

Tantalizing connections between autoimmune rheumatic diseases and cancer have become increasingly evident over the past several decades. These connections are complex, with different relationships in frequency, timing, and types of cancers observed in different diseases or disease subgroups. Several recent advances from disparate fields have begun to illuminate the dynamic and bidirectional interactions occurring at the cancer–immune system interface which may be relevant to understanding the origins of autoimmunity (1). These interactions include the existence of potent anticancer immune responses that limit tumor growth, as well as multiple immune and inflammatory pathways that can contribute to tumor growth and robustness. The striking ability of immune checkpoint inhibitors to reveal powerful anticancer immune responses in patients with cancer highlights the fact that natural immune responses to cancers occur, and may regulate the emergence of cancer (2).
Recent data from patients with systemic sclerosis (SSc, scleroderma) suggest that in some cases, autoimmunity may be initiated by autoantigen mutation in the patient's cancer (3, 4). Interestingly, there are patients with the same form of scleroderma and an identical autoimmune response who do not have a detectable cancer, raising the possibility that in these patients, the disease mechanism is the same except that the antitumor immune response has successfully eliminated the cancer. Similar striking associations with cancer are also apparent in other rheumatic phenotypes, particularly dermatomyositis (DM). The autoimmune rheumatic diseases therefore provide an exceptional opportunity to study cancer–immune system interactions and interrogate the mechanisms of the autoimmune rheumatic diseases, as well as the natural immune response to cancers in humans.
This review highlights the relationships between cancer and rheumatic diseases, focusing on kinetics (how closely in time the cancer and rheumatic disease present) and immune response (the frequency of cancer in rheumatic disease patients with different autoantibody specificities). We will highlight similarities to various paraneoplastic, immune‐mediated processes and will introduce important new tumor‐immunoediting concepts. While space constraints require that this review focus on specific immune responses associated with cancer in SSc and DM, the principles outlined are likely also relevant to other autoimmune rheumatic syndromes.
Increased risk of cancer, and temporal clustering of cancer with rheumatic disease onset, in DM and SSc
Patients with DM and patients with SSc have an increased risk of cancer compared to general population–based controls after adjustment for age and sex, with reported standardized incidence ratios or relative risks ranging from 3.0 to 7.7 for DM and 1.4 to 3.2 for SSc (5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22). Table 1 highlights cancer sites for which these patients are at elevated risk. While men (8, 14, 23), older patients developing myositis and SSc (5, 17, 18, 22, 24, 25, 26, 27), and patients with rapid and severe onset of disease (26, 27), with poor response to therapy, or with diffuse cutaneous SSc may also have a higher risk of malignancy, these have not been consistently identified as risk factors for cancer.
Table 1.
Increased risk of specific tumor types among patients with dermatomyositis and systemic sclerosis
| Disease, cancer site | Reference(s) |
|---|---|
| Dermatomyositis | |
| Ovary | 20, 21, 72 |
| Lung | 20, 21, 72, 73 |
| Pancreas | 21, 72 |
| Stomach | 21 |
| Colorectal | 21, 72 |
| Breast | 21, 72, 73 |
| Breast | 21, 72, 73 |
| Non‐Hodgkin's lymphoma | 21 |
| Nasopharynx (southeast Asians) | 73 |
| Systemic sclerosis | |
| Lung | 5, 7, 8, 9, 11, 13, 14, 15, 16 |
| Hematologic | 8, 12, 13, 14, 15, 16 |
| Esophagus | 6 |
| Oropharynx | 6, 16 |
| Skin | 8, 9 |
| Cervix | 8 |
| Liver | 9, 14, 74 |
| Bladder | 14 |
In both rheumatic diseases, there is a close temporal relationship between malignancy and autoimmunity onset. This is most striking in DM, in which the majority of patients with a malignancy have cancer prior to myositis diagnosis (19, 21, 28), with myositis often being diagnosed within 2 years (21). The risk of malignancy development after diagnosis of myositis is highest in the first year after myositis diagnosis, and then gradually decreases over time (19, 20, 21). In patients with SSc and breast cancer, a similar temporal relationship has been observed (29, 30). This temporal clustering, in conjunction with reports suggesting that cancer therapy may improve outcomes in myositis (31) or SSc (32, 33), suggests a possible mechanistic relationship between malignancy and rheumatic disease. Investigating this relationship is complex because of the significant heterogeneity in clinical phenotypes, age at rheumatic disease onset, tumor types, and cancer and rheumatic disease therapies used in these patients. However, the strong associations between unique autoantibodies and the temporal clustering of cancer diagnosis with rheumatic disease onset suggest that immunologic subsets may be a critical filter in understanding the cancer–autoimmunity relationship.
Association of unique autoantibodies with temporal clustering of cancer and rheumatic disease
Autoantibodies have important diagnostic and prognostic power across the spectrum of the autoimmune rheumatic diseases. Within a given phenotype, different autoantibodies may be associated with distinct clinical phenotypes. Myositis and SSc autoantibodies illustrate this well, and we have therefore focused on these below.
Myositis
Interestingly, within the spectrum of myositis (34), well‐characterized myositis‐specific autoantibodies are associated with distinct phenotypes. For example, antibodies against the aminoacyl transfer RNA synthetases (especially anti–Jo‐1) are found in myositis patients with a common set of clinical features including interstitial lung disease, mechanic's hands, nonerosive arthritis, and fever (the “antisynthetase syndrome”). Mi‐2 antibodies are found exclusively in DM patients; these patients frequently have more severe skin rashes and respond better to steroid therapy. While antibodies against melanoma differentiation–associated gene 5 (MDA‐5) are also DM specific, patients with this specificity typically do not exhibit clinical myopathy and frequently have interstitial lung disease (35). Yet another distinct clinical component of the myositis spectrum is associated with antibodies against 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase—these are a feature in patients with an immune‐mediated necrotizing myopathy (36, 37).
While clinical phenotypes associated with known autoantibodies are widely recognized, myositis autoantibodies have not, until recently, been meaningfully associated with cancer. The usefulness of autoantibodies as predictors of cancer‐associated myositis was examined by Chinoy et al (38) in a study of 282 patients with myositis–connective tissue disease overlap. The investigators showed that patients with myositis‐specific and myositis‐associated antibodies that can be assayed by routinely available clinical tests have a significantly lower risk of an associated cancer compared to autoantibody‐negative DM patients.
Recently emerging data have shown that while this may be the case for the “well‐established/historic” myositis autoantibodies, 2 new specificities do indeed appear to be associated with cancer. These, and evidence of their cancer association, are discussed below.
Transcription intermediary factor 1γ antibodies
A newly recognized DM‐specific autoantibody, found in 13–21% of adult DM patients, was recently reported by 2 groups (39, 40). In both studies, cohorts of patient sera were screened by immunoprecipitation using radiolabeled cell lysates, enabling detection of a 155‐kd protein. Although the cohort sizes and the numbers of antibody‐positive patients were small, in both studies these antibodies were frequently detected in patients with an associated malignancy. The target of this new antibody specificity was identified as transcription intermediary factor 1γ (TIF1γ) (41). This multifunctional protein is a member of the tripartite motif–containing protein family and has complex effects on various cellular pathways. For example, TIF1γ plays a critical role in tissue differentiation through interactions with Smad proteins (42). Thus, in embryonic stem cells, TIF1γ interacts with Smad2/3, allowing this complex to activate specific differentiation genes by promoting transcriptional elongation (43). TIF1γ is also required for proper development of mammary glands, where it inhibits Smad4 by ubiquitinylation (44).
To evaluate the usefulness of TIF1γ antibodies for diagnosing cancer‐associated DM, Trallero‐Araguas et al (45) performed a systematic review and meta‐analysis using data from 6 published studies; immunoprecipitation from lysates was used for antibody detection in all of these studies. The meta‐analysis showed that anti‐TIF1γ–positive DM patients have a 27‐fold higher odds of developing cancer‐associated myositis than their anti‐TIF1γ–negative counterparts (45).
NXP‐2 antibodies
In 1997, the presence of antibodies against a 140‐kd protein (anti‐MJ) in 18% of patients with juvenile DM was reported (46). The targeted autoantigen was subsequently found to be the nuclear matrix protein NXP‐2 (47), a protein that localizes to the promyelocytic leukemia nuclear bodies and the nucleoplasm (38). There are 3 structurally separated, conserved domains (48) with important roles in various functions. For example, Mimura et al (38) showed that NXP‐2 recruits and activates p53, inducing cellular senescence and thereby preventing cell proliferation. Initial studies of this specificity were performed on pediatric DM cohorts (49, 50), confirming a prevalence of ∼23–74%. They are notable for the lack of reported malignancy among the >200 young patients studied. More recent studies in adult myositis populations have demonstrated prevalences of anti–NXP‐2 ranging from 1.6% to 30% of adult DM patients and 1.6% to 8% of adult polymyositis (PM) patients (51, 52). Of interest, Ichimura et al (52) noted that associated cancers were present in 3 of the 7 anti–NXP‐2–positive DM patients in their cohort (43%), with all of the carcinomas being at an advanced stage. Intriguingly, 6 of the 7 anti–NXP‐2–positive DM patients were male (86%), and all 3 of the cancers in the group were in male patients; the cancers were not restricted to male‐specific cancer sites.
Presence of antibodies against NXP‐2 or TIF1γ in >80% of patients with cancer‐associated myositis
Identification of specific antibodies in the 140–155‐kd range following immunoprecipitation from lysates is challenging as there are multiple specificities in this size range (including TIF1γ, NXP‐2, and MDA‐5). We and our colleagues therefore recently developed sensitive, specific assays that unequivocally detect antibodies against NXP‐2 and TIF1γ (53). Using these, antibodies against TIF1γ and NXP‐2 were evaluated in 213 patients from 2 separate, well‐defined DM cohorts (111 patients from the Department of Dermatology, Stanford University [Stanford, California] School of Medicine and 102 from the Myositis Center, Johns Hopkins University).
Antibodies against TIF1γ and NXP‐2 were detected in 82 of 213 DM patients (38%) and 37 of 213 DM patients (17%), respectively. The antibody groups were mostly non‐overlapping, with only 2 patients having both specificities. Cancer‐associated DM was detected in 29 of the DM patients (14%), with 24 of the 29 (83%) having antibodies against either TIF1γ or NXP‐2. The overall frequency of cancer in TIF1γ/NXP‐2–positive patients was 20.5%. In the remaining 96 patients with neither TIF1γ nor NXP‐2 antibodies, there were only 5 cases of cancer (5%). An important relationship between age and cancer frequency in DM patients was noted across all antibody groups: among patients >60 years old, cancer was found in 55% of those with anti–NXP‐2 antibodies, 31% of those with anti‐TIF1γ antibodies, and 17% of patients without either of these antibodies. Additionally, antibodies against NXP‐2 were specifically associated with cancer in male patients (7 of 9 [78%]). This observation is similar to findings in 3 patients reported by Ichimura et al (52). While the numbers in both of these studies are small, the results are intriguing and await confirmation.
The above‐described observations make several important points about the relationship between DM and cancer: 1) where cancers occur, they generally manifest around the time of DM diagnosis, irrespective of antibody response; 2) the prevalence of cancers within 3 years of myositis diagnosis is higher in anti‐TIF1γ and NXP‐2 autoantibody–positive groups; 3) among patients with TIF1γ or NXP‐2 antibodies, however, a sizable proportion do not manifest cancer; 4) the frequency of cancers associated with DM increases at age >60 years, irrespective of serologic status; and 5) the association of NXP‐2 antibodies and cancer in DM may be enhanced in males. Additional data are needed to confirm whether these findings are generalizable (particularly to different ethnic populations). The association of DM and cancer therefore does not appear to be binary but rather to be influenced strongly by several parameters, including autoantibody targets and age, although it is possible that a binary parameter remains to be defined.
Of note, currently reported data regarding immune responses address overall autoantibody responses, but we do not yet understand the nuances of these specificities in terms of epitopes and magnitude, or the specificity of other immune effector pathways for these antigens. It is also possible that no individual covariate will be the key determinant of whether a cancer emerges clinically, but that the additive effects of multiple factors will be explicative. The close temporal clustering of DM and cancer, and the elevated frequency of cancer in patients with TIF1γ/NXP‐2 antibodies irrespective of age, suggests that additional important parameters remain to be defined (see below).
Scleroderma
As in myositis, scleroderma‐specific autoantibodies are associated with distinct clinical phenotypes and are useful for risk stratification and assessment of long‐term prognosis (54, 55, 56, 57). The 3 most common scleroderma‐specific autoantibodies are anticentromere, anti–topoisomerase I (anti–topo I), and anti–RNA polymerase III (anti–RNAP III). Patients who are positive for antibodies targeting centromere proteins B/C tend to have limited cutaneous disease with features of CREST syndrome (calcinosis, Raynaud's phenomenon, esophageal dysmotility, sclerodactyly, telangiectasias), major ischemic digital loss, pulmonary hypertension, and overlap features with Sjögren's syndrome or primary biliary cirrhosis. In contrast, patients with anti–topo I antibodies have a higher risk of diffuse cutaneous disease and interstitial lung disease, and those with anti–RNAP III antibodies usually have aggressive, rapidly progressive diffuse cutaneous disease and a significantly higher risk of scleroderma renal crisis, myopathy, cardiac disease, and gastric antral vascular ectasia. Careful study of patients in these autoantibody subsets has demonstrated the usefulness of autoantibodies as predictors of cancer‐associated scleroderma.
RNAP III antibodies identify patients at risk of cancer‐associated scleroderma
Many investigations probing the relationship between cancer and scleroderma have excluded cancer cases diagnosed around the time of scleroderma onset, to avoid a potential detection bias. Consequently, patients with a close temporal relationship between cancer diagnosis and scleroderma onset have not been studied until recently. We investigated whether clinical characteristics among patients with scleroderma and cancer differed by autoantibody status (4). We demonstrated that in patients with RNAP III antibodies there was a close temporal relationship between malignancy diagnosis and the clinical onset of scleroderma, and patients exhibited a unique nucleolar RNAP III expression pattern in their cancerous tissues. These data suggested that expression of scleroderma antigens in cancers might be associated with scleroderma‐specific autoantibody responses.
The association between RNAP III antibodies and a close cancer–scleroderma interval has subsequently been confirmed by others (24, 30, 58). In an Italian scleroderma cohort, patients with RNAP III antibodies had a higher prevalence of cancer and were more likely to have cancer concurrent with scleroderma onset than patients with other autoantibodies (58). However, the study included only 16 anti–RNAP III–positive patients, 7 of whom had cancer. In a study of 451 Australian scleroderma patients, those with RNAP III antibodies had a 4.2‐fold increased odds of having malignancy diagnosed within 5 years of scleroderma onset compared to patients without this specificity (24). The overall prevalence of cancer (∼13%) was similar between patients with and those without RNAP III antibodies (24). Another investigation in a cohort of 2,177 UK scleroderma patients demonstrated that the prevalence of malignancy was higher in patients with RNAP III antibodies (14.2%) than in patients with topo I antibodies (6.3%; P < 0.0001) or centromere antibodies (6.8%; P < 0.001) (30). Among patients with cancer diagnosed within 36 months of scleroderma onset, 55.3% were positive for RNAP III antibodies, whereas 13.6% were positive for topo I antibodies (P < 0.002) and 23.5% for centromere antibodies (P < 0.008) (30). These data suggest that patients with new‐onset scleroderma and RNAP III antibody positivity may benefit from more aggressive evaluation for an underlying malignancy, given their heightened cancer risk.
Potential relevance of other scleroderma autoantigens
Interestingly, while a close cancer–scleroderma interval is most frequent among patients with RNAP III antibodies, there were patients with a short cancer–scleroderma interval and other autoantibody specificities in all studied cohorts. Since an anticancer immune response may be an important feature in some patients with scleroderma and RNAP III antibodies (see below), the co‐occurrence (though infrequent) of cancer and scleroderma in these other serologic subgroups suggests that cancer may be an important initiator of the immune response in many scleroderma patients, but that an immune response against specific targets might exert more potent anticancer effects. Indeed, centromere proteins and topo I play important roles in cancer fitness and survival. These pathways are targets of potent anticancer therapeutic agents, including inhibitors of topoisomerase and the mitotic spindle, suggesting that immune responses to some pathways may have more deleterious effects on cancer growth and survival than others. In rheumatic diseases in which cancer incidence is low, it is possible that the other targets of the immune response may be effective therapeutic targets in cancers. It is also possible that distinct mechanisms (unrelated to neoplastic transformation), including infections and other cellular states, may underlie the immune targeting of centromere and topo I.
Genetic alteration of autoantigens in cancer may be an antigen source in the rheumatic diseases
It has been hypothesized that patients with cancer and rheumatic disease may develop cancer secondary to 1) target tissue damage from the autoimmune disease, or 2) cytotoxic therapies used to treat aggressive manifestations, or 3) as a consequence of a defective immune system that predisposes to the development of both cancer and autoimmunity (3, 59). However, data demonstrating unique nucleolar RNAP III expression in cancerous tissues from scleroderma patients with RNAP III antibodies (4), and the co‐occurrence of cancer and scleroderma in this patient subset, raised the intriguing possibility that genetic alterations in the POLR3A locus in tumors may trigger autoimmunity.
To address this, we studied tumors from 16 scleroderma patients: 8 with RNAP III antibodies and 8 with either centromere or topo I antibodies (3). The POLR3A locus had genetic alterations in 6 of 8 cancers from patients with RNAP III antibodies, but not in tumors from patients with other scleroderma antibodies.
Interestingly, these genetic alterations took 2 forms: 1) Somatic mutations—3 patients had somatic mutations in POLR3A; in each, the mutation caused a change in a single amino acid (different in all 3 patients). The mutations were present at a diminished frequency in the cancers, suggesting that they arose quite late in cancer development, or that the mutations arose early and were negatively selected during cancer evolution (see loss of heterozygosity, below). It is noteworthy that mutations in POLR3A are very uncommon in cancer, with a frequency of 0.7% in the Cosmic database (P < 10−20), suggesting that these mutations help to initiate the immune response to RNAP III. Indeed, 2 of the 3 patients with mutated forms of RNAP III exhibited mutation‐specific T cell immune responses. The fact that mutations in specific autoantigens such as POLR3A are infrequent in cancers in general suggests that even infrequent mutations, if they are in the right genes (i.e., autoantigens), may initiate autoimmunity when presented to the immune system in the context of the appropriate major histocompatibility complex (MHC) framework. A deeper understanding of these relationships will require much more extensive data on frequency of mutations in additional autoantigens. 2) Loss of heterozygosity—5 of the 8 patients exhibited loss of heterozygosity at the POLR3A locus. This was not a feature in cancers in scleroderma patients with other immune responses, including antibodies to topo I or centromere proteins, strongly indicating that the RNAP III–specific immune response might be shaping the molecular evolution of the cancer, consistent with immunoediting (see below).
Approximately 85% of patients with scleroderma and RNAP III antibodies do not, however, manifest a cancer clinically. We therefore hypothesize that potent antitumor immune responses successfully eradicate an underlying malignancy in most patients with scleroderma who manifest an autoantibody response against RNAP III.
Although the immune response to RNAP III may be initiated against mutated proteins in the patient's cancer, the presence of autoimmune injury to self tissues (which do not express the mutated version) suggests that the immune response spreads to the wild‐type (WT) version of the antigen present in self tissue. Consistent with this, we found that autoantibodies in the patients with mutated RNAP III were cross‐reactive with both WT and mutant RNAP III (3). There is significant evidence demonstrating that a modified version of a self antigen can initiate a T cell immune response to the altered antigen, and that the resulting B cell response recognizes the altered and WT antigens similarly (60). When WT and altered antigens are present at the time of immunization, a T cell response to the WT antigen can be initiated (61). We propose a similar mechanism here, with mutated antigen initiating a mutant‐specific T cell response, a cross‐reactive B cell response, and upon release and autoantibody‐mediated uptake of WT antigen, a T cell response directed against the WT molecule (Figure 1). While 1 patient in our study had CD4 T cells recognizing the WT RNAP III (subunit A) antigen, defining CD4 T cells directed against the WT RNAP III antigen in scleroderma patients with and without cancer remains a high priority.
Figure 1.
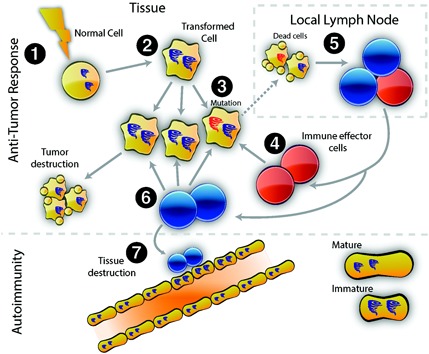
Model for cancer‐induced autoimmunity. Transformation of normal cells (1) may result in gene expression patterns that resemble immature cells involved in tissue healing (2). Occasionally, autoantigens become mutated (3); these are not driver mutations, and not all cancer cells have them. The first immune response is directed against the mutated form of the antigen (4), and may spread to the wild‐type version (5). Immune effector cells directed against the mutant (depicted in red) delete exclusively cancer cells containing the mutation (6). Immune effector cells directed against the wild type (depicted in blue) delete cancer cells without the mutation and also cross‐react with the patient's own tissues (particularly immature cells expressing high levels of antigen, found in damaged/repairing tissue) (7). Once autoimmunity has been initiated, the disease is self‐propagating. Immature cells (expressing high antigen levels) that repair the immune‐mediated injury can themselves become the targets of the immune response, sustaining an ongoing cycle of damage/repair that provides the antigen source that fuels the autoimmune response.
The existence of other cancer‐induced autoimmune syndromes suggests that tumor antigen expression triggers unique autoimmune phenotypes and perhaps improved cancer outcomes
There is significant evidence of activation of the immune response in various cancers. Several tumor antigens have been defined under these circumstances; in most instances in which antibodies to tumor antigens have been defined, these are not associated with damage of normal tissue. However, there are numerous examples of paraneoplastic autoimmune syndromes, where there is immune damage of specific target tissues (e.g., paraneoplastic pemphigus, paraneoplastic neurologic degenerations, Lambert‐Eaton syndrome) (62). Additionally, during cancer immunotherapy, activation of autoimmunity often predicts a beneficial anticancer effect (e.g., vitiligo in melanoma, autoantibodies appearing during interferon [IFN] treatment of metastatic melanoma) (63, 64). Tumor‐infiltrating lymphocytes are also predictive of better cancer prognosis.
Patients with autoimmune paraneoplastic disease often have smaller tumors than patients without paraneoplastic syndromes (65). Indeed, some tumors may not be evident at diagnosis, likely due to a robust immune response. However, the amount of damage to normal tissue that can accrue over time can be high, causing significant morbidity and even mortality. The finding that severe autoimmunity affecting the nervous system sometimes significantly delays tumor diagnosis resembles observations in the autoimmune rheumatic diseases, where cancers may not become manifest for prolonged periods. Interestingly, in several cases, there is shared antigen expression in tumors and in target tissues (65). In one investigation, myositis autoantigen expression was increased in cancer types associated with myositis and in regenerating muscle cells in myositis muscle (66). These data suggest that while malignancies may be an antigen source initiating the immune response, regenerating cells in target tissues may be an antigen source propagating a feedforward loop of tissue damage and autoimmune disease (66).
Principles of cancer immunosurveillance and immunoediting may provide insight into the kinetics and pathogenesis of the rheumatic diseases
Several important immunologic principles that were suggested during development of the discipline of immunology (e.g., cancer immunosurveillance and suppressor T cells) preceded the ability to address their underlying molecular mechanisms, resulting in an initial mistaken conclusion that the principle was incorrect. This was true of the immunosurveillance hypothesis of Burnet in the 1950s (67), which proposed that mutations in cancer might provoke an effective immune response causing regression of the tumor without it ever making its existence known. The hypothesis was difficult to address directly in those early years, due to limited knowledge about molecular mechanisms of immunity and the specific cells mediating immune responses, and an inability to define specific cancer antigens. When an extensive series of experiments in the 1970s failed to show any increase in cancer initiation in immunodeficient mice exposed to mutagen (68), the field of immunosurveillance began to fall out of favor. As the cells, molecules, and pathways of immunology became better defined, enabling investigation of the roles of specific pathways in vivo in various mouse models using genetic approaches, several observations strongly suggested that various immune pathways (e.g., IFNγ, perforin) played central roles in regulating cancer emergence. Additionally, a growing body of knowledge (for review, see ref.1) showed that the immune system could target specific antigens in different cancers, demonstrating that cancers were not immunologically silent but rather were actively recognized by the immune system.
The immunosurveillance hypothesis underwent an important revision in the early 2000s, when it was recognized that the tumor does not remain constant in the presence of immune pressure, but is rather shaped by the immune response such that the resulting tumor is less capable of stimulating the immune system. Dunn and colleagues proposed that immunosurveillance be renamed “immunoediting,” acknowledging the dual “host‐protective and tumor‐promoting actions of immunity on developing cancers” (69). One form of editing involves loss of cancer‐specific antigen expression (similar to the loss of heterozygosity described above for POLR3A). Another form involves the expression of immune checkpoints by the cancer (2). The recent success of immune checkpoint inhibitors (e.g., anti–programmed cell death protein 1) in activating durable tumor‐specific immune responses, with clinically significant anticancer effects, underscores the relevance of preformed immune responses which become silenced during tumor evolution, and emphasizes that the clinical emergence of cancer likely represents a distinct phase of immunoediting.
Indeed, Schreiber and colleagues have proposed that cancer immunoediting has 3 stages: 1) elimination, 2) equilibrium, and 3) escape (1). Although not all stages have been observed in vivo, significant indirect evidence suggests their existence. There is likely considerable heterogeneity among cancers and individuals in terms of whether each stage occurs, and its duration. Conceptually, the stages are defined by the relative dominance of the cancer or the immune response. In the first stage (elimination), the immune response dominates, and antigens in the nascent cancer initiate innate and adaptive immune responses, resulting in elimination of the cancer. In the second stage (equilibrium), the anticancer immune response and the cancer are balanced—the cancer does not grow significantly, nor is the host immune system fully effective at eradication. This highly dynamic stage depends on ongoing matching of the immune response and the cancer. If significant changes occur in either (e.g., immune system weakening, or changes in the cancer that allow it to be less affected by the immune response, or acquired resistance to an immune effector pathway through expression of a checkpoint molecule), the cancer could escape and grow. The third stage (escape) represents the cancer dominating the immune response, evidenced by unregulated cancer growth. Cancers in this stage are likely significantly immunoedited, and are much poorer immune targets than nascent, unedited tumors. Of note, almost all cancers that present clinically are already in stage 3; current cancer immunotherapies targeting immune checkpoints focus on this stage.
A model of cancer‐induced autoimmunity in the rheumatic diseases
The causes of the autoimmune rheumatic diseases remain unclear. Although a minority of patients with autoimmune rheumatic diseases manifest cancers, some intriguing observations about this association suggest that cancers may play important roles in disease initiation. The evolving understanding of cancer immunoediting, clearer definition of the targets of the immune responses in cancer‐associated rheumatic diseases, and the important co‐clustering of cancer and rheumatic diseases in a subgroup of patients suggest a model in which mutations in autoantigens in cancers initiate an autoimmune response against highly specific targets. The initial mutation‐specific immune response subsequently spreads to the WT version of the protein, inducing tissue damage, focused on tissues in which the function and expression of that antigen is prominent. In some cases this immune response challenges the cancer, either eliminating it or maintaining it in equilibrium. Such patients present with the autoimmune rheumatic phenotype, but no cancer. In other cases the anticancer response actively immunoedits the cancer, and the cancer may eventually lose expression of the mutant antigen, and may also evolve to avoid the anticancer effects through other mechanisms. In this minority of patients, cancer emerges.
The critical immune effector pathways, which effect damage and dysfunction of normal tissue or the cancer expressing the WT allele, are not yet defined but likely include CD4 T cells, CD8 T cells, B cells, and autoantibodies. In addition to nontolerized structure, initiation of a primary immune response to cancer autoantigens would require the appropriate MHC scaffold, costimulation, and absence of immune checkpoints. The recent recognition of nonmalignant somatic mutation as a cause of chronic tissue dysfunction (70) also raises the question of whether mutations occurring in benign lesions which become visible to the immune system in the setting of danger might also be relevant to autoimmunity (71).
This model provides numerous testable hypotheses related to early detection of cancer, and possible therapy of cancer as an approach in treating autoimmune diseases. Since the immune response to the autoantigen is initiated and driven by the cancer and cross‐reacts with antigens in normal tissue, effective therapy to remove the cancer could rid the host of the apical immune stimulus, and allow the peripheral immune‐mediated damage to wane once resolution and tissue healing occur. The striking examples of autoimmune diseases disappearing after effective anticancer therapy (31, 32, 33) are consistent with this model. This also focuses attention on the immune response in rheumatic autoimmune diseases as a potentially positive force, and suggests that therapeutic approaches that regulate antigen expression in the target tissue may allow the beneficial anticancer effects of the immune system to remain focused on the cancer, while avoiding damage to self tissues.
The next decade will be an exciting time in terms of understanding the rheumatic autoimmune diseases. It is also likely that these diseases will reveal critical secrets about natural antitumor immunity, and how this might be harnessed for treating cancers.
AUTHOR CONTRIBUTIONS
All authors drafted the article, revised it critically for important intellectual content, and approved the final version to be published.
Acknowledgments
We thank John Hall, PhD for assistance with creating Figure 1, and Paul Rosen for assistance with editing.
REFERENCES
- 1. Schreiber RD, Old LJ, Smyth MJ.Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion.Science 2011;331:1565–70. [DOI] [PubMed] [Google Scholar]
- 2. Pardoll DM.The blockade of immune checkpoints in cancer immunotherapy.Nat Rev Cancer 2012;12:742–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Joseph CG, Darrah E, Shah AA, Skora AD, Casciola‐Rosen LA, Wigley FM, et al.Association of the autoimmune disease scleroderma with an immunologic response to cancer.Science 2014;343:152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shah AA, Rosen A, Hummers L, Wigley F, Casciola‐Rosen L.Close temporal relationship between onset of cancer and scleroderma in patients with RNA polymerase I/III antibodies.Arthritis Rheum 2010;62:2787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abu‐Shakra M, Guillemin F, Lee P.Cancer in systemic sclerosis.Arthritis Rheum 1993;36:460–4. [DOI] [PubMed] [Google Scholar]
- 6. Derk CT, Rasheed M, Artlett CM, Jimenez SA.A cohort study of cancer incidence in systemic sclerosis.J Rheumatol 2006;33:1113–6. [PubMed] [Google Scholar]
- 7. Hill CL, Nguyen AM, Roder D, Roberts‐Thomson P.Risk of cancer in patients with scleroderma: a population based cohort study.Ann Rheum Dis 2003;62:728–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olesen AB, Svaerke C, Farkas DK, Sorensen HT.Systemic sclerosis and the risk of cancer: a nationwide population‐based cohort study.Br J Rheumatol 2010;163:800–6. [DOI] [PubMed] [Google Scholar]
- 9. Rosenthal AK, McLaughlin JK, Gridley G, Nyren O.Incidence of cancer among patients with systemic sclerosis.Cancer 1995;76:910–4. [DOI] [PubMed] [Google Scholar]
- 10. Rosenthal AK, McLaughlin JK, Linet MS, Persson I.Scleroderma and malignancy: an epidemiological study.Ann Rheum Dis 1993;52:531–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peters‐Golden M, Wise RA, Hochberg M, Stevens MB, Wigley FM.Incidence of lung cancer in systemic sclerosis.J Rheumatol 1985;12:1136–9. [PubMed] [Google Scholar]
- 12. Siau K, Laversuch CJ, Creamer P, O'Rourke KP.Malignancy in scleroderma patients from south west England: a population‐based cohort study.Rheumatol Int 2011;31:641–5. [DOI] [PubMed] [Google Scholar]
- 13. Bonifazi M, Tramacere I, Pomponio G, Gabrielli B, Avvedimento EV, La Vecchia C, et al.Systemic sclerosis (scleroderma) and cancer risk: systematic review and meta‐analysis of observational studies.Rheumatology (Oxford) 2013;52:143–54. [DOI] [PubMed] [Google Scholar]
- 14. Onishi A, Sugiyama D, Kumagai S, Morinobu A.Cancer incidence in systemic sclerosis: meta‐analysis of population‐based cohort studies.Arthritis Rheum 2013;65:1913–21. [DOI] [PubMed] [Google Scholar]
- 15. Zhang JQ, Wan YN, Peng WJ, Yan JW, Li BZ, Mei B, et al.The risk of cancer development in systemic sclerosis: a meta‐analysis.Cancer Epidemiol 2013;37:523–7. [DOI] [PubMed] [Google Scholar]
- 16. Kuo CF, Luo SF, Yu KH, Chou IJ, Tseng WY, Chang HC, et al.Cancer risk among patients with systemic sclerosis: a nationwide population study in Taiwan.Scand J Rheumatol 2012;41:44–9. [DOI] [PubMed] [Google Scholar]
- 17. Chen YJ, Wu CY, Huang YL, Wang CB, Shen JL, Chang YT.Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan.Arthritis Res Ther 2010;12:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Airio A, Pukkala E, Isomaki H.Elevated cancer incidence in patients with dermatomyositis: a population based study.J Rheumatol 1995;22:1300–3. [PubMed] [Google Scholar]
- 19. Buchbinder R, Forbes A, Hall S, Dennett X, Giles G.Incidence of malignant disease in biopsy‐proven inflammatory myopathy: a population‐based cohort study.Ann Intern Med 2001;134:1087–95. [DOI] [PubMed] [Google Scholar]
- 20. Chow WH, Gridley G, Mellemkjaer L, McLaughlin JK, Olsen JH, Fraumeni JF Jr. Cancer risk following polymyositis and dermatomyositis: a nationwide cohort study in Denmark.Cancer Causes Control 1995;6:9–13. [DOI] [PubMed] [Google Scholar]
- 21. Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, et al.Frequency of specific cancer types in dermatomyositis and polymyositis: a population‐based study.Lancet 2001;357:96–100. [DOI] [PubMed] [Google Scholar]
- 22. Stockton D, Doherty VR, Brewster DH.Risk of cancer in patients with dermatomyositis or polymyositis, and follow‐up implications: a Scottish population‐based cohort study.Br J Cancer 2001;85:41–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zahr ZA, Baer AN.Malignancy in myositis.Curr Rheumatol Rep 2011;13:208–15. [DOI] [PubMed] [Google Scholar]
- 24. Nikpour M, Hissaria P, Byron J, Sahhar J, Micallef M, Paspaliaris W, et al.Prevalence, correlates and clinical usefulness of antibodies to RNA polymerase III in systemic sclerosis: a cross‐sectional analysis of data from an Australian cohort.Arthritis Res Ther 2011;13:R211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Derk CT.Associations of breast cancer development in patients with systemic sclerosis: an exploratory study.Clin Rheumatol 2007;26:1615–9. [DOI] [PubMed] [Google Scholar]
- 26. Fardet L, Dupuy A, Gain M, Kettaneh A, Cherin P, Bachelez H, et al.Factors associated with underlying malignancy in a retrospective cohort of 121 patients with dermatomyositis.Medicine (Baltimore) 2009;88:91–7. [DOI] [PubMed] [Google Scholar]
- 27. Ponyi A, Constantin T, Garami M, Andras C, Tallai B, Vancsa A, et al.Cancer‐associated myositis: clinical features and prognostic signs.Ann N Y Acad Sci 2005;1051:64–71. [DOI] [PubMed] [Google Scholar]
- 28. Zantos D, Zhang Y, Felson D.The overall and temporal association of cancer with polymyositis and dermatomyositis.J Rheumatol 1994;21:1855–9. [PubMed] [Google Scholar]
- 29. Colaci M, Giuggioli D, Vacchi C, Lumetti F, Iachetta F, Marcheselli L, et al.Breast cancer in systemic sclerosis: results of a cross‐linkage of an Italian Rheumatologic Center and a population‐based Cancer Registry and review of the literature.Autoimmun Rev 2014;13:132–7. [DOI] [PubMed] [Google Scholar]
- 30. Moinzadeh P, Fonseca C, Hellmich M, Shah AA, Chighizola C, Denton CP, et al.Association of anti‐RNA polymerase III autoantibodies and cancer in scleroderma.Arthritis Res Ther 2014;16:R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andras C, Ponyi A, Constantin T, Csiki Z, Szekanecz E, Szodoray P, et al.Dermatomyositis and polymyositis associated with malignancy: a 21‐year retrospective study.J Rheumatol 2008;35:438–44. [PubMed] [Google Scholar]
- 32. Hasegawa M, Sato S, Sakai H, Ohashi T, Takehara K.Systemic sclerosis revealing T‐cell lymphoma.Dermatology 1999;198:75–8. [DOI] [PubMed] [Google Scholar]
- 33. Juarez M, Marshall R, Denton C, Evely R.Paraneoplastic scleroderma secondary to hairy cell leukaemia successfully treated with cladribine.Rheumatology (Oxford) 2008;47:1734–5. [DOI] [PubMed] [Google Scholar]
- 34. Mammen AL.Dermatomyositis and polymyositis: clinical presentation, autoantibodies, and pathogenesis.Ann N Y Acad Sci 2010;1184:134–53. [DOI] [PubMed] [Google Scholar]
- 35. Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola‐Rosen L.The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM‐140): a retrospective study.J Am Acad Dermatol 2011;65:74–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Christopher‐Stine L, Casciola‐Rosen LA, Hong G, Chung T, Corse AM, Mammen AL.A novel autoantibody recognizing 200‐kd and 100‐kd proteins is associated with an immune‐mediated necrotizing myopathy.Arthritis Rheum 2010;62:2757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mammen AL, Chung T, Christopher‐Stine L, Rosen P, Rosen A, Doering KR, et al.Autoantibodies against 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase in patients with statin‐associated autoimmune myopathy.Arthritis Rheum 2011;63:713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mimura Y, Takahashi K, Kawata K, Akazawa T, Inoue N.Two‐step colocalization of MORC3 with PML nuclear bodies.J Cell Sci 2010;123:2014–24. [DOI] [PubMed] [Google Scholar]
- 39. Kaji K, Fujimoto M, Hasegawa M, Kondo M, Saito Y, Komura K, et al.Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy.Rheumatology (Oxford) 2007;46:74–8. [DOI] [PubMed] [Google Scholar]
- 40. Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O'Hanlon TP, et al, for the Childhood Myositis Heterogeneity and International Myositis Collaborative Study Groups .A novel autoantibody to a 155‐kd protein is associated with dermatomyositis.Arthritis Rheum 2006;54:3682–9. [DOI] [PubMed] [Google Scholar]
- 41. Targoff IN, Trieu EP, Levy‐Neto M, Prasertsuntarasai T, Miller FW.Autoantibodies to transcriptional intermediary factor 1‐γ(TIF1‐γ) in dermatomyositis [abstract].Arthritis Rheum 2006;54 Suppl:S518. [Google Scholar]
- 42. Massague J, Xi Q.TGF‐β control of stem cell differentiation genes.FEBS Lett 2012;586:1953–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow‐Tsang LF, Liu JX, et al.A poised chromatin platform for TGF‐β access to master regulators.Cell 2011;147:1511–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hesling C, Lopez J, Fattet L, Gonzalo P, Treilleux I, Blanchard D, et al.Tif1γ is essential for the terminal differentiation of mammary alveolar epithelial cells and for lactation through SMAD4 inhibition.Development 2013;140:167–75. [DOI] [PubMed] [Google Scholar]
- 45. Trallero‐Araguas E, Rodrigo‐Pendas JA, Selva‐O'Callaghan A, Martinez‐Gomez X, Bosch X, Labrador‐Horrillo M, et al.Usefulness of anti‐p155 autoantibody for diagnosing cancer‐associated dermatomyositis: a systematic review and meta‐analysis.Arthritis Rheum 2012;64:523–32. [DOI] [PubMed] [Google Scholar]
- 46. Oddis CV, Fertig N, Goel A, Espada G, Confalone Gregorian M, Maldonado Cocco JA, et al.Clinical and serological characterization of the anti‐MJ antibody in childhood myositis [abstract].Arthritis Rheum 1997;40 Suppl:S139. [Google Scholar]
- 47. Targoff IN, Trieu EP, Levy‐Neto M, Fertig N, Oddis CV.Sera with autoantibodies to the MJ antigen react with NXP2 [abstract].Arthritis Rheum 2007;56 Suppl:S787. [Google Scholar]
- 48. Kimura Y, Sakai F, Nakano O, Kisaki O, Sugimoto H, Sawamura T, et al.The newly identified human nuclear protein NXP‐2 possesses three distinct domains, the nuclear matrix‐binding, RNA‐binding, and coiled‐coil domains.J Biol Chem 2002;277:20611–7. [DOI] [PubMed] [Google Scholar]
- 49. Gunawardena H, Wedderburn LR, Chinoy H, Betteridge ZE, North J, Ollier WE, et al, for the Juvenile Dermatomyositis Research Group, UK and Ireland .Autoantibodies to a 140‐kd protein in juvenile dermatomyositis are associated with calcinosis.Arthritis Rheum 2009;60:1807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Espada G, Maldonado Cocco JA, Fertig N, Oddis CV.Clinical and serologic characterization of an Argentine pediatric myositis cohort: identification of a novel autoantibody (anti‐MJ) to a 142‐kDa protein.J Rheumatol 2009;36:7447–51. [DOI] [PubMed] [Google Scholar]
- 51. Ceribelli A, Fredi M, Taraborelli M, Cavazzana I, Franceschini F, Quinzanini M, et al.Anti‐MJ/NXP‐2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis.Arthritis Res Ther 2012;14:R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al.Anti‐NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy.Ann Rheum Dis 2012;71:710–3. [DOI] [PubMed] [Google Scholar]
- 53. Fiorentino DF, Chung LS, Christopher‐Stine L, Zaba L, Li S, Mammen AL, et al.Most patients with cancer‐associated dermatomyositis have antibodies to nuclear matrix protein NXP‐2 or transcription intermediary factor 1γ.Arthritis Rheum 2013;65:2954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Harris ML, Rosen A.Autoimmunity in scleroderma: the origin, pathogenetic role, and clinical significance of autoantibodies.Curr Opin Rheumatol 2003;15:778–84. [DOI] [PubMed] [Google Scholar]
- 55. Steen VD.The many faces of scleroderma.Rheum Dis Clin North Am 2008;34:1–15, v. [DOI] [PubMed] [Google Scholar]
- 56. Graf SW, Hakendorf P, Lester S, Patterson K, Walker JG, Smith MD, et al.South Australian Scleroderma Register: autoantibodies as predictive biomarkers of phenotype and outcome.Int J Rheum Dis 2012;15:102–9. [DOI] [PubMed] [Google Scholar]
- 57. Shah AA, Wigley FM.My approach to the treatment of scleroderma.Mayo Clin Proc 2013;88:377–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Airo P, Ceribelli A, Cavazzana I, Taraborelli M, Zingarelli S, Franceschini F.Malignancies in Italian patients with systemic sclerosis positive for anti‐RNA polymerase III antibodies.J Rheumatol 2011;38:1329–34. [DOI] [PubMed] [Google Scholar]
- 59. Shah AA, Rosen A.Cancer and systemic sclerosis: novel insights into pathogenesis and clinical implications.Curr Opin Rheumatol 2011;23:530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin RH, Mamula MJ, Hardin JA, Janeway CA Jr. Induction of autoreactive B cells allows priming of autoreactive T cells.J Exp Med 1991;173:1433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mamula MJ, Lin RH, Janeway CA Jr, Hardin JA.Breaking T cell tolerance with foreign and self co‐immunogens: a study of autoimmune B and T cell epitopes of cytochrome c.J Immunol 1992;149:789–95. [PubMed] [Google Scholar]
- 62. Darnell RB, Posner JB.Paraneoplastic syndromes involving the nervous system.N Engl J Med 2003;349:1543–54. [DOI] [PubMed] [Google Scholar]
- 63. Gogas H, Ioannovich J, Dafni U, Stavropoulou‐Giokas C, Frangia K, Tsoutsos D, et al.Prognostic significance of autoimmunity during treatment of melanoma with interferon.N Engl J Med 2006;354:709–18. [DOI] [PubMed] [Google Scholar]
- 64. Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth‐Dreese FA, et al.Tumor regression and autoimmunity after reversal of a functionally tolerant state of self‐reactive CD8+ T cells.J Exp Med 2003;198:569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Albert ML, Darnell RB.Paraneoplastic neurological degenerations: keys to tumour immunity.Nat Rev Cancer 2004;4:36–44. [DOI] [PubMed] [Google Scholar]
- 66. Casciola‐Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, et al.Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy.J Exp Med 2005;201:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Burnet M.Cancer: a biological approach. I. The processes of control.Br Med J 1957;1:779–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stutman O.Immunodepression and malignancy.Adv Cancer Res 1975;22:261–422. [DOI] [PubMed] [Google Scholar]
- 69. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD.Cancer immunoediting: from immunosurveillance to tumor escape.Nat Immunol 2002;3:991–8. [DOI] [PubMed] [Google Scholar]
- 70. Poduri A, Evrony GD, Cai X, Walsh CA.Somatic mutation, genomic variation, and neurological disease.Science 2013;341:1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Matzinger P.Tolerance, danger, and the extended family.Annu Rev Immunol 1994;12:991–1045. [DOI] [PubMed] [Google Scholar]
- 72. Sigurgeirsson B, Lindelof B, Edhag O, Allander E.Risk of cancer in patients with dermatomyositis or polymyositis: a population‐based study.N Engl J Med 1992;326:363–7. [DOI] [PubMed] [Google Scholar]
- 73. Huang YL, Chen YJ, Lin MW, Wu CY, Liu PC, Chen TJ, et al.Malignancies associated with dermatomyositis and polymyositis in Taiwan: a nationwide population‐based study.Br J Rheumatol 2009;161:854–60. [DOI] [PubMed] [Google Scholar]
- 74. Chatterjee S, Dombi GW, Severson RK, Mayes MD.Risk of malignancy in scleroderma: a population‐based cohort study.Arthritis Rheum 2005;52:2415–24. [DOI] [PubMed] [Google Scholar]