

Abstract
Background
Activated PI3K-AKT pathway may contribute to decrease sensitivity to inhibitors of key pathogenetic effectors (mutated BRAF, active NRAS or MEK) in melanoma. Functional alterations are deeply involved in PI3K-AKT activation, with a minimal role reported for mutations in PIK3CA, the catalytic subunit of the PI3K gene. We here assessed the prevalence of the coexistence of BRAF/NRAS and PIK3CA mutations in a series of melanoma samples.
Methods
A total of 245 tumor specimens (212 primary melanomas and 33 melanoma cell lines) was screened for mutations in BRAF, NRAS, and PIK3CA genes by automated direct sequencing.
Results
Overall, 110 (44.9%) samples carried mutations in BRAF, 26 (10.6%) in NRAS, and 24 (9.8%) in PIK3CA. All identified PIK3CA mutations have been reported to induce PI3K activation; those detected in cultured melanomas were investigated for their interference with the antiproliferative activity of the BRAF-mutant inhibitor vemurafenib. A reduced suppression in cell growth was observed in treated cells carrying both BRAF and PIK3CA mutations as compared with those presenting a mutated BRAF only. Among the analysed melanomas, 12/245 (4.9%) samples presented the coexistence of PIK3CA and BRAF/NRAS mutations.
Conclusions
Our study further suggests that PIK3CA mutations account for a small fraction of PI3K pathway activation and have a limited impact in interfering with the BRAF/NRAS-driven growth in melanoma.
Keywords: Melanoma, Mutation analysis, PIK3CA gene, Resistance to BRAF/MEK inhibitors
Introduction
Several cell-signaling pathways participate in development and progression of melanoma. Among others, two RAS-driven signal-transduction networks play a crucial role in melanoma pathogenesis: the PI3K/AKT/mTOR and, mainly, the BRAF/MEK/ERK pathways [1–3]. Activation of the latter one, also known as the mitogen-activated protein kinase (MAPK) pathway, is mostly driven by oncogenic mutations in BRAF and, to a less extent, in NRAS genes; somatic mutations in such two genes are mutually exclusive and able to stimulate cell proliferation and tumor growth, through induction of a constitutive ERK phosphorylation [1–3]. Different events may instead contribute to activate the PI3K/AKT pathway: PI3K stimulation by active GTP-bound RAS, occurrence of activating mutations in PIK3CA (the catalytic subunit of the PI3K gene), or silencing of the PTEN tumor suppressor gene [4,5]. The intracellular accumulation of active AKT results in enhancement of cell survival, migration capability, and resistance to apoptosis in human cancers, including melanoma [6,7].
At present, inhibitors of key effectors into the MAPK pathway (BRAF-mutant inhibitors, as vemurafenib or dabrafenib, MEK inhibitors, as trametinib, and their combination) are allowing to overcome the ineffectiveness of the conventional therapies [8]. In patients treated with such inhibitors, a rapid acquisition of drug resistance, as consequence of reactivation of the MAPK pathway or activation of alternative signaling pathways, has been reported to however limit the survival benefits [9,10]. Nevertheless, a fraction of them are primarily refractory due to an intrinsic resistance to such inhibitors [9,10]. On this regard, an increasing amount of evidence indicates that multiple mechanisms may contribute to the development of resistance in melanoma, including those underlying intratumor heterogeneity, alterations in tumor microenvironment (i.e. growth factors and cytokines that interact with their corresponding receptors as well as hormones and neuropeptides), and the ability of tumor to generate an immunosuppressive environment [3,8,11–13]. Even different levels of intralesional pigmentation may interfere with melanoma pathogenesis and/or affect the behavior of the disease [12,13].
At intracellular and molecular level, crosstalk mechanisms between the MAPK and PI3K/AKT pathways, with the mutated BRAF inducing a negative regulation of the AKT network, have been described [14]. Inactivation of the oncogenic BRAF by targeted inhibitors is thus supposed to increase the intracellular levels of phosphorylated AKT, contributing to the enhancement of cell survival and the development of drug resistance [14]. Suppression of AKT activity by inhibition of either upstream (PI3K) or downstream (mTOR) effectors of this signaling cascade is being proposed as an effective tool for the improvement of the antitumor response to the MAPK-targeted therapies [15–17]. In preclinical studies, combined treatment based on inhibition of BRAF and silencing of AKT3 was found to significantly increase suppression of tumour growth as compared to the result obtained by single agent administration [18,19]. Recently, combination of a BRAF or MEK inhibitor with a PI3K/mTOR inhibitor was found to enhance cell growth inhibition through achievement of ERK hypo-phosphorylation, overcoming the resistance encountered by the use of a single anti-BRAF or anti-MEK agent [17,20].
Overall, identification of melanomas with activated alternative signaling pathways may be helpful in selecting the fraction of patients carrying BRAF mutations primarily refractory to the treatment with either a BRAF or MEK inhibitor. In our case, this raises the question whether a test for detecting the activation of the PI3K/AKT pathway should be routinely used in clinical practice for a more accurate classification of the patients before addressing them to be treated with such inhibitors.
Toward the identification of a more appropriate test for assessing the activated status of the PI3K/AKT pathway, genetic variations, whose assessment is qualitative (detecting the objective presence or absence of each specific sequence variant), can be considered as more reliable predictive markers as compared to expression alterations, whose classification is quantitative or semi-quantitative (for immunohistochemistry, strictly depending on subjective evaluation of both intensity and distribution of tissue protein staining). Since PTEN is mostly inactivated by gene deletions or rearrangements [21] as well as AKT and mTOR are mainly altered at functional level, PI3K - and, particularly, its catalytic subunit PIK3CA - remains the gene mostly affected by activating somatic mutations into this pathway [5].
The aim of this study was to investigate the prevalence and distribution of pathogenetic variants in BRAF, NRAS, and PIK3CA genes among 245 DNA samples from pigmented melanomas of cutaneous origin, defining the fraction of cases harboring coexistent PIK3CA and BRAF or NRAS mutations. Melanoma cell lines with coexistence of PIK3CA and BRAF mutations were also investigated to evaluate the level of interference with the anti-proliferative effects of the BRAF-mutant inhibitor vemurafenib.
Methods
Samples
One hundred and eighty-six patients with histologically-proven diagnosis of pigmented melanoma of cutaneous origin were included into the study. After obtaining their written consent for tissue sampling, patients were enrolled consecutively between March 2010 and November 2012 from centers in Italy, regardless of cancer family history and disease characteristics. Formalin-fixed, paraffin-embedded primary melanoma tissues were obtained from pathological archives for all patients; in 26 of them with multiple primary melanoma, two synchronous or asynchronous primary tumor tissues were collected. Synchronous melanomas were diagnosed in four patients during the same first observation or, at the most, within one month from the first diagnosis, according to previously defined criteria [22]. In the remaining 22 patients with asynchronous multiple melanomas, the subsequent primary tumors were diagnosed at a median time from the first diagnosis of 28 months (range, 6–83 months). Vast majority (189/212; 89%) of primary melanomas included into the study were from intermittently sun-exposed skin.
To improve sensitivity of nucleotide sequencing (sequence variants can be detected when the mutant alleles are at least 15%-20% of the analyzed DNA sample), the neoplastic portion of each tissue section was isolated in order to obtain tumor samples with at least 80% neoplastic cells.
Mutation analysis
Genomic DNA was isolated from tumor tissues, using standard methods. The coding sequence and splice junctions of the mostly mutated domains of candidate genes (exon 15 in BRAF, exons 2 to 4 in NRAS, and exons 9 and 20 in PIK3CA) were screened for mutations by directly sequencing the amplified PCR products, using an automated fluorescence-cycle sequencer (ABIPRISM 3130, Life Technologies/ThermoFisher Scientific, Waltham, MA, USA). Primer sequences were as previously-reported by our group [23,24]. Sequencing analysis was conducted in duplicate (two PCR assays from two different tumor sections) and in both directions (forward and reverse) for all samples. A nucleotide sequence was considered as valid when the quality value (QV) was higher than 20 (<1/100 error probability); in this study, the QV average was 40 (range, 30–45; <1/1000-1/10,000 error probability).
Melanoma cell lines and in vitro proliferation test
For in vitro proliferation assay, four melanoma cell lines were selected: 13443-Mel (with BRAFwild-type and PIK3CAwild-type; as negative control), PNP-Mel (with BRAFV600E and PIK3CAwild-type), M14 (with BRAFV600E and PIK3CAP539R), and M259 (with BRAFV600E and PIK3CAE545K). Cells were plated in triplicate in 96-well plates, at a density of 3–5 × 103 per well, in fresh medium (RPMI 1640; Invitrogen/ThermoFisher Scientific, USA) only, as control, or medium containing different concentrations of vemurafenib (PLX4032, RG7204; Selleck Chemicals, Houston, TX). In particular, melanoma cell lines were treated in triplicate with increasing concentrations (5 to 100 nM) of vemurafenib for 72 hrs. The percentage of melanoma cell proliferation was estimated on day 4 by a colorimetric assay, as we previously described [25].
Statistical analysis
All in vitro data derive from at least three independent experiments and results are expressed as mean values with 95% confidence intervals. The statistical significance of differential findings between experimental and control groups was determined by ANOVA with the Tukey’s multiple comparison test in Graph-Pad Prism 3.0 software (Graph-Pad Software, Inc., San Diego, CA, USA). These findings were considered significant if two-tailed P values were <0.05.
Results and discussion
A total of 245 tumor specimens (212 primary melanomas and 33 melanoma cell lines) was screened for mutations in BRAF, NRAS, and PIK3CA genes. Tumor tissues were collected from 186 patients, since 26 of them presented two synchronous or asynchronous primary melanomas (see Methods for details). Median age of the enrolled patients was 51 years (range, 19–83 years); 99 (53%) were men. Patients’ characteristics are shown in Table 1. For majority (18/33; 55%) of melanoma cell lines, main genotypic features have been previously described [26]; however, an extensive genetic and molecular analysis of candidate genes among the entire series of cell lines is being completed (Sini, manuscript in preparation).
Table 1.
Characteristics of analyzed patients and melanomas
| Characteristics | No. | % |
|---|---|---|
| Total patients analyzed | 186 | |
| Males/Females | 99/87 | 53/47 |
| Median age (years) | 51 | |
| Range | 19-83 | |
| Single/Multiple melanoma | 160/26 | 86/14 |
| Patients’ AJCC stage | ||
| I | 39 | 21 |
| II | 76 | 41 |
| III | 47 | 25 |
| IV | 24 | 13 |
| Total melanoma analyzed | 212 | |
| Primary site | ||
| Head and neck | 35 | 17 |
| Limbs | 79 | 37 |
| Trunk | 98 | 46 |
| Types | ||
| Superficial spreading | 135 | 64 |
| Nodular | 77 | 36 |
AJCC, American Joint Committee on Cancer.
Overall, 160 (65.3%) melanoma samples carried mutations in at least one of such candidate genes: 110 (44.9%) in BRAF, 26 (10.6%) in NRAS, and 24 (9.8%) in PIK3CA (Table 1). For PIK3CA mutations, screening revealed the occurrence of five mutations (p.P539R, p.E542K, p.E545A, p.E545G, and p.E545K in exon 9; no sequence variation was detected in exon 20), which have been widely reported in mutation databases [Human Gene Mutation Database (HGMD) at http://www.hgmd.cf.ac.uk/ac/all.php and Catalogue Of Somatic Mutations In Cancer (COSMIC) at http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/] as commonly associated with human cancer, with a recognized functional role of the corresponding mutated proteins. The variant p.E545A was the mutation with the highest frequency in our series [detected in 14/24 (58.3%) PIK3CA mutants].
Among analyzed melanomas, 12/245 (4.9%) samples presented the coexistence of PIK3CA and BRAF or NRAS mutations [11 cases with BRAF and PIK3CA mutations and 1 case with NRAS and PIK3CA mutations; Table 2). On the other hand, NRAS or BRAF mutations were found in half (12/24; 50%) of patients with PIK3CA mutations, which were conversely detected in less than one tenth (12/136; 8.8%) of cases with NRAS or BRAF mutations (Table 2). Among the 212 tumor tissues of our series, 52 samples were from 26 patients with multiple primary melanoma (all such cases presented two synchronous or asynchronous primary melanomas). As shown in Table 3, about half (12/26; 46.2%) of patients with multiple melanoma showed discrepancies in mutation patterns between first and subsequent primary tumors, further supporting our previous observations that molecular mechanisms underlying the development of multiple melanomas in the same patients are heterogeneous [27,28].
Table 2.
Frequencies of somatic mutations in the three candidate genes
| Sample | BRAF | NRAS | BRAF + PIK3CA | NRAS + PIK3CA | PIK3CA | Wild-type |
|---|---|---|---|---|---|---|
| % | % | % | % | % | % | |
| Primary melanoma (N = 212) | 81 | 21 | 9 | 1 | 10 | 90 |
| 38.2 | 9.9 | 4.2 | 0.5 | 4.7 | 42.5 | |
| Melanoma cell line (N = 33) | 18 | 4 | 2 | 0 | 2 | 7 |
| 54.5 | 12.1 | 6.1 | 0 | 6.1 | 21.2 | |
| Total (N = 245) | 99 | 25 | 11 | 1 | 12 | 97 |
| 40.4 | 10.2 | 4.5 | 0.4 | 4.9 | 39.6 |
Table 3.
Consistency between BRAF/NRAS/PIK3CA mutations in multiple melanomas from same patients, and mutation patterns in those in whom there were discrepancies
| No. of cases | Cases with consistent mutation patterns (second vs first tumor sample), n (%) | Mutation patterns among discrepant paired samples | |||||
|---|---|---|---|---|---|---|---|
| BRAF | NRAS | PIK3CA | |||||
| First tumor | Second tumor | First tumor | Second tumor | First tumor | Second tumor | ||
| 26 | 14 (53.8) | wt | V600E | wt | wt | wt | wt |
| wt | wt | wt | wt | wt | E545A | ||
| V600E | wt | wt | Q61R | E545A | wt | ||
| V600K | wt | wt | wt | wt | wt | ||
| wt | wt | wt | wt | E545A | wt | ||
| V600E | wt | wt | wt | wt | wt | ||
| wt | wt | wt | wt | wt | E542K | ||
| V600E | wt | wt | wt | wt | wt | ||
| V600E | wt | wt | wt | wt | wt | ||
| wt | wt | Q61R | wt | wt | wt | ||
| wt | V600E | wt | wt | E545G | wt | ||
| wt | V600E | wt | wt | wt | wt | ||
Abbreviation: wt wild-type.
Using a panel of four melanoma cell lines carrying constitutive differences in mutational status of both BRAF and PIK3CA genes (including the only two cell lines with coexistence of BRAF and PIK3CA mutations; see Table 2 and Methods), we have investigated the ability of the BRAF-mutant inhibitor vemurafenib to halt cell proliferation. As expected, a remarkable anti-proliferative activity by this drug was observed in the PNP-Mel cells (presenting a mutated BRAF and a wild-type PIK3CA), whereas lack of a significant inhibitory effect on growth was demonstrated in control cells carrying wild-type BRAF and PIK3CA (Figure 1). Suppression in cell proliferation was instead less evident or minimal on BRAF-mutated M14 (presenting the p.P539R mutation in PIK3CA) and M259 (with the p.E545K mutation in PIK3CA) cell lines, respectively (Figure 1). Although experimental assays should be carried extensively on a larger series of melanoma cell lines, our findings permit to speculate that activating mutations of PIK3CA may interfere with the anti-proliferative effects of BRAF inhibitors on melanomas carrying oncogenic variants of BRAF (though levels of interference may vary, strictly depending on the type of sequence alterations at the functional domains of the PIK3CA gene). Although our results are consistent with data from literature - indicating that absence of an activated PI3K-AKT pathway may contribute to increase sensitivity to drugs inhibiting the oncogenically-active MAPK components (either mutated BRAF or MEK) [29–32], it is to keep in mind that other molecular features (BRAF amplification, BRAF splice variants, MEK1-2 mutations, RTK/NRAS activation, PTEN inactivation, RB1 loss, etc.) may be associated with the decreased sensitivity to BRAF inhibitors [10,11,33].
Figure 1.
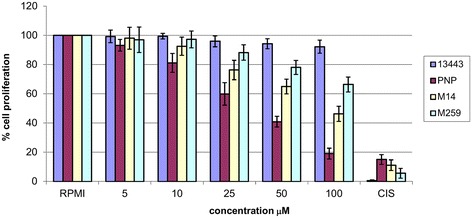
Effects of vemurafenib on the growth of human melanoma cell lines. Four melanoma cell lines were cultured in presence of various concentrations of vemurafenib for 72 hours and cell proliferation was estimated as described in Methods. Results are expressed as percent of cell growth and represent the average (± standard deviation) of triplicate experiments. RPMI, cell cultures in fresh medium only, as control. Cisplatinum (CIS) at concentration of 5 μM was used as cytotoxicity positive control.
In our study, prevalence of pathogenetic PIK3CA mutations in cutaneous melanoma tissues (20/212; 9%) was much higher than that (about 2%) reported in HGMD and COSMIC databases for in vivo melanoma samples. Considering a collection of about 1,000 melanoma samples from the most relevant studies published on this issue [34–38], mutation rates in PIK3CA gene were consistently identical during melanoma progression - from primary cutaneous melanomas (2.3%) to metastatic melanomas (2.5%) and melanoma cell lines (2.5%). For in vitro melanoma samples from our series, frequency of PIK3CA mutations (4/33 melanoma cell lines; 12%) was even higher than that above reported (though BRAF/NRAS mutations remained the most prominent genetic alterations, acting as main drivers of proliferation in vast majority of cultured melanomas). Conversely, the rate of coexistence of PIK3CA and BRAF or NRAS mutations (about 5%) in our series was lower than that previously described (20%; [4]).
Overall, our findings further confirm that PIK3CA is rarely subject to somatic activation through genetic alterations in melanoma. In other words, sequence variations in PIK3CA gene only account for a small fraction of activations of the PI3K-AKT signaling pathway and are thus expected to minimally interfere with the BRAF/NRAS-driven growth. Conversely, activation of AKT, the main downstream component of the PI3K pathway, is mostly determined at functional level as consequence of an increased activity of the upstream effectors (RAS activation, PTEN suppression), with again a minimal role played by genetic alterations (activating mutations in AKT1 and AKT3 genes have been reported in a limited number of melanomas and melanoma cell lines) [14,30,39]. Although it is becoming evident that multiple mechanisms may contribute to AKT activation in melanoma, loss of PTEN - reported in about 10% of melanoma tissues and 30% of cultured melanoma cells [35–37] - is considered the major mechanism for abrogating oncogene-induced senescence in either BRAF or NRAS mutant cells through activation of the PI3K-AKT signaling pathway [6,9,40,41]. In our collection of 33 melanoma cell lines, loss of the PTEN expression was found in eight (24%) of them by immunocytochemistry (data not shown). For this reason, occurrence of PTEN inactivation should be mainly evaluated in melanoma patients harboring activation of the MAPK pathway and undergoing treatment with BRAF and MEK-selective inhibitors in order to determine whether these alterations may be associated with a diminished clinical benefit.
In very next future, more accurate molecular signatures of cancer tissues will be achieved through the advancements of the technologies. Indeed, next-generation sequencing (NGS) approaches are already being demonstrated to better characterize molecular mechanisms underlying resistance to targeted therapies [33,42]. As consequence, more reliable molecular biomarkers might be available for improving the accurateness of the prediction of the treatment outcome among patients with melanoma.
Conclusions
In our study, the presence of a limited but representative fraction of cases with coexistence of somatic mutations in PIK3CA and BRAF or NRAS genes seems to somehow represent an indicator for including mutation analysis of PIK3CA into the initial (prior-to-therapy) strategies for molecular classification of melanoma patients. This could allow to prospectively make correlations between the coexistence of PIK3CA/BRAF/NRAS mutations and rates of intrinsic resistance to BRAF or MEK inhibitors.
Acknowledgments
Authors are grateful to patients for their important contribution to this study. Work was partially supported by the Sardinia Regional Government (Regione Autonoma della Sardegna).
Abbreviations
- COSMIC
Catalogue of somatic mutations in cancer
- HGMD
Human gene mutation database
- MAPK
Mitogen-activated protein kinase
- NGS
Next-generation sequencing
- PCR
Polymerase chain reaction
Footnotes
Competing interests
PAA is consultant of Bristol Myers Squibb, MSD, and Roche-Genentech. He participated into the Advisory Board from Bristol Myers Squibb, MSD, Roche-Genentech, GSK, Amgen, Celgene, Medimmune, and Novartis. He received honoraria from Brystol Myers Squibb, MSD, and Roche-Genentech.
All remaining authors declare the absence of any competing interest.
Authors’ contributions
AM, performed DNA analysis, helped to draft the manuscript; AL, performed quality control of pathological data; MCa, contributed to DNA analysis; PAA, performed data interpretation; GB, CC, and IS participated in patients’ collection and data acquisition; MCo and MS, performed mutation analysis; AC, participated into the study design and data discussion; GP, conceived of the study and participated in its design and coordination, drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Antonella Manca, Email: Antonella.Manca@icb.cnr.it.
Amelia Lissia, Email: a_lissia@yahoo.it.
Mariaelena Capone, Email: marilenacapone@virgilio.it.
Paolo A Ascierto, Email: paolo.ascierto@gmail.com.
Gerardo Botti, Email: gbotti1@aliceposta.it.
Corrado Caracò, Email: corracara@fastwebnet.it.
Ignazio Stanganelli, Email: igstanga@tin.it.
Maria Colombino, Email: colombinom@yahoo.it.
MariaCristina Sini, Email: mc.sini@tiscali.it.
Antonio Cossu, Email: cossu@uniss.it.
Giuseppe Palmieri, Email: gpalmieri@yahoo.com.
References
- 1.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 3.Palmieri G, Capone ME, Ascierto ML, Gentilcore G, Stroncek DF, Casula M, et al. Main roads to melanoma. J Transl Med. 2009;7:86. doi: 10.1186/1479-5876-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS, et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One. 2011;6:e22769. doi: 10.1371/journal.pone.0022769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shull AY, Latham-Schwark A, Ramasamy P, Leskoske K, Oroian D, Birtwistle MR, et al. Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One. 2012;7:e43369. doi: 10.1371/journal.pone.0043369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 7.Plas DR, Thompson CB. Akt-dependent transformation: there is more to growth than just surviving. Oncogene. 2005;24:7435–42. doi: 10.1038/sj.onc.1209097. [DOI] [PubMed] [Google Scholar]
- 8.Ascierto PA, Grimaldi AM, Acquavella N, Borgognoni L, Calabrò L, Cascinelli N, et al. Future perspectives in melanoma research. Meeting report from the “Melanoma Bridge. Napoli, December 2nd-4th 2012”. J Transl Med. 2013;11:137. doi: 10.1186/1479-5876-11-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartsough E, Shao Y, Aplin AE. Resistance to RAF inhibitors revisited. J Invest Dermatol. 2014;134:319–25. doi: 10.1038/jid.2013.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slominski AT, Carlson JA. Melanoma resistance: a bright future for academicians and a challenge for patient advocates. Mayo Clin Proc. 2014;89:429–33. doi: 10.1016/j.mayocp.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slominski A, Kim TK, Brożyna AA, Janjetovic Z, Brooks DL, Schwab LP, et al. The role of melanogenesis in regulation of melanoma behavior: melanogenesis leads to stimulation of HIF-1α expression and HIF-dependent attendant pathways. Arch Biochem Biophys. 2014;563:79–93. doi: 10.1016/j.abb.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slominski A, Zmijewski MA, Pawelek J. L-tyrosine and L-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment Cell Melanoma Res. 2012;25:14–27. doi: 10.1111/j.1755-148X.2011.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Shen Q, Labow M, Gaither LA. Protein kinase D3 sensitizes RAF inhibitor RAF265 in melanoma cells by preventing reactivation of MAPK signaling. Cancer Res. 2011;71:4280–91. doi: 10.1158/0008-5472.CAN-10-3761. [DOI] [PubMed] [Google Scholar]
- 15.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–25. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 17.Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Jr, Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19:657–67. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung M, Sharma A, Madhunapantula SV, Robertson GP. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008;68:3429–39. doi: 10.1158/0008-5472.CAN-07-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran MA, Gowda R, Sharma A, Park EJ, Adair J, Kester M, et al. Targeting V600EB-Raf and Akt3 using nanoliposomal-small interfering RNA inhibits cutaneous melanocytic lesion development. Cancer Res. 2008;68:7638–49. doi: 10.1158/0008-5472.CAN-07-6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11:909–20. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- 21.Xing F, Persaud Y, Pratilas CA, Taylor BS, Janakiraman M, She QB, et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene. 2012;31:446–57. doi: 10.1038/onc.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carli P, De Giorgi V, Chiarugi A, Stante M, Giannotti B. Multiple synchronous cutaneous melanomas: implications for prevention. Int J Dermatol. 2002;41:583–5. doi: 10.1046/j.1365-4362.2002.01605.x. [DOI] [PubMed] [Google Scholar]
- 23.Casula M, Colombino M, Satta MP, Cossu A, Ascierto PA, Bianchi-Scarrà G, et al. BRAF gene is somatically mutated but does not make a major contribution to malignant melanoma susceptibility. J Clin Oncol. 2004;22:286–92. doi: 10.1200/JCO.2004.07.112. [DOI] [PubMed] [Google Scholar]
- 24.Palomba G, Colombino M, Contu A, Massidda B, Baldino G, Pazzola A, et al. Prevalence of KRAS, BRAF, and PIK3CA somatic mutations in patients with colorectal carcinoma may vary in the same population: clues from Sardinia. J Transl Med. 2012;10:178. doi: 10.1186/1479-5876-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manca A, Sini MC, Izzo F, Ascierto PA, Tatangelo F, Botti G, et al. Induction of arginosuccinate synthetase (ASS) expression affects the antiproliferative activity of arginine deiminase (ADI) in melanoma cells. Oncol Rep. 2011;25:1495–502. doi: 10.3892/or.2011.1220. [DOI] [PubMed] [Google Scholar]
- 26.Casula C, Muggiano A, Cossu A, Budroni M, Caracò C, Ascierto PA, et al. Role of key-regulator genes in melanoma susceptibility and pathogenesis among patients from South Italy. BMC Cancer. 2009;9:352. doi: 10.1186/1471-2407-9-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. 2012;30:2522–9. doi: 10.1200/JCO.2011.41.2452. [DOI] [PubMed] [Google Scholar]
- 28.Colombino M, Lissia A, Capone M, De Giorgi V, Massi D, Stanganelli I, et al. Heterogeneous distribution of BRAF/NRAS mutations among Italian patients with advanced melanoma. J Transl Med. 2013;11:202. doi: 10.1186/1479-5876-11-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70:8736–47. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–60. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byron SA, Loch DC, Wellens CL, Wortmann A, Wu J, Wang J, et al. Sensitivity to the MEK inhibitor E6201 in melanoma cells is associated with mutant BRAF and wild-type PTEN status. Mol Cancer. 2012;11:75. doi: 10.1186/1476-4598-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng W, Gopal YN, Scott A, Chen G, Woodman SE, Davies MA. Role and therapeutic potential of PI3K-mTOR signaling in de novo resistance to BRAF inhibition. Pigment Cell Melanoma Res. 2012;25:248–58. doi: 10.1111/j.1755-148X.2011.00950.x. [DOI] [PubMed] [Google Scholar]
- 33.Spagnolo F, Ghiorzo P, Queirolo P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget. 2014;5:10206–21. doi: 10.18632/oncotarget.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curtin JA, Stark MS, Pinkel D, Hayward NK, Bastian BC. PI3-kinase subunits are infrequent somatic targets in melanoma. J Invest Dermatol. 2006;126:1660–3. doi: 10.1038/sj.jid.5700311. [DOI] [PubMed] [Google Scholar]
- 35.Omholt K, Kröckel D, Ringborg U, Hansson J. Mutations of PIK3CA are rare in cutaneous melanoma. Melanoma Res. 2006;16:197–200. doi: 10.1097/01.cmr.0000200488.77970.e3. [DOI] [PubMed] [Google Scholar]
- 36.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–14. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siroy AE, Boland GM, Milton DR, Roszik J, Frankian S, Malke J, et al. Clinical mutation panel testing by next-generation sequencing in advanced melanoma. J Invest Dermatol. 2015;135:508–15. doi: 10.1038/jid.2014.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99:1265–8. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vredeveld LC, Possik PA, Smit MA, Meissl K, Michaloglou C, Horlings HM, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012;26:1055–69. doi: 10.1101/gad.187252.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nogueira C, Kim KH, Sung H, Paraiso KH, Dannenberg JH, Bosenberg M, et al. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene. 2010;29:6222–32. doi: 10.1038/onc.2010.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turajlic S, Furney SJ, Stamp G, Rana S, Ricken G, Oduko Y, et al. Whole genome sequencing reveals complex mechanisms of intrinsic resistance to BRAF inhibition. Ann Oncol. 2014;25:959–67. doi: 10.1093/annonc/mdu049. [DOI] [PMC free article] [PubMed] [Google Scholar]