

Abstract
Mitochondria-generated reactive oxygen species (ROS) play a crucial role in the pathogenesis of aging and age-associated diseases. In this study, we evaluated the effects of XJB-5-131 (XJB), a mitochondria-targeted ROS and electron scavenger, on cardiac resistance to ischemia-reperfusion (IR)-induced oxidative stress in aged rats. Male adult (5-month old, n=17) and aged (29-month old, n=19) Fischer Brown Norway (F344/BN) rats were randomly assigned to the following groups: adult (A), adult+XJB (AX), aged (O), and aged+XJB (OX). XJB was administered 3 times per week (3 mg/kg body weight, IP) for four weeks. At the end of the treatment period, cardiac function was continuously monitored in excised hearts using the Langendorff technique for 30 min, followed by 20-min of global ischemia, and 60-min reperfusion. XJB improved post-ischemic recovery of aged hearts, as evidenced by greater left ventricular developed-pressures and rate-pressure products than the untreated, aged-matched group. The state 3 respiration rates at complexes I, II and IV of mitochondria isolated from XJB-treated aged hearts were 57% (P<0.05), 25% (P<0.05) and 28% (P<0.05), respectively, higher than controls. Ca2+-induced swelling, an indicator of permeability transition pore opening, was reduced in mitochondria of XJB-treated aged rats. In addition, XJB significantly attenuated the H2O2-induced depolarization of the mitochondrial inner membrane as well as total and mitochondrial ROS levels in cultured cardiomyocytes. This study underlines the importance of mitochondrial ROS in aging-induced cardiac dysfunction and suggests that targeting mitochondrial ROS may be an effective therapeutic approach to protect the aged heart against IR injury.
Keywords: heart, aging, ischemia/reperfusion, mitochondria, ROS scavenger, XJB-5-131
1. Introduction
Aging is associated with increased prevalence of cardiovascular diseases. In particular, myocardial infarction is a major cause of chronic disability and mortality in the elderly [1]. Aged hearts exhibit low tolerance to various forms of stress, including oxidative stress induced by ischemia/reperfusion (IR). Aged animals [2–4] and elderly patients [5, 6] are more prone to myocardial damage following IR than their younger adult counterparts, although the precise mechanisms underlying this decreased tolerance are not fully understood. Existing cardioprotective strategies confer minimal or no protection against IR injury in aged hearts. For instance, although ischemic preconditioning with brief periods of IR protects the adult heart from damage induced by subsequent prolonged ischemia [7], this protection is remarkably reduced in aged rodent [8–10] and human hearts [11]. Likewise, neither pharmacological preconditioning, induced by activation of mitochondrial KATP channels [12] and stimulation of adenosine receptors [12, 13], nor anesthetic preconditioning [14, 15] provides protection or reduces infarct size in aged animals. The mechanisms underlying this loss of cardioprotection likely involve 1) inadequate energy metabolism, including impaired oxidative phosphorylation and energy transfer, 2) disruption of redox status and generation of highly reactive oxygen species (ROS), and 3) alterations of protective signaling pathways in cardiac cells. Thus, existing studies highlight the importance of understanding the molecular mechanisms underlying the loss of cardioprotection and identifying new intracellular targets for cardioprotection in the aged heart.
A major contributing factor in the pathogenesis of an aged heart is mitochondrial dysfunction. Indeed, mitochondria are common targets and effectors in cardiac injury of aging hearts [16–18]. The free radical theory of aging implicates ROS as a major factor in senescent-induced cell damage [19]. Many studies have established mitochondria as predominant sources of ROS by demonstrating that diminished electron transport chain (ETC) activity and reduced antioxidant capacity enhance ROS production in aged hearts (reviewed in [17]). Accordingly, high ROS levels alter mitochondrial function and integrity, and increase the number of giant mitochondria in aged hearts due to the reduced effectiveness of mitophagy. Giant mitochondria contain mutated DNA that encodes mutated proteins for ETC complexes, thereby leading to further amplification of mitochondrial ROS production [20, 21]. The role of mitochondrial DNA (mtDNA) mutations in cardiac aging was confirmed in transgenic mice that expressed a proof-reading-deficient version of mtDNA polymerase with a high load of mtDNA mutations and deletions [20]. This increase in mtDNA mutations was associated with a reduced lifespan and the development of aging-related symptoms between 25–40 weeks of age. On the other hand, mice with overexpression of mitochondrial matrix-targeted catalase demonstrated less cardiac mtDNA mutations and protein carbonylation than age-matched wild-type controls. These mice were characterized by an increased lifespan and delayed cardiac aging, suggesting that aging-related mitochondrial and cardiac abnormalities are partially rescued by catalase overexpression in mitochondria [22]. In addition to elevated ROS, aged hearts exhibit diminished mitochondrial Ca2+ handling and increased Ca2+-induced mitochondrial damage, associated with increased calcium vulnerability of senescent cardiac mitochondria [23]. Apparently, the decreased ability to accumulate and retain Ca2+ reduces tolerance of the aged myocardium to IR injury. The increased mitochondrial ROS generation, along with altered Ca2+ handling and energy metabolism (ATP synthesis and energy transfer), predisposes the aging heart to the induction of mitochondrial permeability transition pore (PTP) opening and, thereby, stimulates mitochondria-mediated cell death [24].
Thus, mitochondrial ROS (mitoROS) play a central role in the pathogenesis of cardiac IR injury and senescence. Moreover, cardioprotective signaling pathways that converge on mitochondria are less effective in aged hearts. These observations emphasize the importance of developing and testing new compounds that directly target mitochondria to delay the aging process and increase the tolerance of the aged heart to IR injury. mitoROS scavengers are the most promising pharmacological agents to protect the organelle against ROS induced damage. A recently developed analog of the antibiotic gramicidin S, XJB-5-131 (XJB) [25], has been shown to target mitochondria and provide mitoROS and electron scavenging capacity by virtue of its conjugation to a 4-amino-2,2,6,6-tetramethylpiperidinooxy (4-Amino-TEMPO) moiety [26]. Previous in vitro studies demonstrated that XJB reduced apoptosis and enhanced cell survival in mouse embryonic cells [25]. Further, it attenuated the disease phenotype and improved mitochondrial function in a mouse model of Huntington’s disease [27]. XJB has also been shown to prevent ileal mucosal barrier dysfunction and tissue ischemia in a rat model of hemorrhagic shock. Intravenous administration of the compound significantly prolonged the survival of rats subjected to a large blood loss [28].
The antioxidant effects of XJB are likely to occur through the scavenging of electrons leaking from uncoordinated electron carriers, rather than being caused by SOD-like activity [29]. The one-electron reduction of the nitroxide at the XJB molecule generates a hydroxylamine, which can undergo a two-electron oxidation to the nitroxonium cation (Fig. 1). Single-electron reduction of the nitroxonium cation regenerates the nitroxide, and therefore all of these N-O oxidation states are in an equilibrium determined by the redox state of the environment [30]. In a conversion resembling that of singlet oxygen dismutase (SOD), superoxide radical anion as well as reactive nitrogen species can react with both nitroxide and hydroxylamine species, generating hydroxylamine and nitroxide radical, respectively (Fig. 1, Eq. (a) and (b)) [31]. The nitroxide radical is also a superb single-electron acceptor and both the resulting hydroxylamine as well as the parent nitroxide can quench radicals originating from hydrogen atom abstraction by superoxide, among other pathways (Fig. 1, Eq. (c)–(f)) [32, 33].
Figure 1.

Structure of XJB-5-131, equilibria of nitroxide oxidation states, and pertinent reactions with reactive oxygen species, electrons, and radicals. See 2.2. XJB: mitoROS and electron scavenging capacity for details.
The mitochondrial targeting properties of XJB were assessed by fluorescence imaging, EPR and MS analyses. Within 1 h of incubation, BODIPY-FL-XJB, a derivative of XJB labeled with the fluorescent (FL) boron-di-pyrromethene (BODIPY), crossed the plasma membrane of primary striatal neurons and stained mitochondria, as verified by co-staining with MitoTracker Deep Red [27]. A quantitative determination of integration efficiency was possible with electron spin resonance (ESR) spectroscopy and electrospray-ionization mass spectrometry (ESI-MS). Both methods independently suggested a very high level of mitochondrial enrichment of XJB, up to 600-fold vs the cytosolic fraction [29, 34]. XJB associates closely with the mitochondria-specific phospholipid cardiolipin and is very efficient in preventing its oxidation [29]. While the mitochondrial membrane targeting sequence of the neutral, acyclic XJB is conceptually derived from the cationic, antimicrobial cyclodecapeptide gramicidin S (GS), it does not permeabilize cell membranes or have antibacterial activity, which is not surprising since GS-like cell lysis requires a relatively rigid β-sheet conformation that positions cationic and hydrophobic side chains on opposite faces [35].
In the present study, we determined that XJB exerted cardioprotective effects against IR in adult and aged rats. The results demonstrated that hearts from aged animals treated with XJB showed increased resistance to IR injury, as evidenced by improved post-ischemic recovery of cardiac function and reduced lactate dehydrogenase (LDH) activity in the coronary effluent during IR. In addition, XJB-treated aged rats exhibited higher mitochondrial respiration rates than their untreated, age-matched counterparts.
2. Materials and Methods
2.1. Animals
Male adult (5-month old, n=17) and aged (29-month old, n=19) Fischer Brown Norway (F344/BN) rats were purchased from Charles River (NIA NIH colony at Charles River, Kingston, NY). All experiments were performed according to the protocol approved by the University Animal Care and Use Committee and conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
2.2. Experimental protocols
Animals were randomly assigned to the following four groups: adult (A), adult+XJB (AX), aged (O), and aged+XJB (OX). XJB was dissolved in 0.5 mL of sunflower seed oil (Sigma-Aldrich, St. Louis, MO, USA) and administered 3 times per week (3 mg/kg body weight, IP) for four weeks. Rats in untreated groups (A and O) were injected an equal volume of sunflower without XJB. The body weights of the animals were measured weekly. After four weeks of treatment, animals were sacrificed and the heart was isolated for ex-vivo perfusion.
2.3. Perfusion of isolated rat hearts and ex-vivo model of IR
Isolated hearts were perfused by the Langendorff mode to determine cardiac function [36]. A water-filled latex balloon was inserted into the left ventricle for continuous monitoring of heart rate (HR), left ventricular systolic (LVSP) and end diastolic (LVEDP) pressure, and maximum velocity of contraction (dP/dtmax) and maximum velocity of relaxation (dP/dtmin). All determinations of ventricular performance were obtained using Labscribe2 Data Acquisition Software (iWorx 308T, Dover, NH, USA). Left ventricular developed pressure (LVDP) was calculated as the difference between LVSP and LVEDP (LVDP=LVSP-LVEDP). Cardiac work was estimated by the rate-pressure product (RPP) calculated as RPP=LVDP x HR.
To induce IR, after an equilibration period of 30 min, hearts from all four groups were subjected to 20 min of global normothermic ischemia followed by 60 min of reperfusion. Samples of the effluent were collected prior to ischemia and during reperfusion at indicated time points to measure the activity of LDH [37].
For infarct size measurements, the hearts underwent the corresponding perfusion protocols per group evaluated and were then cut transversely into 1.5-mm-thick slices. Thereafter, slices were stained with 1.0% 2,3,5-triphenyltetrazolium chloride (TTC) at 37°C for 20 min, fixed in 10% neutral buffered formalin [38], and were then mounted on glass plates (1.5 mm thickness) for digital photography. The digital images were analyzed for infarct size (TTC negative) using the NIH ImageJ software. Results were expressed as infarcted area/total area × 100.
2.4. Isolation of mitochondria
Mitochondria were isolated from perfused hearts following IR as described previously [36]. Briefly, the ventricles were cut, weighed and homogenized with a Polytron homogenizer in ice-cold sucrose buffer containing 300 mM sucrose, 10 mM Tris-HCl, and 2 mM EGTA; pH 7.4. Mitochondria were isolated from the homogenate by centrifugation at 2,000g for 2 min to remove cell debris, followed by centrifugation of the supernatant at 10,000g for 5 min. The pellet was then washed two times at 10,000g for 5 min. The final pellet containing mitochondria was used for the measurement of respiratory function, protein carbonylation, and mitochondrial PTP opening.
2.5. Respiratory function of mitochondria
Rates of oxygen consumption in mitochondria isolated from hearts following IR were determined at 30 °C using a YSI Oxygraph (Yellow Springs, OH) model 5300 equipped with a Clark-type oxygen electrode [36]. Briefly, mitochondria were suspended in a buffer containing (in mM): 125 KCl, 20 MOPS, 10 Tris, 0.5 EGTA, and 2 KH2PO4, pH 7.2, supplemented with 2.5 mM 2-oxoglutarate and 1 mM L-malate (complex I) or 2.5 mM succinate and 1 μM rotenone (complex II). Respiration rates were measured in the absence (state 2) and presence (state 3) of 1 mM ADP. To measure state 3 for complex IV, complex III was blocked with 0.5 μM antimycin A, and 10 mM ascorbate and 0.3 mM N,N,N′,N′-tetramethyl-p-phenylendiamine (TMPD) were added in the presence of 1 mM ADP. Citrate synthase activity was determined spectrophotometrically at 412 nm [36].
2.6. Mitochondrial PTP opening
Swelling of de-energized mitochondria as an indicator of PTP opening in the presence or absence of Ca2+ was determined by monitoring the decrease in light scattering at 545 nm as described previously [39]. Mitochondria isolated from hearts following IR were incubated at 37 °C in 1 mL buffer containing 0.2 M sucrose; 10 mM Tris-MOPS, pH 7.4; 5 mM succinate; 1 mM Pi; 2 μM rotenone; 10 μM EGTA-Tris. Mitochondria containing ~0.5 mg of protein were added, and absorbance was monitored in the presence and absence of 100 μM CaCl2.
2.7. Protein carbonylation assay
Protein carbonyls were analyzed as described previously [40]. Briefly, an aliquot of the mitochondrial protein was derivatized with dinitrophenylhydrazine (DNPH, Sigma-Aldrich, St. Louis, MO, USA) under acid denaturing conditions. Proteins were separated by SDS-PAGE and subjected to western blotting with anti-dinitrophenyl primary antibodies (Sigma-Aldrich, St. Louis, MO, USA) at 1:1000 dilution. In order to correct for non-specific binding of antibodies, separate aliquots of the mitochondrial proteins that had been acid-denatured but not treated with DNPH were run in parallel. Blots were scanned and carbonylation was determined as the sum of all band intensities for each track after subtraction of non-specific background signal.
2.8. Isolation and culture of adult rat ventricular cardiomyocytes
Calcium-tolerant adult rat ventricular cardiomyocytes were isolated using a previously described method [41] with minor modifications. Briefly, hearts removed from male Sprague-Dawley rats (225–275 g, Charles River, Wilmington, MA, USA) were retrogradely perfused with a Ca2+-free buffer, and then perfusion was continued with a buffer containing collagenase (Worthington Biochemical, Lakewood, NJ, USA) and proteases. At the end of perfusion, the ventricles were removed, minced into small pieces, and filtered through a nylon mesh. Calcium reintroduction in a stepwise fashion (up to 1 mM) was performed in a 100-mm sterile dish at room temperature. Cells were pre-plated on laminin-coated dishes in culture medium (Life technologies) supplemented with 5% serum, 10 mM 2,3-butanedione monoxime (BDM), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, and incubated in a humidified incubator with 95% air and 5% CO2 at 37°C for 2 h. After the pre-plating period, the medium was replaced with a new medium containing 10 mM BSA, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, without serum and BDM. The viability of cardiomyocytes was greater than 95% of the final cell population. The next day, the cells were used for cell viability and mitochondrial assessment experiments. Cell viability was determined by the trypan blue exclusion assay using a TC20 Automated Cell Counter (Bio-Rad, Hercules, CA).
2.9. Oxidative stress in H9c2 cells and adult rat cardiomyocytes
In adult cardiomyocytes, oxidative stress was induced by the addition of 300 μM H2O2 for 30 min. XJB at concentrations of 2.5, 5, 10 or 20 μM was added to the culture media 1 h before H2O2. H9c2 embryonic rat cardiomyocytes were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM complete cell culture medium (Invitrogen, Carlsbad, CA), supplemented with 10% FBS, 1% penicillin and streptomycin (Hyclone, Thermo Scientific), and maintained in 95% air and 5% CO2 at 37 °C according to the manufacturer’s protocol. Prior to all experiments, cells were serum-starved for 24 h. XJB was added to the culture media 1 h before the induction of oxidative stress in H9c2 cells by 75 μM H2O2, 10 μM dimethoxynaphthoquinone (DMNQ, ROS donor), or 1 μM antimycin A (inhibitor of complex III).
2.10. Mitochondrial membrane potential (ΔΨm) assay
To monitor ΔΨm, H9c2 cells and adult rat cardiomyocytes plated in a 24-well culture plate were incubated for 30 min with the membrane potential-sensitive dye JC-1 (10 μg/mL, 5,5′,6,6′-tetraethyl-benzimidazolylcarbocyanine iodide, Molecular Probes) [42]. The intensity of fluorescence was measured using a Spectramax M3 microplate reader (Molecular Devices) at 527 nm and 590 nm for emission and 488 nm for excitation. Data were calculated as the ratio of red fluorescent intensity (“J-aggregates”, indicator of high ΔΨm) to green fluorescent intensity (“JC-1 monomers”, indicator of low ΔΨm), and presented in arbitrary units.
2.11. Total ROS and mitoROS assays
H9c2 cells were trypsinized with a 0.25% trypsin-EDTA solution (Hyclone) and centrifuged at 1,000 rpm for 5 min at room temperature. For measurement of total ROS, the pellet was resuspended in culture medium, and cells were incubated with 2′,7′-dichlorofluorescein diacetate (DCF-DA; 20 μM, Alexis Biochemicals) for 30 min at 37 °C. To quantify mitoROS, cells (2×105 cells/well) in the 96-well culture plate were incubated with the mitochondrial superoxide-sensitive fluorescent dye MitoSOX Red (1 μM, Invitrogen, Carlsbad CA) for 20 min at 37 °C [42]. Fluorescence intensity was measured using a Spectramax M3 plate reader (Molecular Devices, Sunnyvale, CA) at excitation/emission of 485 nm/530 nm for DCF-DA and 510 nm/580 nm for MitoSOX Red. Antioxidant effects of XJB were evaluated in cells treated with 10 μM DMNQ or 1 μM antimycin A.
2.12. Statistical analysis
Statistical analysis was performed using Sigmaplot 12.5 (Systat Software Inc, San Jose, CA). Differences among multiple groups were assessed using two-way ANOVA for the two factors, age and treatment. One-way repeated measures ANOVA was used to analyze differences between time-points for cardiac function and LDH activity at reperfusion. Differences between treated and untreated groups in in vitro studies were determined by 2-tailed Student’s t-test. Data are presented as means±SEM of 8–10 rats per group. Differences were considered to be statistically significant when P<0.05.
3. Results
3.1. Mitochondria-targeted ROS scavenging improves cardiac function after global IR in aged rat hearts
Analysis of gravimetric parameters showed that XJB did not affect the body weight (BW) or heart weight (HW) of either adult or aged rats (Fig. 2A,B). Consequently, the HW to BW ratio remained unchanged in all groups (Fig. 2C). No animal died during the treatment.
Figure 2.
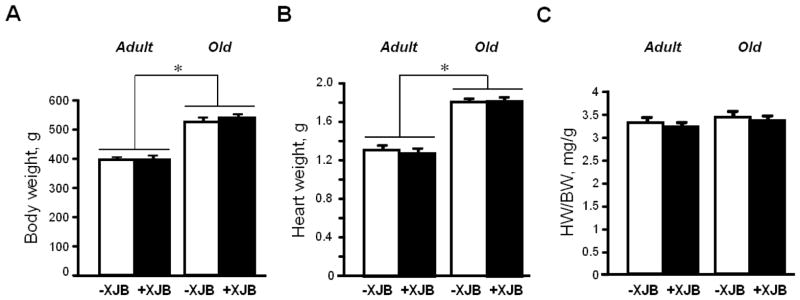
Gravimetric parameters of adult and aged rats. A, Body weight (BW); B, Heart weight (HW); and C, HW/BW. *P<0.01 O and OX vs. A and AX, respectively.
We subjected the hearts of adult and aged rats to ex-vivo IR using the Langendorff-mode perfusion technique to determine whether XJB enhanced the tolerance of adult and aged hearts to oxidative stress. There was no difference between the 4 groups with regard to pre-ischemic values of cardiac function (Table 1). However, IR exerted distinct effects on the untreated hearts of adult and aged rats; untreated aged rats demonstrated 56% (P<0.001) and 54% (P<0.001) less recovery of LVDP and RPP, respectively, than their adult counterparts after 60 min of reperfusion (Fig. 3). Maximal contraction (dP/dtmax) and relaxation (dP/dtmin) velocities upon reperfusion were significantly lower in untreated aged rats than adult rats (Table 1). XJB had no significant effects on post-ischemic LVDP and RPP in adult rats. However, XJB-treated aged rats demonstrated a 2.40-fold (P<0.05) and 1.95-fold (P<0.05) higher recovery of LVDP and RPP, respectively, than their untreated counterparts at the end of reperfusion (Fig. 3A,C). No significant differences in LVSP measurements with XJB were observed between adult and aged rats, suggesting that the increased LVDP, dP/dt and RPP observed in aged rats with XJB result from improvements in diastolic function. Interestingly, the hearts of XJB-treated aged rats developed twofold less ischemic contracture than untreated animals (O: 6.9±1.7 mmHg; OX: 3.2±1.6 mmHg; P<0.05). It is likely that post-ischemic, diastolic dysfunction develops secondary to calcium-induced contracture because both of these processes are XJB-sensitive.
Table 1.
The effects of global IR on hemodynamic parameters of Langendorff-perfused hearts.*
| Parameters | Pre-ischemia | Ischemia | Reperfusion | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | A+XJB | O | O + XJB | A | A+XJB | O | O+ XJB | A | A+XJB | O | O + XJB | |
| LVSP, mmHg | 79.7±6.6 | 75.5±5.7 | 79.9±6.3 | 80.4±4.1 | 71.9±5.8 | 77.8±2.9 | 70.3±7.8 | 63.2±6.4 | ||||
| LVEDP, mmHg | 3.8±1.6 | 4.2±1.4 | 3.5±1.9 | 3.4±3.1 | 49.7±5.6 | 52.9±3.3 | 62.5±4.8 | 39.8±5.0+ | ||||
| LVDP, mmHg | 80.3±7.4 | 75.9±5.7 | 76.4±4.9 | 81.5±8.1 | 21.5±1.6 | 25.6±2.7 | 9.4±1.9++ | 22.6±4.4* | ||||
| HR, beats/min | 241±14 | 226±19 | 235±13 | 213±12 | 250±7 | 256±7 | 230±7 | 219±10& | ||||
| RPP(LVDPxHR), mmHgxbeats/sec | 329±42 | 282±26 | 296±14 | 288±30 | 90±8 | 108±9 | 41±9++ | 80±16* | ||||
| dP/dtmax, mmHg/sec | 1628±149 | 1522±110 | 1546±76 | 1673±163 | 432±31 | 523±60 | 148±38+ | 412±78* | ||||
| dP/dtmin, mmHg/sec | 1104±128 | 992±96 | 1005±53 | 1069±132 | 308±25 | 369±41 | 156±22++ | 295±47* | ||||
| Ischemic contracture, mmHg | 8.8±2.0 | 7.2±0.9 | 6.9±1.7 | 3.2±1.6*& | ||||||||
Results shown in the table present the values of indicated parameters at the end of the pre-ischemia, ischemia and reperfusion periods.
P<0.01;
P<0.001 O vs. A;
P<0.05 OX vs. O;
P<0.05 OX vs. AX
Figure 3.


Post-ischemic recovery of cardiac function, and LDH activity in the coronary effluent of adult and aged rats. A, LVDP at the end (at 60 min) of reperfusion, shown as percent of pre-ischemia values; B, LVEDP at the end (at 60 min) of reperfusion, given in mmHg; C, RPP recovery during the reperfusion period, shown as percent of pre-ischemia values; D, LDH activity in the coronary effluent released during the reperfusion period, shown in munits/mL per gram heart weight (HW); E, Myocardial infarction size after IR injury. Upper panel: typical cross-sections of hearts stained with TTC for infarct (white) and viable tissue (red). Lower panel: Quantitative data of infarction size expressed in percent of total area (whole heart). All data are shown as means ± SE. n=2–3 per group for infarction size experiments. *P<0.05 OX vs. O; +P<0.05, ++P<0.01 O vs. A.
Analysis of LDH revealed a significant difference between untreated adult and aged hearts during the first 10 min of reperfusion (Fig. 3D). Surprisingly, LDH activity in the coronary effluent released from aged hearts was lower than the effluent from adult hearts by 43% (P<0.05), 38% (P<0.001) and 29% (P<0.001) at 1, 3, and 5 min of reperfusion, respectively. However, there were no significant differences in LDH activity between adult and aged hearts from 20 min until the end of reperfusion. Both adult and aged XJB-treated rats exhibited significantly lower LDH activity in the coronary effluent than their age-matched counterparts at 40, 50 and 60 min of reperfusion. Pre-ischemic values of LDH were 2.18±0.48, 2.14±0.38, 1.24±0.23, and 0.89±0.19 mU/ml per g heart weight for A, AX, O, and OX groups, respectively. There were no differences on infarction size between groups (Fig. 3E, see. 5. Limitations of the study).
These data demonstrate that mitochondria-targeted ROS scavenging by XJB exerted cardioprotective effects and remarkably improved post-ischemic recovery of cardiac function but did not affect myocardial infarction size in aged hearts.
3.2. XJB treatment attenuates respiratory dysfunction and prevents PTP opening in mitochondria of aged rats
Following IR, cardiac mitochondria were isolated and utilized to determine oxygen consumption rates and mitochondrial PTP opening. Analysis of mitochondrial respiration rates, including state 2, state 3 and RCI for complexes I, II and IV, revealed no differences between untreated adult and aged hearts (Fig. 4). XJB failed to improve the mitochondrial respiratory function of adult rats. No difference was observed between treated and untreated adult groups with regard to state 3 for complexes I and II. However, treatment promoted a 32% (P<0.05) increase of state 3 for complex IV in adult rats. The effects of XJB were more consistent in aged rats. Treated, aged mitochondria demonstrated a 57%, 19% and 19% (P<0.05 for all) higher state 3 for complexes I, II and IV substrate, respectively, compared to untreated, aged mitochondria. These results for state 3 in XJB-treated groups were statistically significant even after normalization of respiration rates to citrate synthase activity. These data suggest that the differences between treated and untreated aged rats were not due to a loss of fragile mitochondria by homogenization and centrifugation during the isolation procedure. Activity of citrate synthase, a marker of mitochondrial mass (abundance), was 14% (P<0.01) lower in aged mitochondria than in adults (Fig. 5). Treatment with XJB did not affect this parameter. Similarly, no significant differences between groups with regard to the P/O ratio (Fig. 4F) were observed.
Figure 4.

Respiratory rates of cardiac mitochondria isolated from adult and aged rats. A, B, C, State 3 respiration rates for ETC complexes I, II and IV, respectively, measured in the presence 1 mM ADP in a buffer containing 2.5 mM 2-oxoglutarate and 1 mM L-malate (complex I), 2.5 mM succinate and 1 μM rotenone (complex II) or 0.5 μM antimycin A, 10 mM ascorbate and 0.3 mM TMPD (complex IV). Data are given in nmol O/min per mg of mitochondrial protein. The solubility of oxygen in the buffer was 230 nmol of oxygen per mL of water. D, E, RCI for complexes I and II calculated as the ratio of state 3 to state 2. F, P/O ratio for complex I calculated as the ratio of ATP produced per consumed oxygen. *P<0.05 OX vs. O. &P<0.05, &&P<0.01 OX vs. AX, +P<0.05 O vs. A.
Figure 5.

Citrate synthase activity in mitochondria of adult and aged rats. +P<0.01 O vs. A; &P<0.05 OX vs. AX.
Next, we determined the sensitivity of mitochondria to Ca2+-induced swelling and used it as an indicator of in vitro PTP opening. Mitochondria of aged hearts exhibited high vulnerability to PTP opening and had a 22% (P<0.05) higher Ca2+-induced swelling rate than mitochondria in adult hearts (Fig. 6A,B). Mitochondria isolated from the XJB-treated hearts of adult and aged rats demonstrated 10% (P<0.05) and 14% (P<0.05) less swelling, respectively, indicating that XJB was an effective treatment in aged hearts. In addition, mitochondria isolated from XJB-treated young and old rats exhibited a trend toward low protein carbonylation, a marker of protein oxidation, in response to oxidative stress (Fig. 6C,D).
Figure 6.

Mitochondrial PTP opening and protein carbonylation levels in the hearts of adult and aged rats. A,B, PTP opening was determined by monitoring the Ca2+-induced decrease of light scattering at 545 nm in de-energized mitochondria. Representative curves (A), and quantitative data (B) shown as a ratio of optical density (ΔA) values in the presence or absence of Ca2+. C, D, Protein carbonylation determined by western blotting using anti-dinitrophenyl antibodies. Representative immunoblots (C), and quantitative data (D) presented as the sum of all band intensities for each track after subtraction of non-specific background signal. DNPH, dinitrophenylhydrazine. *P<0.05 AX and OX vs. A and O, respectively; +P<0.05 O vs. A.
Overall, these data reveal no significant differences between adult hearts and aged hearts with regard to mitochondrial respiratory function following IR. However, XJB treatment improved mitochondrial respiratory function and inhibited PTP opening in aged rats.
3.3. The beneficial effects of XJB on oxidative stress-induced mitochondrial dysfunction of cultured cardiomyocytes are associated with abrogation of ROS production
In order to establish whether XJB exerts a direct effect on cardiomyocytes and prevents oxidative stress-induced mitochondrial dysfunction, cultured H9c2 cells were subjected to H2O2 in the presence or absence of XJB. As shown in Fig. 7A, XJB alone, at concentrations of 2.5, 5 and 10 μM, had no effect on Δψmit in control cells. Oxidative stress depolarized the mitochondrial inner membrane (IMM) as evidenced by a reduction in Δψmit.(Fig. 7B). However, pre-treatment of the cells with 10 μM XJB significantly attenuated the H2O2-induced depolarization. In addition, the ROS inducer DMNQ and the complex III inhibitor, antimycin A were tested as a positive control. As expected, exposure to either antimycin A or DMNQ elevated total ROS and mitoROS in cells. In both scenarios, pre-treatment with XJB significantly reduced total ROS (Fig. 7C), and mitoROS (Fig. 7D) levels. In addition, XJB significantly increased cell survival in adult rat cardiomyocytes exposed to oxidative stress, and this effect was associated with attenuation of mitochondrial membrane depolarization (Fig. 8A,B). Notably, similar to H9c2 cells (Fig. 7A), XJB had no effect on Δψmit in mitochondria isolated from healthy adult rat heart (Fig. 8C). Taken together, these data demonstrate that XJB exerts beneficial effects against oxidative stress in cultured cardiomyocytes and prevents cell death and mitochondrial dysfunction.
Figure 7.

The effects of XJB on mitochondrial membrane potential and total ROS levels in H9c2 cells. A, Different concentrations of XJB on untreated (C, control) cells; B, Cells treated with 75 μM H2O2 in the presence of 10 μM XJB. Data represent the ratio of fluorescent intensity of “J-aggregates” (high ΔΨm) to fluorescent intensity of “JC-1 monomers” (low ΔΨm) as an indicator of healthy mitochondria. C, D, Total ROS and mitoROS levels in cells treated with 1 μM antimycin A (Ant) or 10 μM dimethoxynaphthoquinone (DMNQ) in the presence or absence of 10 μM XJB. Total ROS and mitoROS levels were determined using the fluorescence dyes DCF-DA and MitoSOX Red, respectively. Data are given in fluorescence intensity per 10000 cells. n=4–6 per group. *P<0.05, **P<0.01 H2O2, Ant or DMNQ vs. C; +P<0.05, ++P<0.01 H2O2+XJB, Ant+XJB and DMNQ+XJB vs. H2O2, Ant and DMNQ, respectively.
Figure 8.

The effects of XJB on cell survival and mitochondrial membrane potential in adult rat cardiomyocytes. Cell survival (A) and mitochondrial membrane potential (B) were assessed in cultured cardiomyocytes isolated from healthy (control) adult rats. The cells were exposed to 300 μM H2O2 in the presence and absence of XJB. Cell viability was assessed by the trypan blue exclusion method, and JC-1 was used to quantify the mitochondrial membrane potential. Direct effects of XJB on state 3 respiration rate (C) in mitochondria isolated from healthy adult rats. n=4–6 per group. *P<0.05, **P<0.01 vs. control (C); +P<0.05, ++P<0.01 vs. H2O2 (not treated with XJB).
4. Discussion
The present study is the first to demonstrate that i) the mitochondria-targeted ROS scavenger, XJB, enhances cardiac tolerance to oxidative stress in aged rats and, as such, significantly improves post-ischemic recovery of cardiac function, ii) following IR in XJB-treated aged rats, cardiac mitochondria exhibit a high state 3 respiration rate for complexes I, II and IV, iii) XJB inhibits PTP opening in aged hearts subjected to IR, as evidenced by the reduction of mitochondrial Ca2+-induced swelling, and iv) in cultured cardiomyocytes, XJB attenuates oxidative stress-induced loss of δψmit and, total and mitoROS production. Our data on reduced post-ischemic recovery of cardiac function in aged rats are consistent with previous studies obtained on rodents [2–4], with the exception of guinea pigs [43]. Hearts from old (52 weeks) guinea pigs were better preserved and demonstrated greater recovery and lower infarct size than young (4 weeks) counterparts following IR. Interestingly, despite a higher cytoplasmic [Ca2+], aged guinea pig hearts contained significantly less mitochondrial [Ca2+] than young hearts [43]. Most likely, reduced [Ca2+] uptake in cardiac mitochondria of aged guinea pigs plays a key role in the enhanced resistance of the hearts to oxidative stress. In contrast to the human heart that has markedly low (<20%) collateral flow, the guinea pig heart demonstrates significant collateral circulation [44]. Apparently, adaptive mechanisms developed with aging in old guinea pig hearts provide better cardioprotection against IR compared to young animals.
MitoROS play a crucial role in the pathogenesis of cardiac diseases and heart function in aging. High ROS levels due to diminished ETC activity and reduced antioxidant capacity cause oxidative damage to proteins, lipids and mtDNA. In the absence of protective histones, mtDNA is highly susceptible to oxidative stress, and, consequently, mutated mtDNA increases synthesis of defective proteins. This further amplifies mitoROS production, which ultimately leads to mitochondria-mediated cell death [20, 21]. Indeed, mitochondria are the end-targets and effectors of cardioprotective pathways, many of which entail ROS signaling.
Pharmacological approaches that prevent total cellular ROS in the heart are sometimes ineffective, and mitochondrial dysfunction is most likely responsible for the failure of the cardioprotection against oxidative stress in aged rats. This is due to the vital role that ROS play in the mediation of cardioprotective signaling [45]. Hence, the cardioprotective effects of current therapeutic strategies against myocardial infarction and IR, such as pharmacological and ischemic preconditioning, are lost in aged hearts [12–15]. Consequently, the development of ROS scavengers that primarily target mitoROS could be of clinical relevance in the treatment of cardiac diseases and the possible delay of aging processes. In this line, a newly developed mitochondria-targeting ROS scavenger, XJB, has shown to possess anti-apoptotic effects and enhances cell survival in mouse embryonic cells [25]. It also attenuates oxidative DNA damage, mitochondrial dysfunction and disease phenotypes in a mouse model of Huntington’s disease [27]. Treatment of human intervertebral disc cells with XJB blunts the adverse effects of oxidative stress, suggesting a potentially important role for ROS scavengers in preventing aging-related intervertebral disc degeneration [46]. It also ameliorates tissue ischemia and prolonged survival in a rodent model of hemorrhagic shock [28].
The beneficial effects of XJB may, in part, originate from its channel forming action across the IMM, similar to the mechanism implicated in the antibacterial effects of gramicidin. It is likely that this action of XJB could lead to mild membrane depolarization and protect mitochondria from Ca2+-overload induced by oxidative stress. PKC, PKG, cytosolic ROS, and other signaling molecules induce opening of mitKATP channels leading to depolarization of the IMM, reductions of [Ca2+] in the mitochondrial matrix and induce short burst of mitoROS. mitoROS released into the cytoplasm activate several survival mechanisms that resist subsequent sustained IR injury [47]. These pathways underlie the cardioprotective actions of ischemic and pharmacological preconditioning against sustained IR. However, in contrast to ischemic preconditioning, XJB, at different concentrations, had no effect on ΔΨmit in control cardiomyocytes (Fig. 7A) and isolated mitochondria (Fig. 8C), thus, excluding a possible role for “mild depolarization” in the protective actions of XJB. The drug, however, remarkably attenuated oxidative stress-induced depolarization of the mitochondrial membrane and reduced both mitochondrial and total ROS levels in cardiomyocytes. Finally, ability of XJB to alleviate antimycin A-induced ROS production, together with our previous findings that it accumulates in mitochondria, indicates that XJB scavenges mitoROS by virtue of its nitroxide functionality and the prevention of cardiolipin peroxidation. XJB ameliorated peroxidation of cardiolipin in the brain after experimental traumatic brain injury [29] and ileal mucosal samples from rats subjected to lethal hemorrhagic shock [28]. The anti-ROS capacity of XJB could involve scavenging of electrons leaking from uncoordinated electron carriers, rather than an SOD-like activity [29].
It is not yet clear whether increased ROS levels decrease the respiratory function of mitochondria in the aged heart [48]. Studies have reported either a decline of state 3 in mitochondria of aged hearts [23, 49] or no changes in the respiration rates of these organelles [50, 51]. Although we have not investigated respiratory function of mitochondria in non-IR adult and aged hearts, analysis of mitochondrial oxygen consumption rates after IR did not reveal a significant difference between adult and aged hearts. This may be secondary to the preparation of mitochondria used for oxygen consumption studies because we did not separate the mitochondrial subpopulations present in cardiomyocytes. Indeed, these cells contain two functionally distinct mitochondrial subpopulations; the subsarcolemmal mitochondria, which reside beneath the plasma membrane, and the interfibrillar mitochondria, which are located between the myofibrils. The subpopulations of mitochondria differ in state 3, Ca2+ sensitivity and ROS production [16, 18], and previous studies demonstrated that aging decreases state 3, oxidative phosphorylation and the activity of ETC complexes in interfibrillar mitochondria, whereas subsarcolemmal mitochondria of the heart remain unaltered [52, 53].
In aged rats, treatment with XJB improved the state 3 mitochondrial respiration rates, particularly those of complexes I and IV. This preserved ETC activity may be due to XJB-induced reduction of mitoROS damage to mtDNA. Out of 13 mtDNA-encoded proteins, 10 are subunits of complexes I and III. Therefore, a reduction of mitoROS prevents mtDNA damage and presumably improves ETC activity in aged hearts. In addition, the enhanced susceptibility of the aged hearts to IR injury could be associated with a greater and more prolonged accumulation of Ca2+ in cardiomyocytes during ischemia and early reperfusion compared to young rats [54]. Ca2+ overload and increased levels of ROS trigger the opening of the mitochondrial PTP in aged hearts. IR further increased pore opening in both adult and aged rats. XJB’s inhibition of PTP opening in adult and aged hearts and its reduction of ischemic contracture in aged hearts indicate that this agent regulates Ca2+ homeostasis in the cytoplasm and also in the mitochondria. In this way the drug could also reduce damage secondary to calcium-induced contracture.
5. Limitations of the study
The present study has several limitations to be indicated. There could be sex-associated differences in the post-ischemic recovery of cardiac and mitochondrial function in adult and aged rats. Gender differences could play an important role in the effect of XJB and this area was not evaluated in the present study. In addition, our study involves treatment of adult and aged rats with XJB for one month which could limit the observed cardiac responses to the drug. Likewise, the administration of XJB prior to IR hinders the observation of acute effects of the drug during or after global ischemia. This could be of significance in the clinical setting where reperfusion injury is observed. Finally, we did not observed significant effects of XJB on infarct size in adult and aged animals (n=2–3), although LDH release was significantly reduced by the drug during the reperfusion period (40–60 min) of these groups. It is possible that this discrepancy between LDH release and infarct size determinations stems from the small sample size used in the latter studies. Further studies are needed to correlate cardiac and mitochondrial function with infarct size using the TTC method, and to establish the cause-and-effect relationship between improved mitochondrial respiration and post-ischemic recovery of cardiac function in aged rats treated with XJB.
6. Conclusion
Mitochondrial dysfunction during aging renders the heart more susceptible to oxidative stress. It is likely that metabolic and functional abnormalities in mitochondria, associated with increased mitoROS production, reduce the effectiveness of cardioprotective therapies in aged hearts. The present study demonstrates that pharmacological intervention targeting mtROS generation attenuates the age-induced loss of cardioprotection and enhances cardiac resistance to IR injury in aged rats, suggesting that improved treatment options for myocardial infarction in elderly patients should specifically address intracellular oxidative stress.
Highlights.
The mitochondria-targeted ROS scavenger, XJB-5-131 improves post-ischemic recovery of aged hearts;
XJB-5-131 increases state 3 for cardiac mitochondrial ETC complexes I, II and IV after IR in aged rats;
XJB-5-131 inhibits mitochondrial PTP opening in aged hearts subjected to IR;
XJB-5-131 attenuates H2O2-induced loss of Δψmit, and ROS production in cardiomyocytes.
Acknowledgments
Funding
This study was supported by the Puerto Rico Science and Technology Trust Grant (to WF), the NHLBI NIH Grant SC1HL118669 (to SJ), and, in part, by the RCMI National Center for Research Resources NIH Grant G12RR-003051 and the NIH P50 CMLD Program (GM067082 to PW).
The authors thank Miriam Castro and Jessica Soto Hernandez for their invaluable technical assistance in performing of experiments, and Bryan Agostini for his critical reading of the manuscript.
Abbreviations
- BW
body weight
- DMNQ
dimethoxynaphthoquinone
- dP/dtmax
maximum velocity of contraction
- −dP/dtmin
maximum velocity of relaxation
- ETC
electron transport chain
- HR
heart rate
- HW
heart weight
- IR
ischemia-reperfusion
- LDH
lactate dehydrogenase
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end diastolic pressure
- LVSP
left ventricular systolic pressure
- mitoROS
mitochondrial ROS
- PTP
permeability transition pore
- RCI
respiratory control index
- ROS
reactive oxygen species
- RPP
rate-pressure product
- XJB
XJB-5-131 (L-ornithinamide, 1-[(2S,3E,5S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-7-methyl-1-oxo-2-(phenylmethyl)-3-octen-1-yl]-L-prolyl-L-valyl-N5-[(phenylmethoxy)carbonyl]-N-(2,2,6,6-tetramethyl-1-oxy-4-piperidinyl)-)
Footnotes
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Odding E, Valkenburg HA, Stam HJ, Hofman A. Determinants of locomotor disability in people aged 55 years and over: the Rotterdam Study. Eur J Epidemiol. 2001;17:1033–41. doi: 10.1023/a:1020006909285. [DOI] [PubMed] [Google Scholar]
- 2.Azhar G, Gao W, Liu L, Wei JY. Ischemia-reperfusion in the adult mouse heart influence of age. Exp Gerontol. 1999;34:699–714. doi: 10.1016/s0531-5565(99)00031-5. [DOI] [PubMed] [Google Scholar]
- 3.Lucas DT, Szweda LI. Declines in mitochondrial respiration during cardiac reperfusion: age-dependent inactivation of alpha-ketoglutarate dehydrogenase. Proc Natl Acad Sci U S A. 1999;96:6689–93. doi: 10.1073/pnas.96.12.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abete P, Cioppa A, Calabrese C, Pascucci I, Cacciatore F, Napoli C, et al. Ischemic threshold and myocardial stunning in the aging heart. Exp Gerontol. 1999;34:875–84. doi: 10.1016/s0531-5565(99)00060-1. [DOI] [PubMed] [Google Scholar]
- 5.Lesnefsky EJ, Lundergan CF, Hodgson JM, Nair R, Reiner JS, Greenhouse SW, et al. Increased left ventricular dysfunction in elderly patients despite successful thrombolysis: the GUSTO-I angiographic experience. J Am Coll Cardiol. 1996;28:331–7. doi: 10.1016/0735-1097(96)00148-9. [DOI] [PubMed] [Google Scholar]
- 6.Mariani J, Ou R, Bailey M, Rowland M, Nagley P, Rosenfeldt F, et al. Tolerance to ischemia and hypoxia is reduced in aged human myocardium. J Thorac Cardiovasc Surg. 2000;120:660–7. doi: 10.1067/mtc.2000.106528. [DOI] [PubMed] [Google Scholar]
- 7.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 8.Fenton RA, Dickson EW, Meyer TE, Dobson JG., Jr Aging reduces the cardioprotective effect of ischemic preconditioning in the rat heart. J Mol Cell Cardiol. 2000;32:1371–5. doi: 10.1006/jmcc.2000.1189. [DOI] [PubMed] [Google Scholar]
- 9.Ebrahim Z, Yellon DM, Baxter GF. Ischemic preconditioning is lost in aging hypertensive rat heart: independent effects of aging and longstanding hypertension. Exp Gerontol. 2007;42:807–14. doi: 10.1016/j.exger.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, et al. Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol. 2007;292:H1764–9. doi: 10.1152/ajpheart.01071.2006. [DOI] [PubMed] [Google Scholar]
- 11.Lee TM, Su SF, Chou TF, Lee YT, Tsai CH. Loss of preconditioning by attenuated activation of myocardial ATP-sensitive potassium channels in elderly patients undergoing coronary angioplasty. Circulation. 2002;105:334–40. doi: 10.1161/hc0302.102572. [DOI] [PubMed] [Google Scholar]
- 12.Schulman D, Latchman DS, Yellon DM. Effect of aging on the ability of preconditioning to protect rat hearts from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2001;281:H1630–6. doi: 10.1152/ajpheart.2001.281.4.H1630. [DOI] [PubMed] [Google Scholar]
- 13.Willems L, Ashton KJ, Headrick JP. Adenosine-mediated cardioprotection in the aging myocardium. Cardiovasc Res. 2005;66:245–55. doi: 10.1016/j.cardiores.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Sniecinski R, Liu H. Reduced efficacy of volatile anesthetic preconditioning with advanced age in isolated rat myocardium. Anesthesiology. 2004;100:589–97. doi: 10.1097/00000542-200403000-00019. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen LT, Rebecchi MJ, Moore LC, Glass PS, Brink PR, Liu L. Attenuation of isoflurane-induced preconditioning and reactive oxygen species production in the senescent rat heart. Anesth Analg. 2008;107:776–82. doi: 10.1213/ane.0b013e318180419d. [DOI] [PubMed] [Google Scholar]
- 16.Lesnefsky EJ, Hoppel CL. Ischemia-reperfusion injury in the aged heart: role of mitochondria. Arch Biochem Biophys. 2003;420:287–97. doi: 10.1016/j.abb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 17.Boengler K, Schulz R, Heusch G. Loss of cardioprotection with ageing. Cardiovasc Res. 2009;83:247–61. doi: 10.1093/cvr/cvp033. [DOI] [PubMed] [Google Scholar]
- 18.Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Heart Circ Physiol. 2013;305:H459–76. doi: 10.1152/ajpheart.00936.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harman D. Free radical theory of aging: an update: increasing the functional life span. Ann N Y Acad Sci. 2006;1067:10–21. doi: 10.1196/annals.1354.003. [DOI] [PubMed] [Google Scholar]
- 20.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 21.Choksi KB, Papaconstantinou J. Age-related alterations in oxidatively damaged proteins of mouse heart mitochondrial electron transport chain complexes. Free Radical Bio Med. 2008;44:1795–805. doi: 10.1016/j.freeradbiomed.2008.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–44. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jahangir A, Ozcan C, Holmuhamedov EL, Terzic A. Increased calcium vulnerability of senescent cardiac mitochondria: protective role for a mitochondrial potassium channel opener. Mech Ageing Dev. 2001;122:1073–86. doi: 10.1016/s0047-6374(01)00242-1. [DOI] [PubMed] [Google Scholar]
- 24.Wojtovich AP, Nadtochiy SM, Brookes PS, Nehrke K. Ischemic preconditioning: the role of mitochondria and aging. Exp Gerontol. 2012;47:1–7. doi: 10.1016/j.exger.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, et al. Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J Am Chem Soc. 2005;127:12460–1. doi: 10.1021/ja053679l. [DOI] [PubMed] [Google Scholar]
- 26.Fink MP, Macias CA, Xiao J, Tyurina YY, Jiang J, Belikova N, et al. Hemigramicidin-TEMPO conjugates: novel mitochondria-targeted anti-oxidants. Biochem Pharmacol. 2007;74:801–9. doi: 10.1016/j.bcp.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 27.Xun Z, Rivera-Sanchez S, Ayala-Pena S, Lim J, Budworth H, Skoda EM, et al. Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington’s disease. Cell Rep. 2012;2:1137–42. doi: 10.1016/j.celrep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macias CA, Chiao JW, Xiao J, Arora DS, Tyurina YY, Delude RL, et al. Treatment with a novel hemigramicidin-TEMPO conjugate prolongs survival in a rat model of lethal hemorrhagic shock. Ann Surg. 2007;245:305–14. doi: 10.1097/01.sla.0000236626.57752.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. 2012;15:1407–13. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soule BP, Hyodo F, Matsumoto K, Simone NL, Cook JA, Krishna MC, et al. The chemistry and biology of nitroxide compounds. Free Radical Bio Med. 2007;42:1632–50. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldstein S, Samuni A, Hideg K, Merenyi G. Structure-activity relationship of cyclic nitroxides as SOD mimics and scavengers of nitrogen dioxide and carbonate radicals. J Phys Chem. 2006;110:3679–85. doi: 10.1021/jp056869r. [DOI] [PubMed] [Google Scholar]
- 32.Lam MA, Pattison DI, Bottle SE, Keddie DJ, Davies MJ. Nitric oxide and nitroxides can act as efficient scavengers of protein-derived free radicals. Chem Res Toxicol. 2008;21:2111–9. doi: 10.1021/tx800183t. [DOI] [PubMed] [Google Scholar]
- 33.Nilsson UA, Olsson LI, Carlin G, Bylund-Fellenius AC. Inhibition of lipid peroxidation by spin labels. Relationships between structure and function. J Biol Chem. 1989;264:11131–5. [PubMed] [Google Scholar]
- 34.Jiang J, Kurnikov I, Belikova NA, Xiao J, Zhao Q, Amoscato AA, et al. Structural requirements for optimized delivery, inhibition of oxidative stress, and antiapoptotic activity of targeted nitroxides. J Pharmacol Exp Ther. 2007;320:1050–60. doi: 10.1124/jpet.106.114769. [DOI] [PubMed] [Google Scholar]
- 35.Abraham T, Prenner EJ, Lewis RN, Mant CT, Keller S, Hodges RS, et al. Structure-activity relationships of the antimicrobial peptide gramicidin S and its analogs: aqueous solubility, self-association, conformation, antimicrobial activity and interaction with model lipid membranes. Biochim Biophys Acta. 2014;1838:1420–9. doi: 10.1016/j.bbamem.2013.12.019. [DOI] [PubMed] [Google Scholar]
- 36.Javadov S, Choi A, Rajapurohitam V, Zeidan A, Basnakian AG, Karmazyn M. NHE-1 inhibition-induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc Res. 2008;77:416–24. doi: 10.1093/cvr/cvm039. [DOI] [PubMed] [Google Scholar]
- 37.Barreto-Torres G, Parodi-Rullan R, Javadov S. The Role of PPARalpha in metformin-induced attenuation of mitochondrial dysfunction in acute cardiac ischemia/reperfusion in rats. Int J Mol Sci. 2012;13:7694–709. doi: 10.3390/ijms13067694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Tsuchida A, Cohen MV, Downey JM. Pretreatment with angiotensin II activates protein kinase C and limits myocardial infarction in isolated rabbit hearts. J Mol Cell Cardiol. 1995;27:883–92. doi: 10.1016/0022-2828(95)90038-1. [DOI] [PubMed] [Google Scholar]
- 39.Zamzami N, Maisse C, Metivier D, Kroemer G. Measurement of membrane permeability and the permeability transition of mitochondria. Methods Cell Biol. 2007;80:327–40. doi: 10.1016/S0091-679X(06)80016-6. [DOI] [PubMed] [Google Scholar]
- 40.Khaliulin I, Clarke SJ, Lin H, Parker J, Suleiman MS, Halestrap AP. Temperature preconditioning of isolated rat hearts--a potent cardioprotective mechanism involving a reduction in oxidative stress and inhibition of the mitochondrial permeability transition pore. J Physiol. 2007;581:1147–61. doi: 10.1113/jphysiol.2007.130369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kilic A, Javadov S, Karmazyn M. Estrogen exerts concentration-dependent pro-and anti-hypertrophic effects on adult cultured ventricular myocytes. Role of NHE-1 in estrogen-induced hypertrophy. J Mol Cell Cardiol. 2009;46:360–9. doi: 10.1016/j.yjmcc.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 42.Hernandez JS, Barreto-Torres G, Kuznetsov AV, Khuchua Z, Javadov S. Crosstalk between AMPK activation and angiotensin II-induced hypertrophy in cardiomyocytes: the role of mitochondria. J Cell Mol Med. 2014;18:709–20. doi: 10.1111/jcmm.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhodes SS, Camara AK, Heisner JS, Riess ML, Aldakkak M, Stowe DF. Reduced mitochondrial Ca2+ loading and improved functional recovery after ischemia-reperfusion injury in old vs. young guinea pig hearts. Am J Physiol Heart Circ Physiol. 2012;302:H855–63. doi: 10.1152/ajpheart.00533.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hearse DJ. Species variation in the coronary collateral circulation during regional myocardial ischaemia: a critical determinant of the rate of evolution and extent of myocardial infarction. Cardiovasc Res. 2000;45:213–9. doi: 10.1016/s0008-6363(99)00331-4. [DOI] [PubMed] [Google Scholar]
- 45.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–70. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 46.Nasto LA, Robinson AR, Ngo K, Clauson CL, Dong Q, St Croix C, et al. Mitochondrial-derived reactive oxygen species (ROS) play a causal role in aging-related intervertebral disc degeneration. J Orthop Res. 2013;31:1150–7. doi: 10.1002/jor.22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Rourke B. Myocardial K(ATP) channels in preconditioning. Circ Res. 2000;87:845–55. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- 48.Judge S, Leeuwenburgh C. Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol. 2007;292:C1983–92. doi: 10.1152/ajpcell.00285.2006. [DOI] [PubMed] [Google Scholar]
- 49.Chen JC, Warshaw JB, Sanadi DR. Regulation of mitochondrial respiration in senescence. J Cell Physiol. 1972;80:141–8. doi: 10.1002/jcp.1040800115. [DOI] [PubMed] [Google Scholar]
- 50.Manzelmann MS, Harmon HJ. Lack of age-dependent changes in rat heart mitochondria. Mech Ageing Dev. 1987;39:281–8. doi: 10.1016/0047-6374(87)90067-4. [DOI] [PubMed] [Google Scholar]
- 51.Cocco T, Sgobbo P, Clemente M, Lopriore B, Grattagliano I, Di Paola M, et al. Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radical Bio Med. 2005;38:796–805. doi: 10.1016/j.freeradbiomed.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 52.Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999;372:399–407. doi: 10.1006/abbi.1999.1508. [DOI] [PubMed] [Google Scholar]
- 53.Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–21. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 54.O’Brien JD, Ferguson JH, Howlett SE. Effects of ischemia and reperfusion on isolated ventricular myocytes from young adult and aged Fischer 344 rat hearts. Am J Physiol Heart Circ Physiol. 2008;294:H2174–83. doi: 10.1152/ajpheart.00058.2008. [DOI] [PubMed] [Google Scholar]