of MTHFRC677T; APOE ε4 = Apolipoprotein E (0 = ε33, 1= ε 34 or ε 44). The data on genetic variants were available on 145 participants (144 for CRP), at baseline, and 88 persons at follow up. R2 = Proportion of variance attributed to all covariates. All volumes were divided by 1000 prior to analyses (to avoid estimation problems). Volumes of right and left CbH were divided by 10 because of the large variance of the variable. Bold figures correspond to Bonferroni-adjusted significance α’ =.01.
4.2.2.2. The effects of covariates on volume change
The LCSM results with respect to covariate effects on change in volumes are presented in Table 5. Note that these models are the same as those presented in Table 4. For the sake of clarity, we discuss the results for predicting baseline volume and change in volume separately. The analyses revealed no associations between age differences in the rate of brain shrinkage, apart from a positive effect on Cd (p = .03) that was eliminated by Bonferroni correction (α’ = .01). Diagnosis of hypertension was associated with increased Cd shrinkage (p = .01). The Cd model, which was re-estimated after exclusion of the outlier and the effects of age (p =.02) and hypertension (p =.01) retained similar strength of associations with brain shrinkage. We found no significant effects of any covariates on the PFw and Hc volumes in the models that were replicated after exclusion of the outliers.
Table 5.
Standardized Estimates of Covariates Effects on Changes in Regional Brain Volume.
| ROI | Age | HBP | IL1-β T | CRP T | MTHFRT | APOE ε4 | R2 |
|---|---|---|---|---|---|---|---|
| LPFC | .082 (.166) | .107 (.157) | −.062 (.149) | .104 (.152) | .135 (.109) | −.166 (.133) | .087 (.079) |
| PFw | −.079 (.145) | −.116 (.136) | −.167 (.126) | .094 (.132) | .030 (.094) | .055 (.117) | .072 (.076) |
| Hc | .145 (.142) | −.145 (.142) | −.165 (.137) | .083 (.134) | −.128 (.128) | −.065 (.122) | .161 (.061) |
| PhG | .190 (.142) | −.212 (.133) | −.313 (.122) | .177 (.127) | −.247 (.096) | .168 (.114) | .234 (.113) |
| CbH | .143 (.121) | .049 (.120) | −.255 (.114) | .004 (.114) | −.063 (.109) | −.023 (.103) | .086 (.057) |
| Cd | .393 (.183) | −.440 (.172) | −.172 (.164) | .113 (.167) | .058 (.119) | −.271 (.152) | .305 (.170) |
| Pt | .111 (.190) | −.073 (.178) | −.194 (.172) | .058 (.173) | .060 (.122) | −.180 (.153) | .088 (.091) |
| In | .051 (.126) | .052 (.126) | .056 (.122) | −0.037 (.120) | .033 (.115) | .238 (.106) | .063 (.052) |
| VC | −.174 (.124) | .046 (.126) | −.052 (.122) | .143 (.118) | .046 (.114) | −.144 (.106) | .067 (.056) |
Note. LPFC = Lateral Prefrontal Cortex; PFw = Prefrontal white matter; Hc = Hippocampus; PhG = Parahippocampal Gyrus; CbH = Cerebellar hemispheres; Cd = Caudate; Pt = Putamen; In = Insula; VC = Visual Cortex; Age = age at baseline; HBP = Diagnosis of hypertension at baseline (0 = no, 1 = yesj; IL1-βT = presence of the T allele of IL1-βC-511T ; CRPT = presence of the T allele of CRP −286 C>A>T; MTHFRT = presence of the T allele of MTHFRC677T; APOE = Apolipoprotein E (0 = ε33, 1= ε 34 or ε 44). The data on genetic variants were available on 145 participants (144 for CRP), at baseline, and 88 persons at follow up. R2 = Percentage of variance predicted by all covariates; Volumes of the left and right CbH were divided by 10 because of the large variance of the variable. Bold figures correspond to Bonferroni-adjusted significance α’ =.01.
Several genetic variants were related to the observed variance in regional volume change. Possession of two T alleles of the IL-1β C-511T polymorphism predicted increased shrinkage of CbH (p = .03) and PhG (p = .01). Carriers of at least one T allele of MTHFR C677T polymorphism evidenced increased shrinkage of PhG (p =.01). The directions of the effects were the same in the subsample of normotensive participants. The effects of MTHFR C677T and IL-1β C-511T on shrinkage in PhG survived Bonferroni correction (α’ =.01), in both the full sample and the normotensive subgroup. Comparison of estimated latent change scores between the allelic groups revealed that PhG shrinkage differed between allelic groups, and was essentially zero among the participants with lower genetic predisposition to inflammation (see Figure 5). The effect of the IL-1β C-511T polymorphism on shrinkage in CbH was rendered non-significant by the family-wise error correction in the full sample, but remained significant in the normotensive subgroup (p =.01, α’=.01; see Figure 6). After Bonferroni correction, the effect of the APOEε4 allele on volume change was not present in any group.
Figure 5.
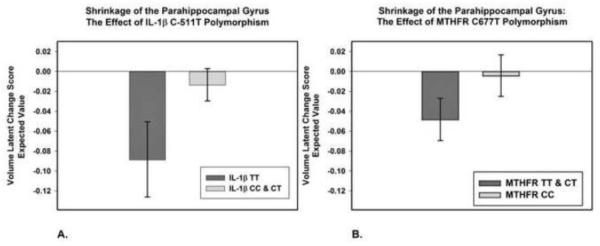
Effects of two pro-inflammatory single-nucleotide polymorphisms, IL-1β C-511T (A) and MTHFR C677T (B), on shrinkage of the parahippocampal gyrus. Shrinkage is indexed by the mean expected value of latent change scores computed from the latent change score models (see text). In panel A, homozygotes for the IL-1β -511T allele, which is associated with the increased circulating levels of interleukin -1β are compared to carriers of the C allele. In panel B, carriers of the MTHFR 677T allele associated with higher plasma homocysteine levels are compared to the C homozygotes. The bars represent 95% confidence intervals around the means. The data on genetic variants were available on 145 participants at baseline, and 88 persons at follow up.
Figure 6.

Effect of a pro-inflammatory single-nucleotide polymorphism, IL-1β C-511T, on shrinkage of the cerebellar hemispheres in normotensive participants. Shrinkage is indexed by the mean expected value of latent change scores computed from the estimated means of the models while taking into account the effects of covariates (see text). Homozygotes for the T allele, which is associated with the increased circulating levels of interleukin -1β are compared to the carriers of the C allele. The bars represent 95% confidence intervals around the means. The data on genetic variants were available on 145 participants at baseline, and 88 persons at follow up.
The models were re-estimated in the subsample with complete SNP data (N = 145 at baseline, N = 88 at follow up), and MTHFR C677T (β = −.35, SE =.17=, p = .03) and IL-1β C-511T (β = −.29, SE = .12, p = .01) still predicted shrinkage of PhG. The effect of IL-1β C-511T on CbH was also present (β = −.38, SE = .13, p =.004).
No associations were found between the CRP polymorphism and regional brain shrinkage. According to the estimates of total magnitude of these effects, the only significant prediction in change in volume was obtained for PhG, because here two covariates (IL-1β C-511T and MTHFR C677T polymorphisms) were significant.
4.3. Non-linearity in associations between volume and age
Visual inspection of the distributions suggested nonlinear relationships between age and some regional volumes. Separate cross-sectional analyses with second-order polynomials (quadratic effects) showed nonlinearities in associations between age and three ROI volumes: PFw (β = −.05, SE = .03, p = .05, α’ = .03), PhG (β = −.05, SE = .03, p = .002, α’ = .03), and VC (β = −.06, SE = .03, p = .045, α’ = .03), although only the non-linear effect on PhG survived Bonferroni correction in the models with outliers. After two bivariate outliers in the associations of age with PFw and PhG volumes were removed, the observed curvilinear associations between age and baseline volumes were still present (PhG) and even strengthened (PFw). The latter was strengthened to such a degree that it survived the Bonferroni correction (β = −.06, SE = .03, p =.021, α’ = .03).
Longitudinal analysis revealed a significant age-related acceleration of PhG shrinkage: (β = −.01, SE = .01, p = .014, α’ = .03). The association was still significant after exclusion of the outlier (p = .02). No other ROIs showed a similar age-related acceleration. The finding was replicated among the normotensive participants (β = −.18, SE = .08, p = .029, α’ = .03).
5. Discussion
In healthy adults, we found mild but significant mean shrinkage in 5 out of 10 examined brain regions over a period of about two years. More importantly, in all but one of these regions, we observed significant individual differences in shrinkage, a result that would have been missed by analytic approaches focusing solely on mean change. In some regions, the individual differences in shrinkage were explained in part by pro-inflammatory genetic variants, independent of cardiovascular risk factors. Carriers of the T allele of the MTHFR C677T and T homozygotes for the IL-1β C-511T evidenced increased shrinkage of the parahippocampal gyrus. The T allele homozygotes of the IL-1β C-511T polymorphism also evidenced greater shrinkage of the cerebellum than C carriers. The association was stronger for the cerebellum in normotensive participants. To the best of our knowledge, this is the first report of the effects of pro-inflammatory genetic variants on volumetric brain changes in healthy adults.
5.1. Heterogeneity of brain shrinkage
The pattern of brain shrinkage observed here on a 4T scanner replicated reasonably well the findings from previous longitudinal studies conducted on weaker systems (1.5T). Substantial shrinkage of the cerebellar hemispheres replicates previous results from longitudinal studies (Raz et al., 2003b, 2005, 2010, 2013; Tang et al., 2001), as does the decline in hippocampal volume (Du et al., 2006; Fjell et al., 2009; Raz et al, 2005, 2010, 2013; Scahill et al., 2003). The observed shrinkage of the parahippocampal and orbitofrontal cortices and of the neostriatal nuclei are also in agreement with previous longitudinal studies (Fjell et al., 2009; Raz et al., 2003c; 2005, 2010, 2013), as is the observed relative stability of the primary visual cortex volume (Fjell et al., 2009; Raz et al., 2005, 2010, 2013).
A notable exception to the general pattern of replication is the lack of mean shrinkage in the lateral prefrontal cortex and the adjacent white matter. Also, the magnitude of insular shrinkage we found is substantially greater than that previously reported (Raz et al., 2010). The absence of mean change in the prefrontal regions contradicts the results of several longitudinal studies of brain aging with inter-scan intervals ranging from 6 months to 5 years (Driscoll et al., 2009; Fjell et al., 2009; Liu et al., 2003; Pfefferbaum et al., 1998; Raz et al., 2005, 2013; Resnick et al., 2003). However, the observed lack of mean change in LPFC volume is in agreement with a recent report (Raz et al., 2010) and suggests that, contrary to the prefrontal aging hypothesis (e.g., West, 1996), prefrontal decline may not be a sine qua non of healthy aging. Because the lateral prefrontal cortex is one of the last brain regions to develop, its stability found here presents a challenge to the “last in, first out” view of brain aging.
The stability of mean basal ganglia volumes observed in this sample contradicts some previous reports (Fjell et al., 2009; Raz et al., 2003c, 2013), but replicates findings from another sample (Raz et al., 2010). In that three-occasion study, we found no change in volumes of the caudate and putamen within the first 15-month period, only to observe significant shrinkage of the caudate nucleus during the second follow-up (Raz et al., 2010). The two-occasion repeated measurement design employed here covers a relatively narrow time window, similar to the first and second intervals in the latter study, and could have been insufficient to detect such changes.
Whereas the magnitude of volume change varies among the regions of the brain within the sample, there are also significant individual differences in the rate of change. The latent change score models enabled us to estimate not only mean change, but also variance in change, which was significant in all measured regions, except the orbito-frontal cortex. Such heterogeneity of change replicates our findings in two other samples, with individual differences in change being most pronounced in the insula and cerebellum, followed by PhG, Hc, OF, Pt, Cd, LPFC, VC, and PFw, in descending order (Raz et al., 2005, 2010).
The mechanisms of the observed regional pattern of brain shrinkage remain unclear and their elucidation is hampered by lack of an established neurobiological basis of volume differences and changes observed on MRI. Volume variation in some regions has been linked to attrition of neurons in patients’ brains who succumb to neurodegenerative disease (Bobinski et al., 2000). Converging evidence suggests that MRI-derived volume differences may reflect neuropil changes; as shown in rodents, volume changes closely track expansion and shrinkage of neuropil during the estrous cycle (Qiu et al., 2013) and are associated with loss of neuropil induced by chronic administration of antipsychotic drugs (Vernon et al., 2014) or cardiac arrest (Suzuki et al., 2013). Loss of Purkinje cells in a murine model of autoimmune encephalomyelitis has been related to smaller cerebellar volume observed on MRI (MacKenzie-Graham et al., 2009). The latter finding underscores the possibility of myelin damage as a potential source of cortical volume variation. The role of myelin reduction in brain aging has been hypothesized by several researchers (Bartzokis, 2011; Courchesne et al., 2000) and is in accord with the observed association between myelination precedence order according to Flechsig (1901) and regional cortical shrinkage (Raz, 2001; Raz & Kennedy, 2009). However, in this study, we combined gray and white matter in CbH volume, and therefore could not examine a differential effect on its compartments.
5.2. Demographic and VR modifiers of shrinkage
Our attempts to explain the observed variance in change by influence of selected moderators were only partially successful. As discussed above, two genetic variants were linked to individual differences in shrinkage of two brain regions. However, other hypothesized moderators of change were not influential. Unlike previous studies (e.g. Raz et al., 2005, 2010), we found no association between hypertension and individual differences in the rates of change in tertiary cortices and hippocampus. Notably, chronological age, which was reliably associated with regional brain volumes at baseline, did not affect volume changes within the two-year time window of this study. The observed lack of correspondence between cross-sectional age-related differences and longitudinal change reinforces the notion that the latter cannot always be inferred from the former. Age-heterogeneous cross-sectional designs are characterized by high commonality between age and multiple measures of interest, thereby increasing the likelihood of confounds with between-person age trends (Hofer & Sliwinski, 2001; Lindenberger & Pötter, 1998), which can lead to spurious inferences concerning interdependencies among age-related functions (Hofer et al., 2003).
In search for potential modifiers of individual differences in the rate of change, we inquired whether our data could lend some support to two popular views of brain resilience: brain reserve (Satz, 1993) and cognitive reserve (Stern, 2002). The first postulates that bigger brains are somehow protected from multiple insults and, by inference, should show reduced change over time compared to smaller brains. The second posits cognitive attainment as a predictor of brain resistance to neurodegenerative changes (Coffey et al., 1999). We found no support for the brain reserve hypothesis, and observed no advantage for larger brains vis-à-vis brain shrinkage. Moreover, larger baseline volumes of two regions (cerebellum and insula) were associated with increased shrinkage over time. In some samples, we have observed similar associations between larger brain volumes at baseline and faster rates of shrinkage in selected (and different) brain regions (e.g., for the hippocampus and LPFC in Raz et al., 2010), whereas in other studies, no such correlations were found (Raz et al., 2005). Such lack of consistency across studies conducted with similar methods, albeit in different populations, does not bode well for the brain reserve hypothesis.
In a test of the second proposition, we found no effect of education on the rate of change across the measured brain regions. This conforms to our previous findings (Raz et al., 2005, 2010). It is possible that reserve works only on a population level where a wide range of brain volumes and educational attainment is observed. Our participants, on average, reported about 15 years of formal schooling, which corresponds to almost full college education. In that narrow range of educational attainment, specific tests of cognitive abilities may provide more sensitive means of testing the cognitive reserve hypothesis. In addition, our study participants were intentionally selected for good health and were not affected by dementia. Both types of reserve may operate as proxies for health disparities that reflect a complex and confounded patchwork of influences such as socio-economic status, culture, and cognitive abilities. The full capacity and importance of such reserves may also become clearer and more crucial in the context of disease. In a selective sample of healthy participants, the influence of these factors might have been attenuated by constriction of range, which precluded showing associations as described.
5.3. Non-linear age trends
Consistent with previous findings (Raz et al., 2005), we observed non-linear associations between age and baseline volume of two regions: prefrontal white matter and parahippocampal gyrus. The youngest and the oldest participants had smaller volume of white matter in the prefrontal region than their middle-aged counterparts. An inverted U-shape conforms to the notion of late myelination continuing into the third decade of life (Benes et al., 1994), and decline of myelin volume in the late part of the lifespan (Bartzokis, 2011). However, we observed no acceleration of PFw shrinkage in the longitudinal analysis, although it is unclear whether this null finding is due to an insufficient interval between the baseline and the follow-up. We did find accelerated shrinkage of parahippocampal gyrus over the same two-year period; although, contrary to previous reports (Fjell et al., 2009, 2013; Raz et al., 2005, 2010), there was no accelerated change in the hippocampal volume. It is possible that parahippocampal gyrus, which includes entorhinal cortex, is more sensitive to age-related declines than the hippocampus. Several studies have found that the parahippocampal gyrus is particularly vulnerable to clinical and subclinical factors that are associated with aging (Bregant et al., 2013; Yau et al., 2014) and have reported particularly fast thinning of the parahippocampal cortex in late-onset Alzheimer’s disease (Cho et al., 2013).
5.4. Genetic modifiers of shrinkage
We found that genetic predisposition to inflammation moderated shrinkage in some brain regions, with only persons who have a strong genetic predisposition to pro-inflammatory response showing significant shrinkage. It is unclear why shrinkage in the parahippocampal gyrus and cerebellum was limited to carriers and homozygotes of two of the examined proinflammatory genetic variants. The literature on regional effects of pro-inflammatory cytokines on the brain is sparse. Response to neuroinflammation has been linked to shrinkage of the entorhinal cortex, which constitutes the anterior part of the PhG (Hauss-Wegrzyniak et al., 2000), and IL-1β may affect glutaminergic and GABAergic transmission in the cerebellum (Mandolesi et al., 2013). In a sample of patients with schizophrenia and normal controls, the MTHFR 677 T allele has been associated with decreased density of parahippocampal and prefrontal grey matter (Zhang et al., 2013). The IL-1β C-511T variant has been linked to smaller prefrontal and temporal cortices in schizophrenia, but is unrelated to regional brain volumes in normal controls (Meisenzahl et al., 2001). Those, however, were cross-sectional studies, and their findings are not straightforwardly comparable to longitudinal observations reported here. Our findings are consistent both with a possibility that the major alleles of IL-1β and MTHFR polymorphisms have neuroprotective influence on the parahippocampal cortex, and the option that the variant alleles promote shrinkage. The observational nature and limited statistical power of this study preclude a more definitive conclusion.
Notably, we observed no APOE ε4-related difference in shrinkage of the regions of interest, including the Hc and the PhG, both of which are particularly vulnerable to Alzheimer-type pathology (Esiri et al., 1990). In contrast to its established role as a risk factor for Alzheimer’s disease, the influence of APOE ε4 variant in healthy adults is far from clear, with a wide range of findings reported in the literature (e.g., Cherbuin et al., 2007; Reinvang et al., 2013). Positive findings from previous studies might have been confounded by inclusion of persons with less than optimal health and prodromal Alzheimer pathology.
We found no effects of the T allele of another polymorphism previously linked to systemic inflammation and vascular risk (CRP-286C>A>T) on any of the examined regional brain volumes. The literature on the role of CRP polymorphisms in brain morphometry is meager. In one study on a sample with a partial overlap with the current one, we found significantly larger white matter hyperintensities burden in carriers of the CRP-286 T allele (Raz et al., 2012), whereas other studies revealed no such associations (Ladenvall et al., 2006; Reitz et al., 2007).
5.5. Limitations
The findings reported here should be interpreted in the context of several limitations. First, we relied on a sample of convenience recruited through advertisements. The participants were healthier and better educated than expected for a representative sample. For instance, prevalence of hypertension in American adults (age 20 and above) is 33% compared to 20% in this sample, and prevalence of diabetes in the same population is 8% compared to 0% in this sample (Go et al., 2014). Stringent selection served our goal to examine brain aging in a sub-population that is likely to attain “successful aging” (Rowe & Khan, 1987). Moreover, by excluding participants with multiple age-related medical conditions, we have eliminated many potential confounding factors that could affect brain morphology. However, the downside of this sampling strategy is reduced generalizability and a possible selection bias. For example, in addition to insufficient statistical power, lack of significant genetic effects may reflect selection of people with risky alleles who nonetheless have additional mitigating factors that were not included in the models.
On the other hand, despite exclusion of the participants with clear cognitive impairment and ascertainment of at least gross cognitive stability over the follow-up period, we still cannot entirely eliminate a risk of inclusion of individuals at the early prodromal stages of dementia. This issue can be addressed only by additional longitudinal measurements.
The extant longitudinal studies of brain aging differ in many respects: sample composition and screening, scanner parameters, duration of delays between follow-ups, and methods of image analysis, to name a few. Thus, the reasons for the observed discrepancies are difficult to pinpoint. Regional volumes are determined in any given moment by a dynamic equilibrium of multiple influences, and expansion and contraction of the parenchyma may occur in a quasi-periodic fashion (Raz et al., 2010). It is unclear how frequently such dynamically equilibrated process should be sampled to be faithfully reproduced. This study, which is a part of an ongoing longitudinal project, was limited to two measurement occasions. Such low measurement density, although allowing for the identification of both mean change and variance in change, precluded examination of the trajectories of change and suffers from reduced statistical power compared to studies with several measurement intervals (e.g., Hertzog et al., 2006; Muthén & Curran, 1997). Nevertheless, despite such reduced power, we did uncover some important effects.
Although our selection of potential modifiers of change was predicated on the extant literature, it was limited. We selected genetic polymorphisms with known pro-inflammatory properties, but the number of genetic and physiological markers of inflammation is vast (Wilson et al., 2002) and awaits further examination. Moreover, it is likely that in healthy populations, interactions between risky alleles of specific genes and other risk factors are responsible for individual variation in brain aging (e.g., Meda et al., 2013). However, in the analyses presented here, we could not introduce interactive terms into the models due to the small number of participants per category and a prohibitively high number of parameters that would have ensued. Studies with greater statistical power are needed to investigate the epistatic effects of multiple genes and interactions of genetic and physiological risks factors.
5.6. Conclusion
In sum, we demonstrated that in healthy adults, multiple brain regions shrink within a two-year period, but the magnitude of shrinkage varies across the brain regions and among individuals. Two genetic variants that promote inflammation exacerbated shrinkage in the parahippocampal gyrus and the cerebellum. These findings encourage further inquiries into the role of neuroinflammation in brain aging.
Supplementary Material
Acknowledgements
This research was supported by the National Institute on Aging grant R37 AG-11230 to NR. NP was supported by grants FOA11H-090, FOA13H-090, FO2011-0504 and FO2013-0189 from Swedish Royal Academia of Sciences, Lars Hiertas Memorial Foundation and Solstickan Foundation and by the Department of Psychology at the Stockholm University (Ann-Charlotte Smedler, Head of the Department). We acknowledge Awantika Deshmukh’s contribution to tracing of the ROIs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement There is no conflict of interest for any of the authors of this paper.
References
- Allison PD. Sage University Papers Series on Quantitative Applications in the Social Sciences. Sage; Thousand Oaks. CA: 2001. Missing data; pp. 107–136. [Google Scholar]
- Banks WA. Blood-brain barrier transport of cytokines: A mechanism for neuropathology. Curr Pharm Des. 2005;11:973–984. doi: 10.2174/1381612053381684. [DOI] [PubMed] [Google Scholar]
- Banks WA, Farr SA, La Scola ME, Morley JE. Intravenous human interleukinalpha impairs memory processing in mice: Dependence on blood-brain barrier transport intoposterior division of the septum. J Pharmacol Exp Ther. 2001;299:536–541. [PubMed] [Google Scholar]
- Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brainbarrier. Neuroimmunomodulation. 1995;2:241–248. doi: 10.1159/000097202. [DOI] [PubMed] [Google Scholar]
- Bardou I, Brothers HM, Kaercher RM, Hopp SC, Wenk GL. Differential effects of duration and age on the consequences of neuroinflammation in the hippocampus. Neurobiol Aging. 2013;34:2293–2301. doi: 10.1016/j.neurobiolaging.2013.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging. 2011;32:1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathum L, Petersen I, Christiansen L, Konieczna A, Sorensen TIA, Kyvik KO. Genetic and environmental influences on plasma homocysteine: results from a Danish twin study. Clinical Chemistry. 2007;53:971. doi: 10.1373/clinchem.2006.082149. [DOI] [PubMed] [Google Scholar]
- Baune BT, Konrad C, Grotegerd D, Suslow T, Birosova E, Ohrmann P, Bauer J, Arolt V, Heindel W, Domschke K, Schöning S, Rauch AV, Uhlmann C, Kugel H, Dannlowski U. Interleukin-6 gene (IL-6): a possible role in brain morphology in the healthy adult brain. J. Neuroinflammation. 2012;9:1–9. doi: 10.1186/1742-2094-9-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Benes FM, Turtle M, Khan Y, Farol P. Myelination of a key relay zone in the hippocampal formation occurs in the human brain during childhood, adolescence, and adulthood. Arch Gen Psychiatry. 1994;51:477–484. doi: 10.1001/archpsyc.1994.03950060041004. [DOI] [PubMed] [Google Scholar]
- Bettcher BM, Wilheim R, Rigby T, Green R, Miller JW, Racine CA, Yaffe K, Miller BL, Kramer JH. C-reactive protein is related to memory and medial temporal brain volume in older adults. Brain Behav Immun. 2012;26:103–108. doi: 10.1016/j.bbi.2011.07.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobinski M, de Leon MJ, Wegiel J, Desanti S, Convit A, Saint Louis LA, Rusinek H, Wisniewski HM. The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer’s disease. Neuroscience. 2000;95:721–725. doi: 10.1016/s0306-4522(99)00476-5. [DOI] [PubMed] [Google Scholar]
- Bregant T, Rados M, Vasung L, Derganc M, Evans AC, Neubauer D, Kostovic I. Region-specific reduction in brain volume in young adults with perinatal hypoxicischaemic encephalopathy. Eur. J. Paediatr. Neurol. 2013;17:608–614. doi: 10.1016/j.ejpn.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Brooks-Wilson AR. Genetics of healthy aging and longevity. Hum. Genet. 2013;132:1323–1338. doi: 10.1007/s00439-013-1342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne M, Cudeck R. Alternate ways of assessing model fit. In: Bollen KA, Long JS, editors. Testing Structural Equation Models. Sage, CA: 1993. pp. 136–162. [Google Scholar]
- Burns JM, Honea RA, Vidoni ED, Hutfles LJ, Brooks WM, Swerdlow RH. Insulin is differentially related to cognitive decline and atrophy in Alzheimer’s disease and aging. Biochim Biophys Acta. 2012;1822:333–339. doi: 10.1016/j.bbadis.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbuin N, Leach LS, Christensen H, Anstey KJ. Neuroimaging and APOE genotype: a systematic qualitative review. Dement. Geriatr Cogn Disord. 2007;24:348–362. doi: 10.1159/000109150. [DOI] [PubMed] [Google Scholar]
- Cho H, Jeon S, Kang SJ, Lee JM, Lee JH, Kim GH, Shin JS, Kim CH, Noh Y, Im K, Kim ST, Chin J, Seo SW, Na DL. Longitudinal changes of cortical thickness in early- versus late-onset Alzheimer’s disease. Neurobiol. Aging. 2013;34:921. doi: 10.1016/j.neurobiolaging.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Choe YM, Sohn BK, Choi HJ, Byun MS, Seo EH, Han JY, Kim YK, Yoon EJ, Lee JM, Park J, Woo JI, Lee DY. Association of homocysteine with hippocampal volume independent of cerebral amyloid and vascular burden. Neurobiol Aging. 2014;35:1519–1525. doi: 10.1016/j.neurobiolaging.2014.01.013. [DOI] [PubMed] [Google Scholar]
- Coffey CE, Saxton JA, Ratcliff G, Bryan RN, Lucke JF. Relation of education to brain size in normal aging: implications for the reserve hypothesis. Neurology. 1999;53:189–196. doi: 10.1212/wnl.53.1.189. [DOI] [PubMed] [Google Scholar]
- Collins LM, Schafer JL, Kam CM. A comparison of inclusive and restrictive strategies in modern missing-data procedures. Psychol Methods. 2001;6:330–351. [PubMed] [Google Scholar]
- Courchesne E, Chisum HJ, Townsend J, Cowles A, Covington J, Egaas B, Harwood M, Hinds S, Press GA. Normal brain development and aging: quantitative analysis at in vivo MR imaging in healthy volunteers. Radiol. 2000;216:672–682. doi: 10.1148/radiology.216.3.r00au37672. [DOI] [PubMed] [Google Scholar]
- Cronbach LJ, Furby L. How we should measure “change” - or should we? Psychol Bull. 1970;74:68–80. [Google Scholar]
- Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- de Lau LM, van Meurs JB, Uitterlinden AG, Smith AD, Refsum H, Johnston C, Breteler MM. Genetic variation in homocysteine metabolism, cognition, and white matter lesions. Neurobiol Aging. 2010;31:2020–2022. doi: 10.1016/j.neurobiolaging.2008.10.004. [DOI] [PubMed] [Google Scholar]
- den Heijer T, Vermeer SE, Clarke R, Oudkerk M, Koudstaal PJ, Hofman A, Breteler MM. Homocysteine and brain atrophy on MRI of non-demented elderly. Brain. 2003;126:170–175. doi: 10.1093/brain/awg006. [DOI] [PubMed] [Google Scholar]
- Driscoll I, Davatzikos C, An Y, Wu X, Shen D, Kraut M, Resnick SM. Longitudinal pattern of regional brain volume change differentiates normal aging from MCI. Neurology. 2009;72:1906–1913. doi: 10.1212/WNL.0b013e3181a82634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du AT, Schuff N, Chao LL, Kornak J, Jagust WJ, Kramer JH, Reed BR, Miller BL, Norman D, Chui HC, Weiner MW. Age effects on atrophy rates ofentorhinal cortex and hippocampus. Neurobiol. Aging. 2006;7:733–740. doi: 10.1016/j.neurobiolaging.2005.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn OJ. Multiple comparisons among means. J Am Stat Assoc. 1961;56(293):52–64. [Google Scholar]
- Eritaia J, Wood SJ, Stuart GW, Bridle N, Dudgeon P, Maruff P, Velakoulis D, Pantelis C. An optimized method for estimating intracranial volume from magnetic resonance images. Magn Reson Med. 2000;44:973–977. doi: 10.1002/1522-2594(200012)44:6<973::aid-mrm21>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Esiri MM1, Pearson RC, Steele JE, Bowen DM, Powell TP. A quantitative study of the neurofibrillary tangles and the choline acetyltransferase activity in the cerebral cortex and the amygdala in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 1990;53:161–165. doi: 10.1136/jnnp.53.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Isaac V, Sim S, Ng TP, Krishnan KR, Chee MW. Associations between elevated homocysteine, cognitive impairment, and reduced white matter volume in healthy old adults. Am J Geriatr.Psychiatry. 2013;21:164–172. doi: 10.1016/j.jagp.2012.10.017. [DOI] [PubMed] [Google Scholar]
- Finch CE. Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1718–24. doi: 10.1073/pnas.0909606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Brewer JB, Dale AM. One-year brain atrophy evident in healthy aging. J Neurosci. 2009;29:15223–15231. doi: 10.1523/JNEUROSCI.3252-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Grydeland H, Amlien I, Espeseth T, Reinvang I, Raz N, Holland D, Dale AM, Walhovd KB. Critical ages in the life course of the adult brain: nonlinear subcortical aging. Neurobiol Aging. 2013;34:2239–2247. doi: 10.1016/j.neurobiolaging.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flechsig P. Developmental (myelogenetic) localisation of the cerebral cortex in the human subject. The Lancet. 1901;158:1027–1030. [Google Scholar]
- Folstein M, Folstein S, McHugh P. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics Committee and Stroke Statistics Subcommittee Executive summary: heart disease and stroke statistics-2014 update: a report from the American Heart Association. Circulation. 2014;129:399–410. doi: 10.1161/01.cir.0000442015.53336.12. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Neurol Clin. 2006;24:521–538. doi: 10.1016/j.ncl.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Grimm KJ, An Y, McArdle JJ, Zonderman AB, Resnick SM. Recent Changes Leading to Subsequent Changes: Extensions of Multivariate Latent Difference Score Models. Struct Equ Modeling. 2012;19:268–292. doi: 10.1080/10705511.2012.659627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak BI, Galons JP, Wenk GL. Quantitative volumetric analyses of brain magnetic resonance imaging from rat with chronic neuroinflammation. Exp Neurol. 2000;165:347–54. doi: 10.1006/exnr.2000.7469. [DOI] [PubMed] [Google Scholar]
- Hertzog C, Lindenberger U, Ghisletta P, Oertzen TV. On the power of multivariate latent growth curve models to detect correlated change. Psychol Methods. 2006;11:244–252. doi: 10.1037/1082-989X.11.3.244. [DOI] [PubMed] [Google Scholar]
- Hoaglin DC, Iglewicz B. Fine Tuning Some Resistant Rules for Outlier Labeling. J Am Stat Assoc. 1987;82:1147–1149. [Google Scholar]
- Hofer SM, Berg S, Era P. Evaluating the interdependence of aging-related changes in visual and auditory acuity, balance, and cognitive functioning. Psychol Aging. 2003;18:285–305. doi: 10.1037/0882-7974.18.2.285. [DOI] [PubMed] [Google Scholar]
- Hofer SM, Sliwinski MJ. Understanding ageing: An evaluation of research designs for assessing the interdependence of ageing-related changes. Gerontology. 2001;47:341–352. doi: 10.1159/000052825. [DOI] [PubMed] [Google Scholar]
- Hu L-T, Bentler PM. Fit indices in covariance structure modeling: Sensitivity to underparameterized model misspecification. Psychol. Methods. 1998;3:424–453. [Google Scholar]
- Hu L, Bentler PM. Cutoff criteria in fix indexes in covariance structure analysis: Conventional criteria versus new alternatives. Struct Equ Modeling. 1999;6:1–55. [Google Scholar]
- Jack CR, Jr, Twomey CK, Zinsmeister AR, Sharbrough FW, Petersen RC, Cascino GD. Anterior temporal lobes and hippocampal formations: normative volumetric measurements from MR images in young adults. Radiology. 1989;172:549–554. doi: 10.1148/radiology.172.2.2748838. [DOI] [PubMed] [Google Scholar]
- Jagust W. Vulnerable neural systems and the borderland of brain aging and eurodegeneration. Neuron. 2013;77:219–234. doi: 10.1016/j.neuron.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson AL, Massaro JM, Wolf PA, Seshadri S, Au R, Vasan RS, Larson MG, Meigs JB, Keaney JF, Jr, Lipinska I, Kathiresan S, Benjamin EJ, DeCarli C. Inflammatory biomarkers are associated with total brain volume: the Framingham Heart Study. Neurology. 2007;68:1032–1038. doi: 10.1212/01.wnl.0000257815.20548.df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katan MB. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet. 1986;1:507–508. doi: 10.1016/s0140-6736(86)92972-7. [DOI] [PubMed] [Google Scholar]
- Korf ES, White LR, Scheltens P, Launer LJ. Midlife blood pressure and the risk of hippocampal atrophy: The Honolulu Asia Aging Study. Hypertension. 2004;44:29–34. doi: 10.1161/01.HYP.0000132475.32317.bb. [DOI] [PubMed] [Google Scholar]
- Ladenvall C, Jood K, Blomstrand C, Nilsson S, Jern C, Ladenvall P. Serum C-reactive protein concentration and genotype in relation to ischemic stroke subtype. Stroke. 2006;37:2018–2023. doi: 10.1161/01.STR.0000231872.86071.68. [DOI] [PubMed] [Google Scholar]
- Lindenberger U, Pötter U. The complex nature of unique and shared effects in hierarchical linear regression: Consequences for cross-sectional developmental research. Psychol Methods. 1998;3:218–230. [Google Scholar]
- Lindenberger U, Singer T, Baltes PB. Longitudinal selectivity in aging populations: Separating mortality-associated versus experimental components in the Berlin Aging Study (BASE) J. Gerontol. B Psychol. Sci. Soc. Sci. 2002;57:474–482. doi: 10.1093/geronb/57.6.p474. [DOI] [PubMed] [Google Scholar]
- Lindenberger U, von Oertzen T, Ghisletta P, Hertzog C. Cross-sectional age variance extraction: what’s change got to do with it? Psychol. Aging. 2011;26:34–47. doi: 10.1037/a0020525. [DOI] [PubMed] [Google Scholar]
- Little RJA. Modeling the dropout mechanism in repeated-measures studies. J Am Stat Assoc. 1995;90:1112–1121. [Google Scholar]
- Liu RS, Lemieux L, Bell GS, Sisodiya SM, Shorvon SD, Sander JW, Duncan JS. A longitudinal study of brain morphometrics using quantitative magnetic resonance imaging and difference image analysis. NeuroImage. 2003;20:22–33. doi: 10.1016/s1053-8119(03)00219-2. [DOI] [PubMed] [Google Scholar]
- MacKenzie-Graham A, Tiwari-Woodruff SK, Sharma G, Aguilar C, Vo KT, Strickland LV, Morales L, Fubara B, Martin M, Jacobs RE, Johnson GA, Toga AW, Voskuhl RR. Purkinje cell loss in experimental autoimmune encephalomyelitis. NeuroImage. 2009;48:637–651. doi: 10.1016/j.neuroimage.2009.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Rall SC. Apolipoprotein E: Far more than a lipid transport protein. Annual Review of Genomics and Humn Genetics. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- Mandolesi G, Musella A, Gentile A, Grasselli G, Haji N, Sepman H, Fresegna D, Bullitta S, De Vito F, Musumeci G, Di Sanza C, Strata P, Centonze D. Interleukin-1β alters glutamate transmission at Purkinje cell synapses in a mouse model of multiple sclerosis. J Neurosci. 2013;33:12105–12121. doi: 10.1523/JNEUROSCI.5369-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsland AL, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR. Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol Psychiatry. 2008;64:484–490. doi: 10.1016/j.biopsych.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell SE, Cole DA. Bias in cross-sectional analyses of longitudinal mediation. Psychol Methods. 2007;12:23–44. doi: 10.1037/1082-989X.12.1.23. [DOI] [PubMed] [Google Scholar]
- McArdle JJ. A latent difference score approach to longitudinal dynamic structural analyses. In: Cudeck R, du Toit S, Sorbom D, editors. Structural equation modeling: Present and future. Scientific Software International; Lincolnwood, IL: 2001. pp. 342–380. [Google Scholar]
- McArdle JJ. Latent variable modeling of longitudinal data. Annu Rev Psychol. 2008;60:577–605. doi: 10.1146/annurev.psych.60.110707.163612. [DOI] [PubMed] [Google Scholar]
- McArdle JJ. Latent variable modeling of differences and changes with longitudinal data. Annu Rev Psychol. 2009;60:577–605. doi: 10.1146/annurev.psych.60.110707.163612. [DOI] [PubMed] [Google Scholar]
- McArdle JJ, Hamagami F, Jones K, Jolesz F, Kikinis R, Spiro R, Albert MS. Structural modeling of dynamic changes in memory and brain structure using longitudinal data from the Normative Aging Study. J. Gerontol. B Psychol. Sci. Soc. Sci. 2004;59:P294–304. doi: 10.1093/geronb/59.6.p294. [DOI] [PubMed] [Google Scholar]
- McArdle JJ, Nesselroade JR. Using multivariate data to structure developmental change. In: Cohen SH, Reese HW, editors. Life-span developmental psychology: methodological contributions. LEA; Hillsdale, NJ: 1994. [Google Scholar]
- Meda SA, Koran ME, Pryweller JR, Vega JN, Thornton-Wells TA, Alzheimer’s Disease Neuroimaging Initiative Genetic interactions associated with 12-month atrophy in hippocampus and entorhinal cortex in Alzheimer’s Disease Neuroimaging Initiative. Neurobiol. Aging. 2013;34:1518.e9–1518.e18. doi: 10.1016/j.neurobiolaging.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisenzahl EM, Rujescu D, Kirner A, Giegling I, Kathmann N, Leinsinger G, Maag K, Hegerl U, Hahn K, Möller HJ. Association of an interleukin-1beta genetipolymorphism with altered brain structure in patients with schizophrenia. Am J Psychiatry. 2001;158:1316–1319. doi: 10.1176/appi.ajp.158.8.1316. [DOI] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR, Jr., Schuff N, Weiner MW, Thompson PM, Alzheimer’s Disease Neuroimaging Initiative Automated 3D mapping of hippocampal atrophy and its clinical correlates in 400 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. Hum Brain Mapp. 2009;30:2766–88. doi: 10.1002/hbm.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthén BO, Curran P. General longitudinal modeling of individual differences in experimental designs: a latent variable framework for analysis and power estimation. PsycholMethods. 1997;2:371–402. [Google Scholar]
- Oldfield RC. The assessment and analysis of handedness. Neuropsychologia. 1971;9:97–113. doi: 10.1016/0028-3932(71)90067-4. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Rosenbloom MJ, Mathalon DH, Lim KO. A controlled study of cortical gray matter and ventricular changes in alcoholic men over a five year interval. Arch Gen Psychiatry. 1998;55:905–912. doi: 10.1001/archpsyc.55.10.905. [DOI] [PubMed] [Google Scholar]
- Qiu LR, Germann J, Spring S, Alm C, Vousden DA, Palmert MR, Lerch JP. Hippocampal volumes differ across the mouse estrous cycle, can change within 24 hours, and associate with cognitive strategies. NeuroImage. 2013;83:593–598. doi: 10.1016/j.neuroimage.2013.06.074. [DOI] [PubMed] [Google Scholar]
- Radloff LS. The CES-D scale: a self-report depression scale for research in thegeneral population. Applied Psychological Measurement. 1977;1:385–401. [Google Scholar]
- Rajagopalan PI, Hua X, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. Homocysteine effects on brain volumes mapped in 732 elderly individuals. Neuroreport. 2011;22:391–395. doi: 10.1097/WNR.0b013e328346bf85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan PI, Jahanshad N, Stein JL, Hua X, Madsen SK, Kohannim O, Hibar DP, Toga AW, Jack CR, Jr, Saykin AJ, Green RC, Weiner MW, Bis JC, Kuller LH, Riverol M, Becker JT, Lopez OL, Thompson PM. Common folate gene variant, MTHFR C677T, is associated with brain structure in two independent cohorts of people with mild cognitive impairment. Neuroimage Clin. 2012;4:179–187. doi: 10.1016/j.nicl.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudenbush SW, Bryk AS. Hierarchical linear models. Sage; Thousand Oaks, CA: 2002. [Google Scholar]
- Raz N. Encyclopedia of Life Sciences. Nature Publishing Group; London: 2001. Ageing and the brain. [Google Scholar]
- Raz N, Ghisletta P, Rodrigue KM, Kennedy KM, Lindenberger U. Trajectories of brain aging in middle-aged and older adults: Regional and individual differences. NeuroImage. 2010;51:501–511. doi: 10.1016/j.neuroimage.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Gunning-Dixon F, Head D, Williamson A, Rodrigue K, Acker JD. Aging, sexual dimorphism, and hemispheric asymmetry of the cerebral cortex: replicability of regional differences in volume. Neurobiol Aging. 2004;25:377–396. doi: 10.1016/S0197-4580(03)00118-0. [DOI] [PubMed] [Google Scholar]
- Raz N, Kennedy KM. Pattern of normal age-related regional differences in white matter microstructure is modified by vascular risk. Brain Res. 2009;10:41–56. doi: 10.1016/j.brainres.2009.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Lindenberger U, Rodrigue KM, Kennedy KM, Head D, Williamson A, Dahle C, Gerstorf D, Acker JD. Regional brain changes in aging healthy adults: general trends, individual differences, and modifiers. Cereb Cortex. 2005;15:1676–1689. doi: 10.1093/cercor/bhi044. [DOI] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM. Differential aging of the brain: patterns, cognitive correlates and modifiers. Neurosci Biobehav Rev. 2006;30:730–748. doi: 10.1016/j.neubiorev.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Acker JD. Hypertension and the brain: vulnerability of the prefrontal regions and executive functions. Behav Neurosci. 2003a;117:1169–1180. doi: 10.1037/0735-7044.117.6.1169. [DOI] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Kennedy KM, Dahle C, Head D, Acker JD. Differential age-related changes in the regional metencephalic volumes in humans: A five-year follow-up. Neurosi Lett. 2003b;349:163–166. doi: 10.1016/s0304-3940(03)00820-6. [DOI] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Kennedy KM, Head D, Gunning-Dixon FM, Acker JD. Differential aging of the human striatum: Longitudinal evidence. AJNR Am J Neuroradiol. 2003c;24:1849–1856. [PMC free article] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Haacke EM. Brain aging and its modifiers: insights from in vivo neuromorphometry and susceptibility weighted imaging. Ann N Y Acad Sci. 2007a;1097:84–93. doi: 10.1196/annals.1379.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Kennedy KM, Acker JD. Vascular health and longitudinal changes in brain and cognition in middle-aged and older adults. Neuropsychology. 2007b;21:149–157. doi: 10.1037/0894-4105.21.2.149. [DOI] [PubMed] [Google Scholar]
- Raz N, Schmiedek F, Rodrigue KM, Kennedy KM, Lindenberger U, Lövdén M. Differential brain shrinkage over six months shows limited association with cognitive practice. Brain Cogn. 2013;82:171–180. doi: 10.1016/j.bandc.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Yang Y, Dahle CL, Land S. Volume of white matter hyperintensities in healthy adults: Contribution of age, vascular risk factors, and inflammation-related genetic variants. Biochim Biophys Acta. 2012;1822:361–369. doi: 10.1016/j.bbadis.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinvang I, Espeseth T, Westlye LT. APOE-related biomarker profiles in non-pathological aging and early phases of Alzheimer’s disease. Neurosci Biobehav Rev. 2013;37:1322–1335. doi: 10.1016/j.neubiorev.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Reitz C, Berger K, de Maat MP, Stoll M, Friedrichs F, Kardys I, Witteman JC, Breteler MM. CRP gene haplotypes, serum CRP, and cerebral small-vessel disease: the Rotterdam Scan Study and the MEMO Study. Stroke. 2007;38:2356–2359. doi: 10.1161/STROKEAHA.107.482661. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Pham DL, Kraut MA, Zonderman AB, Davatzikos C. Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J Neurosci. 2003;23:3295–3301. doi: 10.1523/JNEUROSCI.23-08-03295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses AD. Apolipoprotein E and Alzheimer’s disease. A rapidly expanding field with medical and epidemiological consequences. Ann N Y Acad Sci. 1996;802:50–57. doi: 10.1111/j.1749-6632.1996.tb32598.x. [DOI] [PubMed] [Google Scholar]
- Rowe JW, Kahn RL. Human aging: usual and successful. Science. 1987;10:143–149. doi: 10.1126/science.3299702. [DOI] [PubMed] [Google Scholar]
- Rubin DB. Inference and missing data. Biometrika. 1976;63:581–592. [Google Scholar]
- Satizabal CL, Zhu YC, Mazoyer B, Dufouil C, Tzourio C. Circulating IL-6 and CRP are associated with MRI findings in the elderly: the 3C-Dijon Study. Neurology. 2012;78:720–727. doi: 10.1212/WNL.0b013e318248e50f. [DOI] [PubMed] [Google Scholar]
- Satz P. Brain reserve capacity on symptom onset after brain injury: a formulation and review of evidence for threshold theory. Neuropsychology. 1993;7:273–295. [Google Scholar]
- Scahill RI, Frost C, Jenkins R, Whitwell JL, Rossor MN, Fox NC. A longitudinal study of brain volume changes in normal aging using serial registered magnetic resonance imaging. Arch Neurology. 2003;60:989–994. doi: 10.1001/archneur.60.7.989. [DOI] [PubMed] [Google Scholar]
- Schafer JL, Graham JW. Missing data: overview of the state of the art. Psychol Methods. 2002;7:147–177. [PubMed] [Google Scholar]
- Scott TM, Tucker KL, Bhadelia A, Benjamin B, Patz S, Bhadelia R, Liebson E, Price LL, Griffith J, Rosenberg I, Folstein MF. Homocysteine and B vitamins relate to brain volume and white-matter changes in geriatric patients with psychiatric disorders. Am J Geriatr Psychiatry. 2004;12:631–638. doi: 10.1176/appi.ajgp.12.6.631. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Wolf PA, Beiser AS, Selhub J, Au R, Jacques PF, Yoshita M, Rosenberg IH, D’Agostino RB, DeCarli C. Association of plasma total homocysteine levels with subclinical brain injury: cerebral volumes, white matter hyperintensity, and silent brain infarcts at volumetric magnetic resonance imaging in the Framingham Offspring Study. Arch Neurol. 2008 doi: 10.1001/archneur.65.5.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura T, Anan F, Masaki T, Umeno Y, Eshima N, Saikawa T, Yoshimatsu H, Fujiki M, Kobayashi H. Homocysteine levels are associated with hippocampus volume in type 2 diabetic patients. Eur J Clin Invest. 2011;417:751–758. doi: 10.1111/j.1365-2362.2010.02464.x. [DOI] [PubMed] [Google Scholar]
- Shrout PE, Fleiss JL. Intraclass correlations: uses in assessing raters reliability. Psychol Bull. 1979;86:420–428. doi: 10.1037//0033-2909.86.2.420. [DOI] [PubMed] [Google Scholar]
- Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc. 2002;8:448–460. [PubMed] [Google Scholar]
- Suzuki H, Sumiyoshi A, Taki Y, Matsumoto Y, Fukumoto Y, Kawashima R, Shimokawa H. Voxel-based morphometry and histological analysis for evaluating hippocampal damage in a rat model of cardiopulmonary resuscitation. NeuroImage. 2013;77:215–221. doi: 10.1016/j.neuroimage.2013.03.042. [DOI] [PubMed] [Google Scholar]
- Taki Y, Thyreau B, Kinomura S, Sato K, Goto R, Wu K, Kakizaki M, Tsuji I, Kawashima R, Fukuda H. Correlation between high-sensitivity C-reactive protein and brain gray matter volume in healthy elderly subjects. Hum Brain Mapp. 2013;34:2418–2424. doi: 10.1002/hbm.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamnes CK, Walhovd KB, Dale AM, Østby Y, Grydeland H, Richardson G, Westlye LT, Roddey JC, Hagler DJ, Jr, Due-Tønnessen P, Holland D, Fjell AM. Brain development and aging: overlapping and unique patterns of change Alzheimer’s Disease Neuroimaging Initiative. NeuroImage. 2013;68:63–74. doi: 10.1016/j.neuroimage.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Whitman GT, Lopez I, Baloh RW. Brain volume changes on longitudinal magnetic resonance imaging in normal older people. J Neuroimaging. 2001;11:393–400. doi: 10.1111/j.1552-6569.2001.tb00068.x. [DOI] [PubMed] [Google Scholar]
- Van Dijk EJ, Prins ND, Vermeer SE, Vrooman HA, Hofman, Koudstaal APJ, Breteler MMB. C-reactive protein and cerebral small-vessel disease: the Rotterdam scan study. Circulation. 2005;112:900–905. doi: 10.1161/CIRCULATIONAHA.104.506337. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Crum WR, Lerch JP, Chege W, Natesan S, Modo M, Cooper JD, Williams SC, Kapur S. Reduced cortical volume and elevated astrocyte density in rats chronically treated with antipsychotic drugs-linking Magnetic Resonance Imaging findings to cellular pathology. Biol Psychiatry. Biol Psychiatry. 2014;2014:75, 982–990. doi: 10.1016/j.biopsych.2013.09.012. [DOI] [PubMed] [Google Scholar]
- West RL. An application of prefrontal cortex function theory to cognitive aging. Psycholl Bull. 1996;120:272–292. doi: 10.1037/0033-2909.120.2.272. [DOI] [PubMed] [Google Scholar]
- West SG. New approaches to missing data in psychological research: Introduction to the special section. PsycholMethods. 2001;6:315–316. doi: 10.1037/1082-989x.6.4.315. [DOI] [PubMed] [Google Scholar]
- Williams JH, Pereira EA, Budge MM, Bradley KM. Minimal hippocampal width relates to plasma homocysteine in community-dwelling older people. Age Ageing. 2002;31(6):440–444. doi: 10.1093/ageing/31.6.440. [DOI] [PubMed] [Google Scholar]
- Wilson C-J, Finch CE, Cohen HJ. Cytokines and Cognition-The Case for A Head-to-Toe Inflammatory Paradigm. JAGS. 2002;50:2041–2056. doi: 10.1046/j.1532-5415.2002.50619.x. [DOI] [PubMed] [Google Scholar]
- Yarlagadda A, Alfson E, Clayton AH. The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry (Edgmont) 2009;6:18–22. [PMC free article] [PubMed] [Google Scholar]
- Yau PL, Kluger A, Borod JC, Convit A. Neural substrates of verbal memory impairments in adults with type 2 diabetes mellitus. J Clin Exp Neuropsychol. 2014;36:74–87. doi: 10.1080/13803395.2013.869310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yan H, Tian L, Wang F, Lu T, Wang L, Yan J, Liu Q, Kang L, Ruan Y, Zhang D, Yue W. Association of MTHFR C677T polymorphism with schizophrenia and its effect on episodic memory and gray matter density in patients. Behav. Brain Res. 2013;243:146–152. doi: 10.1016/j.bbr.2012.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.