

Abstract
Subcutaneous route of administration is highly desirable for protein therapeutics. It improves patient compliance and quality of life1,2, while reducing healthcare cost2. Recent evidence also suggests that sc administration of protein therapeutics can increase tolerability to some treatments such as intravenous immunoglobulin therapy (IVIG) by administering it subcutaneously (subcutaneous immunoglobulin therapy SCIG), which will reduce fluctuation in plasma drug concentration3. Furthermore, sc administration may reduce the risk of systemic infections associated with iv infusion1,2. This route, however, has its challenges especially for large multi-domain proteins. Poor bioavailability and poor scalability from preclinical models are often cited. This commentary will discuss barriers to sc absorption as well as physiological and experimental factors that could affect pharmacokinetics of subcutaneously administered large protein therapeutics in preclinical models. A mechanistic pharmacokinetic model is proposed as a potential tool to address the issue of scalability of sc pharmacokinetic from preclinical models to humans
Introduction
Protein therapeutics are classified based on their pharmacological function into i) proteins with enzymatic/regulatory function or ii) proteins with targeting function (monoclonal antibodies)4. The first class contains proteins ranging in size from small peptide-hormones such as insulin and erythropoietin to the large multi-domain proteins such as FVIII and Acid Alpha-Glucosidase (GAA). These therapeutics are designed to: i) replace lacking or aberrantly formed endogenous counterparts to ameliorate disease conditions such as the use of Insulin in diabetes. ii) Augment existing pathways such as the use of human follicle-stimulating hormone (FSH) for infertility. iii) Provide a novel function such as Hyaluronidase5,6. The second class contains monoclonal antibodies (mAb) and their derivatives. This class of protein therapeutics is characterized by unique pharmacokinetics due to their high target-binding affinity and the presence of the Fc fragment (in the case of mAb), which imparts the prolonged half-life of this class of biologics.
The wide range in the size and properties of protein therapeutics makes it difficult to treat them as a single class of therapeutics especially when discussing sc absorption. Furthermore, the classification of protein therapeutics based on pharmacological function may be irrelevant when discussing absorption from the subcutaneous space. This necessitates a different categorization system based on size rather than function of these therapeutics. The following sections discusses the physical barriers to sc absorption of protein therapeutics, which should help in classifying protein therapeutics, based on size, into i) small proteins < 10 nm in diameter ii) large proteins > 10 nm in diameter iii) and mAbs. Next we discuss pre-systemic degradation as a contributing factor to incomplete bioavailability before presenting possible experimental artifacts in preclinical models that can further contribute to poor scalability to humans.
2. Barrier to sc absorption of protein therapeutics
2.1 Physical barriers
After a drug is deposited in the sc space, it must traverse the extracellular matrix to reach an entry point into systemic circulation. Entry can be directly into the blood stream or by transiting through the lymphatics 7.
2.1.1 Direct uptake into blood
Uptake into blood requires entry at the post-capillary bed or by traversing the basal membrane of blood vesicles, both of which are size limiting. The post-capillary bed is involved in blood/tissue fluid exchange, it is also the primary site of leukocytes and plasma protein leakage8. These capillaries preferentially reabsorb particles up to 10 nm9. Alternatively, the drug enters systemic circulation by crossing the basal membrane of blood vessels via the para-cellular or trans-cellular pathway. The former is limited by the size of the fenestrations in the basal membrane reported to be 6–12 nm for most non-sinusoidal blood capillaries 10.
The trans-cellular pathway may not be a major player in protein uptake. Indeed, large proteins have been shown to have poor trans-cellular trafficking11. Those therapeutic proteins are generally hypdrophilic, which prevents them from traversing the cell membrane. Protein entering through pinocytosis or phagocytosis will likely be degraded leading to the loss of protein. One exception is monoclonal antibodies. Trans-cellular transport of mAbs has been recognized since the early 70’s11. This is mediated by FcRn receptors on the surface of endothelia cells. FcRn not only facilitates the bidirectional transport of mAbs12,13, but it also protects the antibody during fluid phase pinocytosis by binding the antibody and sorting it away from the lysosomal pathways14–16. FcRn mediated transport explains the high bioavailability and the saturable nature17 of mAb uptake from sc.
Physicochemical properties of antibodies that can potentially affect trans-cellular trafficking of mAbs such as isotype, FcRn binding affinity, charge, hydrophobicity and solubility have been investigated by a number of researchers in the field with conflicting results. For example, Khawli et al showed no effect of charge variants of IgG1 on their pharmacokinetics after sc administration in rats18. This, however, is in contrast to other reports showing that alteration to the isoelectric point of mAbs altered the bioavailability after sc administration in mice19. Interestingly, both groups reported no change in FcRn binding affinity as a function of changes in pI of the protein18,19.
The role of pI and protein surface charge in sc uptake could be explained by charge-charge interaction during fluid phase pinocytosis. It has been shown that IgG’s with higher pI have higher cellular uptake20,21. This suggests that a positively charged IgG interacts more favorably with the negatively charged cell surface allowing for more uptake of mAb during fluid phase pinocytosis21,19,20, the IgG will then bind to FcRn which will protect it from degradation. Negatively charged IgG, on the other hand, will have lower uptake due to the repulsion between the negative protein and the negative cell surface19. However, in the context of the sc space, the repulsive forces between the negative protein and the negative extracellular matrix could enhance convective movement of the protein through the extracellular matrix and improve lymphatic trafficking, as we will discuss below.
2.1.2 Uptake by the lymphatics
Uptake by lymphatics is less restrictive. The initial lymphatics, where interstitial fluid enters the lymphatic system and becomes lymph fluid, do not have a continuous basal membrane 9,22. Rather, the endothelial cells of the initial lymphatic overlap while being anchored by collagen VII to the extra-cellular matrix 9,22. The lack of the basal membrane allows for large proteins as well as small cells, bacteria and viruses to enter the lymphatics 22. Anchorage to the extra-cellular matrix, on the other hand, allows for the transmission of mechanical forces from the extra-cellular matrix to the lymphatic lumen 22. This can allow the initial lymphatics to open up in response to mechanical movement; this can explain the improved lymph flow in response to massaging or movement.
Despite the lax size limitation of lymphatic uptake, there are still a number of other impediments to absorption of biologics via this route. After injection, the protein must navigate the extra-cellular matrix to reach a point of entry into the lymphatics. The density of the initial lymphatics at the injection site7 will affect the proficiency of lymphatic uptake of protein from the injection site. This process can also be affected by the size and charge of the proteins 9. Larger proteins are selectively taken up by the lymphatics, however, the larger the protein the slower the uptake 9 due to increased resistance to convective and/or diffusive movement. Also, electrostatic interaction with glycosaminoglycanes, the negatively charged component of the glycocalyx matrix8,9, can hider or promote the movement of the protein through the extracellular matrix9,23. Indeed, positively charged proteins have been reported to reach the lymphatics at a delayed time as compared to negatively charged protein of comparable size 24.
It is important to note that the above-mentioned uptake pathways are not mutually exclusive, and protein absorption can occur via one or more of the pathways discussed above. For example; small protein therapeutics can utilize the post-capillary bed as well as the fenestrations in the basal membrane of blood vessels; this explains their good bioavailability. mAbs can utilize FcRn receptors on the surface of endothelial cells as well as lymphatic uptake. Large protein therapeutics, however, must utilize lymphatic uptake.
Strategies that can overcome one or more of the above mentioned physical barriers could enhance bioavailability of protein therapeutics. For example the use of hyaluronidase to “loosen” the extra-cellular matrix enhances the diffusion of the co-administered biologics6. Another example is the use of albumin to manipulate the oncotic pressure, and by extension the interstitial fluid volume in sc space, to enhance sc bioavailability25,26. Other strategies to manipulate the environment in the sc space to improve overall bioavailability of protein therapeutics such as viscosity, osmolarity and volume of injection have been recognized by a number of workers in the field and are discussed in a number of reviews7,27.
In our own work, we found a relationship between increased buffer tonicity and improved bioavailability of subcutaneously administered rituximab in a mouse model28. Furthermore, our data suggests that the effect of buffer hypertonicity on rituximab bioavailability is excipient specific. We found that mannitol, a neutral excipient, performed better than the negatively charged O-Phoshpo-L-Serine at equal tonicities. The enhanced bioavailability was associated with enhanced lymph node uptake of rituximab28. We propose that hypertonic buffers perturb the isotonicity of the interstitial space altering the formation and reabsorption of interstitial fluid at the post-capillary beds. The draining of excess interstitial fluid by the lymphatics enhances bulk movement of fluid through the sc space carrying with it the protein from the injection site through to the lymphatic28.
2.2 Pre-systemic degradation
Another complicating factor for sc absorption of protein therapeutics is pre-systemic elimination7,17. This could be due to degradation at the injection site by proteolytic enzymes. The presence of such enzymes is supported by reports showing this proteolytic activity in the sc space to be saturable by administering high doses of the drug as well as inhibited by co-administering protease inhibitors 17,29,30. This proteolytic activity has been proposed as a reason for incomplete bioavailability of biologics. Another form of pre-systemic elimination is the uptake and processing of protein therapeutics by professional antigen presenting cells in the skin. Upon sc administration, dermis and epidermis resident dendritic cells migrate to the injection site to sample the injected protein31. This leads to maturation of these cells, which is accompanied by the release of pro-inflamatory cytokine and chemokines 31–33. This can recruit more antigen presenting cells to the injection site for sustained sampling of the injected drug for later presentation in the lymph node 31–33. This sustained sampling of the drug can be a significant form of elimination at the injection site. Reducing the residence time of the drug in the subcutaneous space could reduce the effect of the aforementioned degradation pathways. This could be achieved by enhancing lymphatic uptake, which will siphon the drug away from the injection site. Such a strategy could also prove particularly effective in situations where saturable uptake, such as FcRn uptake of mAbs, is in effect. Trafficking the drug effectively through the lymphatic may alleviate the load on uptake transporters and enhance overall bioavailability of injected therapeutic proteins.
3. Preclinical models and experimental artifacts
3.1 Anatomy and physiology of preclinical models
The complexity of absorption processes after sc administration, as discussed in the previous section, and the variety of pathways that are involved in this process are key contributing factors to the poor scalability and poor predictive power of preclinical species. This section will examine some physiological and experimental factors that can further contribute to this issue. The suitability of rodent models for predicting pharmacokinetics of subcutaneously administered biotherapeutic in humans has been questioned recently. Anatomical and physiological differences in rodent sc space viz a viz humans are often cited. For example, rodent models exhibits a wide lateral expansion of an sc injected dose due to loose connective tissue in the sc space 7. This lateral expansion means a wider surface area for the drug to diffuse through and can result in better absorption. Another commonly cited difference is the panniculus carnosus, a muscular layer embedded in the hypodermis under the superficial adipose tissue and above the membranous layer (figure 1A). This layer is less pronounced in larger non-furred animals and almost lacking in humans 1.
Figure 1.
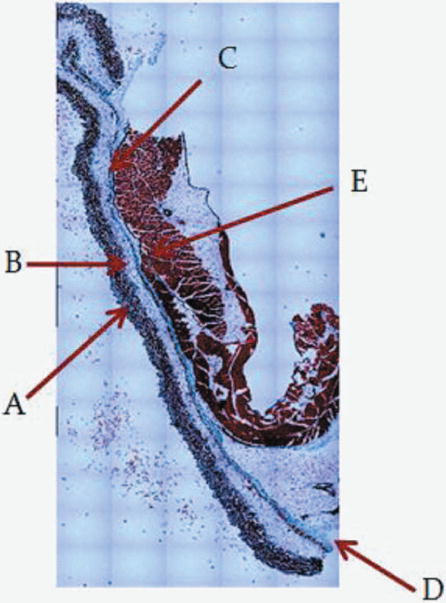
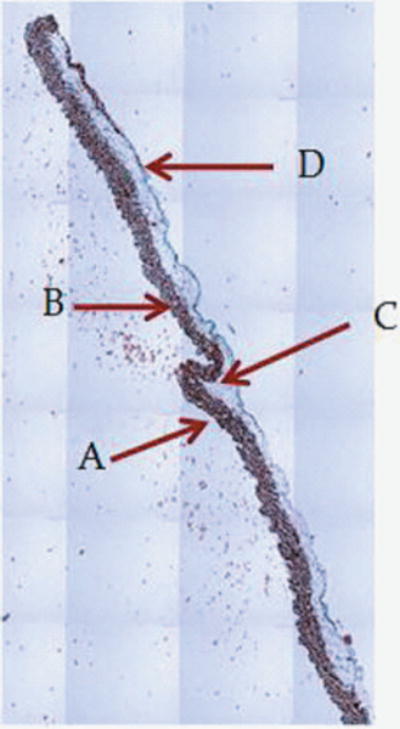
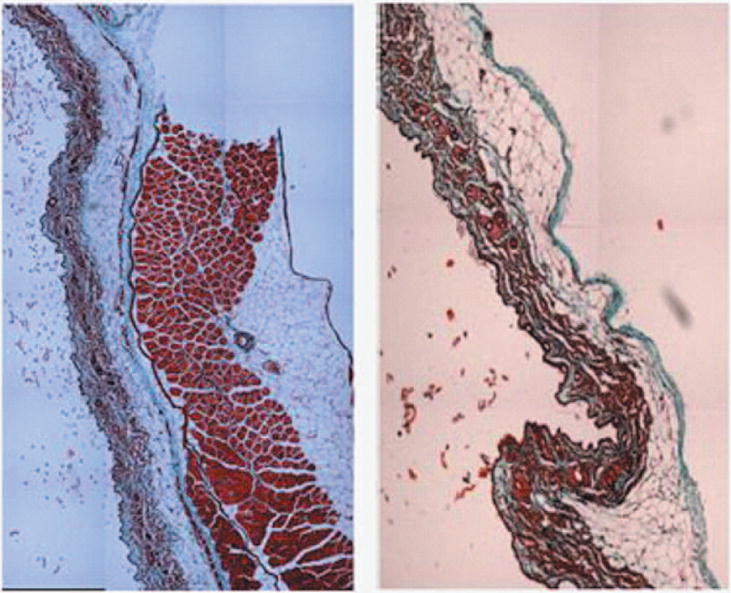
1A: 5X magnification of skin and abdomen samples. On the left Trichrom staining of the abdominal intact wall-skin samples obtained from HA mice. On the right is the skin sample obtained after a simulated sc injection using the “pinch” method. This figures show that the “pinch” method is separating the skin at the membranous layer, which results in a much deeper deposit of the sc dose than expected in humans. Arrow are pointing at: A: Dermis. B: Epidermis. C: Superficial adipose tissue D: Deep adipose tissue E: Membranous layer. 1B: 40X magnification of skin and abdomen samples. On the left Trichrom staining of the abdominal wall-skin samples obtained from HA mice we can see the panniculus carnosus (red intermittent muscular layer). On the right is the skin sample obtained from the “pinch” method. Here we can see the membranous layer (blue) appearing thinner when compared to the image to the right.
Other preclinical models, such as swine models, have a fixed skin with a tight attachment to the subcutaneous tissues34. This may reduce the issue of wide lateral expansion after sc administration. In general, swine models are regarded suitable for experimental toxicologic, dermatologic and wound healing studies 35 and are gaining popularity in pharmacokinetics studies. This is mainly due to structural and biochemical similarities between human and pig skin 34–36. There are, however, potentially important differences that can affect the absorption of biotherapeutics from sc space in pigs as compared to humans. Reduced vasculature in pig’s skin and a unique inverted architecture of swine lymph nodes 34,37 may affect the uptake and trafficking of drug from sc space.
Despite the anatomical similarities between swine models and human skin, the fact remains that there is no reliable preclinical model to predict pharmacokinetics of sc administered protein therapeutics in humans 1. Indeed, a comparison of the bioavailability of 13 biotherapeutic in human vs monkeys, dogs, rats, mice and pigs, showed no strong correlation between results in humans and any one species 7. If anatomy and physiology, per se, cannot account for the differences in sc uptake and bioavailability, then one must examine some experimental artifacts that could help explain the observed differences.
3.2: Experimental artifacts affecting PK of sc administered protein therapeutics
One might argue that the experimental procedure may significantly contribute to the poor scalability of bioavailability estimates obtained in rodents or large animals to humans. One such artifact is the volume of injection used in preclinical models. Those volumes could create super-physiological pressure in the sc space that will not translate into humans. For example; a 100-ul sc injection into a 20 g mouse, corresponds to a volume of 350 ml for an average 70 kg human if scaled based on body weight. Or 23 ml if scaled up based on surface area. Both of which are un-realistic super-physiological volumes. This indicates that rodent models are being injected with massive volumes creating ultra-physiological pressure in the interstitial space. As the volume spreads laterally in the sc space it may cause shear stress. This is known to cause alteration in the glycocalyx structure and the release of Nitric Oxide8, a potent intracellular signaling molecule, altering the permeability of the endothelial barrier8.
The effect of large injection volumes (relative to body size) could be a major contributor to the altered rate of uptake in small animals and is not usually accounted for in typical PK models. Furthermore, this effect is species independent since it depends on the size of the animals. One could argue that this artifact could plague data from other pre-clinical models with more relevant sc anatomy and physiology but small body size and surface area such as mini-pigs. This is a very important question to investigate especially with the increased interest in using mini-pigs as preclinical models for sc administration of protein based therapeutics38. Swine models not only have similar sc anatomy and physiology compared to humans, but their surface area is also comparable to that of an adult human34. This makes them a good candidate for a preclinical model of sc absorption. However, due to difficulty in handling, housing and experimenting with typical “farm” pigs, miniature breeds called minipigs were developed out of necessity by selective breeding 35. Those breeds, however, are one fifth to one tenth the size of their “farm” or domestic counterparts depending on the breed (Hanford or Yucatan, vs Sinclair or Göttingen)35. This means that mini-pigs have a smaller body surface area and as such could be susceptible to the effects of large volume of injection (compared to body surface area) as other “small” preclinical models. Indeed, a study comparing the bioavailability of 9 mAb in Göttingen minipigs (which can grow to about 10 Kg) showed poor correlation between bioavailability and absorption rate obtained in those pigs and humans38. Values obtained from mini-pigs were higher for the majority of the tested mAbs38. This is despite good correlation in clearance and similar FcRn binding properties in both species.
Injection technique is also recognized as a determining factor in the bioavailability of sc administered biologics1. When we examined the pinch method in mouse models, one of the more common methods of sc injection into mice, we observed that this method results in separation of the membranous layer of the sc space in mice (Figure 1A and B). This indicates that the injected dose is being deposited much deeper in rodents as opposed to humans. In humans the dose is expected to deposit in the superficial adipose tissue and not the membranous layer. This deeper deposit in rodent models can have two effects: i) the drug has to traverse a shorter distance to reach an uptake site and ii) the drug may be taken up by initial lymphatics of different compliance and elasticity affecting its propulsion. Negrini et al 22 highlight the effects of the local surroundings of initial lymphatics on lymph drainage, retention and propulsion. Their model predicts that vessels surrounded by loose compliant tissue are themselves compliant and as such act as reservoirs of drained lymphatics, but have a slower lymphatic propulsion a compared to those surrounded by stiff tissue22.
Another experimental artifact that can account for the differences between sc data obtained in rodent models vs humans is the site of injection. Active areas in the body or those with cyclical compression and expansion, such as the thoracic region, tend to have higher lymph flow due to high mechanical stress imposed on the lumen of the initial lymphatics 22. In our lab we observed marked differences in the amount of FVIII recovered in plasma after 3 hours of injecting FVIII sc in the ventral (abdominal skin) vs the dorsal side (scruff) of HA mice (table 1), highlighting the effects of injection site on absorption in small animals. The rapid breathing of small rodents, such as mice, results in rapid cyclical motion in the thoracic and abdominal region of the animal. Since humans have slower breathing rate and less pronounced movement of the abdominal region with every breath, we do not expect marked difference in bioavailability between abdominal injection and injections in the thigh for example. Another common experimental practice in preclinical settings is the injection into the paw of the hind leg. This is done for ease of collection of lymph data. This practice is also seen when larger animals are used such as sheep and dogs. Those anatomical sites have no equivalent in humans; this practice may also result in an exaggerated response not seen in humans.
Table 1.
FVIII recovery from plasma 3 hours after sc administration of 5IU/g of FVIII into the lower dorsal region or the abdominal region of HA mice.
| Injection site | Plasma IU/ml |
|---|---|
| Dorsal Side | 0.25 |
| 0 | |
| 0 | |
| Abdominal | 2.20 |
| 0.28 | |
| 1.18 |
Limitations, notwithstanding, we argue that small preclinical models, such as rodent models, are suitable for mechanistic studies and can help shed light on factors affecting uptake from sc space. Also, we must acknowledge that due to logistic and practical considerations, especially in academic setting, rodent models will not be replaced anytime soon by larger animals. However, by understanding their limitations we can cautiously extrapolate into higher species and humans. Minipigs are also emerging as a possible practical model for sc absorption; however, caution must be practiced to account for some physiological and experimental effects as mentioned above.
We further propose that a mechanistic pharmacokinetic model (such as the one proposed in figure 2) for sc uptake that takes into account the effects of different barriers, limitation, and experimental artifacts on sc absorption should help us better predict sc bioavailability in humans from preclinical data. In the model presented in figure 2, the skin in perfused with arterial blood at a rate “Q”, and drained by lymph flow and venular blood flow at rates QL and Q-QL respectively. Within the skin, the blood and interstitial compartments are presented separately, interstitial fluid is formed and reabsorbed based on known physiological parameters at steady state. Excess interstitial fluid is drained by the lymph flow. Such a model will allow for physiological parameter to change as we extrapolate from one species to another. Depositing a dose in the interstitial space will perturb steady state conditions. This will result in changes in permeability, interstitial volume and flow. The magnitude and extent of such changes will differ from one species to another. This could be determined from in vitro and in vivo studies and then incorporated into the model.
Figure 2.
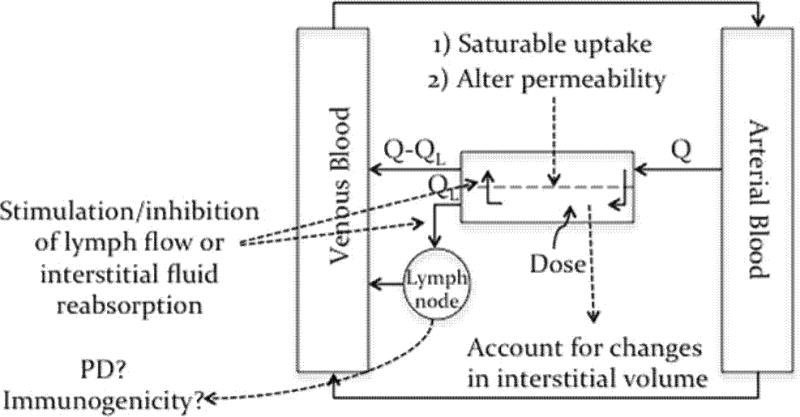
A proposed physiological model for sc absorption, see text for discussion and details.
For example studying the effect of skin biopsy homogenates on protein therapeutic stability in vitro can shed light on the rate of protein degradation post sc injection. This rate could differ depending on the preclinical model being used. Another useful parameter to determine from in vitro analysis of skin biopsy is the Glycoaminoglycan content of the sc space in different animal models and how it related to humans sc space. Swabb et al showed a relationship between Glycoaminoglycan content in different tissue and the dominance of convective vs diffusive forces on macromolecule movement in those tissue39. Indeed, the disruption of the Glycoaminoglycan matrix by enzymes such as hyaluronidase can alter the dispersion of co-injected therapeutic proteins as well as reduce interstitial pressure5,40 further highlighting the importance of the glycosaminoglycan matrix in macromolecule transport and absorption from the sc space. The rate of interstitial fluid flow in the proposed mechanistic model can be modulated to account for interspecies differences in Glycosaminoglycan content. The binding affinity of the therapeutic protein to possible ligand in the sc space could also be determined from in vitro studies. If such a ligand exists in the sc space, it can hinder the migration and absorption of the therapeutic protein by trapping it in the skin. This is especially important in the case of monoclonal antibodies. mAbs are designed to bind their target with high affinity, if the target is abundant in the sc space, this can hinder the absorption of mAbs. Furthermore, FcRn binding and uptake is key in mAb absorption from sc space17. Thus, determining the affinity and abundance of mAb ligands in different preclinical models as well as mAb/FcRn binding affinity as compared to humans can further improve the predictive power of preclinical data.
Similar all-encompassing models for oral absorption exist with successful commercial software applications capable of incorporating a wide range of information such as drug properties, in vitro-in vivo correlation (IVIVC) of transport and metabolism data as well as physiological data. Similar efforts are needed to develop such models that can better predict the absorption and bioavailability of this important class of therapeutics after sc administration.
Acknowledgments
The authors would like to acknowledge Krithika Shetty for her input and help during the revision of this manuscript. The authors are grateful for the financial support from National Heart, Lung and Blood Institute, National Institute of Health, HL-70227 to Dr Sathy Balu-Iyer.
Footnotes
Conflict of Interest:
The authors declare that there is no financial conflict of interest.
References
- 1.McDonald TA, Zepeda ML, Tomlinson MJ, Bee WH, Ivens IA. Subcutaneous administration of biotherapeutics: current experience in animal models. Curr Opin Mol Ther. 2010;12(4):461–470. [PubMed] [Google Scholar]
- 2.Dychter SS, Gold DA, Haller MF. Subcutaneous drug delivery: a route to increased safety, patient satisfaction, and reduced costs. J Infus Nurs. 2012;35(3):154–160. doi: 10.1097/NAN.0b013e31824d2271. [DOI] [PubMed] [Google Scholar]
- 3.Kobrynski L. Subcutaneous immunoglobulin therapy: a new option for patients with primary immunodeficiency diseases. Biologics. 2012;6:277–287. doi: 10.2147/BTT.S25188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nature reviews Drug discovery. 2008;7(1):21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 5.Bookbinder LH, Hofer A, Haller MF, Zepeda ML, Keller GA, Lim JE, Edgington TS, Shepard HM, Patton JS, Frost GI. A recombinant human enzyme for enhanced interstitial transport of therapeutics. Journal of controlled release : official journal of the Controlled Release Society. 2006;114(2):230–241. doi: 10.1016/j.jconrel.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 6.Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert opinion on drug delivery. 2007;4(4):427–440. doi: 10.1517/17425247.4.4.427. [DOI] [PubMed] [Google Scholar]
- 7.Richter WF, Bhansali SG, Morris ME. Mechanistic determinants of biotherapeutics absorption following SC administration. Aaps J. 2012;14(3):559–570. doi: 10.1208/s12248-012-9367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan SYRR. In: Structure and Function of Exchange Regulation of Endothelial Barrier Function. San Rafael Morgan & Claypool Life Sciences, editor. 2010. [PubMed] [Google Scholar]
- 9.Swartz MA. The physiology of the lymphatic system. Advanced Drug Delivery Reviews. 2001;50(1,Äì2):3–20. doi: 10.1016/s0169-409x(01)00150-8. [DOI] [PubMed] [Google Scholar]
- 10.Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2(14):14. doi: 10.1186/2040-2384-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones EA, Waldmann TA. The mechanism of intestinal uptake and transcellular transport of IgG in the neonatal rat. J Clin Invest. 1972;51(11):2916–2927. doi: 10.1172/JCI107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickinson BL, Badizadegan K, Wu Z, Ahouse JC, Zhu X, Simister NE, Blumberg RS, Lencer WI. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J Clin Invest. 1999;104(7):903–911. doi: 10.1172/JCI6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claypool SM, Dickinson BL, Wagner JS, Johansen FE, Venu N, Borawski JA, Lencer WI, Blumberg RS. Bidirectional transepithelial IgG transport by a strongly polarized basolateral membrane Fcgamma-receptor. Molecular biology of the cell. 2004;15(4):1746–1759. doi: 10.1091/mbc.E03-11-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. European journal of immunology. 1996;26(3):690–696. doi: 10.1002/eji.1830260327. [DOI] [PubMed] [Google Scholar]
- 15.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93(11):5512–5516. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tzaban S, Massol RH, Yen E, Hamman W, Frank SR, Lapierre LA, Hansen SH, Goldenring JR, Blumberg RS, Lencer WI. The recycling and transcytotic pathways for IgG transport by FcRn are distinct and display an inherent polarity. The Journal of cell biology. 2009;185(4):673–684. doi: 10.1083/jcb.200809122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kagan L, Turner MR, Balu-Iyer SV, Mager DE. Subcutaneous absorption of monoclonal antibodies: role of dose, site of injection, and injection volume on rituximAb pharmacokinetics in rats. Pharm Res. 2012;29:9. doi: 10.1007/s11095-011-0578-3. [DOI] [PubMed] [Google Scholar]
- 18.Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, Yao Z, Sreedhara A, Cano T, Tesar D, Nijem I, Allison DE, Wong PY, Kao YH, Quan C, Joshi A, Harris RJ, Motchnik P. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. mAbs. 2010;2(6):613–624. doi: 10.4161/mabs.2.6.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, Hattori K. Reduced elimination of IgG antibodies by engineering the variable region. Protein engineering, design & selection : PEDS. 2010;23(5):385–392. doi: 10.1093/protein/gzq009. [DOI] [PubMed] [Google Scholar]
- 20.Devenny JJ, Wagner RC. Transport of immunoglobulin G by endothelial vesicles in isolated capillaries. Microcirculation, endothelium, and lymphatics. 1985;2(1):15–26. [PubMed] [Google Scholar]
- 21.Hong G, Chappey O, Niel E, Scherrmann JM. Enhanced cellular uptake and transport of polyclonal immunoglobulin G and fab after their cationization. Journal of drug targeting. 2000;8(2):67–77. doi: 10.3109/10611860008996853. [DOI] [PubMed] [Google Scholar]
- 22.Negrini D, Moriondo A. Lymphatic anatomy and biomechanics. J Physiol. 2011;589(Pt 12):2927–2934. doi: 10.1113/jphysiol.2011.206672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy ST, Berk DA, Jain RK, Swartz MA. A sensitive in vivo model for quantifying interstitial convective transport of injected macromolecules and nanoparticles. J Appl Physiol. 2006;101(4):1162–1169. doi: 10.1152/japplphysiol.00389.2006. [DOI] [PubMed] [Google Scholar]
- 24.Xie DDHV. Factors affecting the lymphatic absorption of macromolecules following extravascular administration. Pharm Res. 1996;13(S396) [Google Scholar]
- 25.Bocci V, Muscettola M, Grasso G, Magyar Z, Naldini A, Szabo G. The lymphatic route. 1) Albumin and hyaluronidase modify the normal distribution of interferon in lymph and plasma. Experientia. 1986;42(4):432–433. doi: 10.1007/BF02118644. [DOI] [PubMed] [Google Scholar]
- 26.Bocci V, Muscettola M, Naldini A. The lymphatic route–III. Pharmacokinetics of human natural interferon-beta injected with albumin as a retarder in rabbits. General pharmacology. 1986;17(4):445–448. doi: 10.1016/0306-3623(86)90189-8. [DOI] [PubMed] [Google Scholar]
- 27.Porter CJ, Edwards GA, Charman SA. Lymphatic transport of proteins after s.c. injection: implications of animal model selection. Adv Drug Deliv Rev. 2001;50(1–2):157–171. doi: 10.1016/s0169-409x(01)00153-3. [DOI] [PubMed] [Google Scholar]
- 28.Fathallah AM, Turner MR, Mager DE, Balu-Iyer SV. Forthcoming. Effects of hypertonic buffer composition on lymph node uptake and bioavailability of rituximab after subcutaneous administration. Biopharmaceutics & Drug Disposition. 2014 doi: 10.1002/bdd.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parsons JA, Rafferty B, Stevenson RW, Zanelli JM. Evidence that protease inhibitors reduce the degradation of parathyroid hormone and calcitonin injected subcutaneously. Br J Pharmacol. 1979;66(1):25–32. doi: 10.1111/j.1476-5381.1979.tb16093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mager DE, Neuteboom B, Efthymiopoulos C, Munafo A, Jusko WJ. Receptor-mediated pharmacokinetics and pharmacodynamics of interferon-beta1a in monkeys. J Pharmacol Exp Ther. 2003;306(1):262–270. doi: 10.1124/jpet.103.049502. [DOI] [PubMed] [Google Scholar]
- 31.Fathallah AM, Bankert RB, Balu-Iyer SV. Immunogenicity of Subcutaneously Administered Therapeutic Proteins-a Mechanistic Perspective. Aaps J. 2013 doi: 10.1208/s12248-013-9510-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langenkamp A, Messi M, Lanzavecchia A, Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nature immunology. 2000;1(4):311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 33.Hwang ST. Homeward bound: how do skin dendritic cells find their way into the lymph system? The Journal of investigative dermatology. 2012;132(4):1070–1073. doi: 10.1038/jid.2012.39. [DOI] [PubMed] [Google Scholar]
- 34.Swindle MM. Porcine Integumentary System Models: Part 1 – Dermal Toxicology. 2006 ed., http://www.sinclairresearch.com.
- 35.Swindle MM, Makin A, Herron AJ, Clubb FJ, Jr, Frazier KS. Swine as models in biomedical research and toxicology testing. Veterinary pathology. 2012;49(2):344–356. doi: 10.1177/0300985811402846. [DOI] [PubMed] [Google Scholar]
- 36.Eaglstein WH, Mertz PM. New methods for assessing epidermal wound healing: the effects of triamcinolone acetonide and polyethelene film occlusion. The Journal of investigative dermatology. 1978;71(6):382–384. doi: 10.1111/1523-1747.ep12556814. [DOI] [PubMed] [Google Scholar]
- 37.McFarlin DE, Binns RM. Lymph node function and lymphocyte circulation in the pig. Advances in experimental medicine and biology. 1973;29(0):87–93. doi: 10.1007/978-1-4615-9017-0_13. [DOI] [PubMed] [Google Scholar]
- 38.Zheng Y, Tesar DB, Benincosa L, Birnbock H, Boswell CA, Bumbaca D, Cowan KJ, Danilenko DM, Daugherty AL, Fielder PJ, Grimm HP, Joshi A, Justies N, Kolaitis G, Lewin-Koh N, Li J, McVay S, O’Mahony J, Otteneder M, Pantze M, Putnam WS, Qiu ZJ, Ruppel J, Singer T, Stauch O, Theil FP, Visich J, Yang J, Ying Y, Khawli LA, Richter WF. Minipig as a potential translatable model for monoclonal antibody pharmacokinetics after intravenous and subcutaneous administration. mAbs. 2012;4(2):243–255. doi: 10.4161/mabs.4.2.19387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swabb EA, Wei J, Gullino PM. Diffusion and convection in normal and neoplastic tissues. Cancer Res. 1974;34(10):2814–2822. [PubMed] [Google Scholar]
- 40.Kang DW, Oh DA, Fu GY, Anderson JM, Zepeda ML. Porcine model to evaluate local tissue tolerability associated with subcutaneous delivery of protein. Journal of pharmacological and toxicological methods. 2013;67(3):140–147. doi: 10.1016/j.vascn.2013.01.011. [DOI] [PubMed] [Google Scholar]