

Abstract
Objectives
Antiretroviral interruption is associated with liver fibrosis progression in HIV/hepatitis C virus (HCV) coinfection. It is not known what level of HIV viraemia affects fibrosis progression.
Methods
We evaluated 288 HIV/HCV-coinfected cohort participants with undetectable HIV RNA (< 50 HIV-1 RNA copies/mL) on two consecutive visits while on combination antiretroviral therapy (cART) without fibrosis [aspartate aminotransferase to platelet ratio index (APRI) < 1.5], end-stage liver disease or HCV therapy. An HIV blip was defined as a viral load of ≥ 50 and < 1000 copies/mL, preceded and followed by undetectable values. HIV rebound was defined as: (i) HIV RNA ≥ 50 copies/mL on two consecutive visits, or (ii) a single HIV RNA measurement ≥ 1000 copies/mL. Multivariate discrete-time proportional hazards models were used to assess the effect of different viraemia levels on liver fibrosis progression (APRI ≥ 1.5).
Results
The mean age of the patients was 45 years, 74% were male, 81% reported a history of injecting drug use, 51% currently used alcohol and the median baseline CD4 count was 440 [interquartile range (IQR) 298, 609] cells/μL. Fifty-seven (20%) participants [12.4/100 person-years (PY); 95% confidence interval (CI) 9.2−15.7/100 PY] progressed to an APRI ≥ 1.5 over a mean 1.1 (IQR 0.6, 2.0) years of follow-up time at risk. Virological rebound [hazard ratio (HR) 2.3; 95% CI 1.1, 4.7] but not blips (HR 0.5; 95% CI 0.2, 1.1) predicted progression to APRI ≥ 1.5. Each additional 1 log10 copies/mL HIV RNA exposure (cumulative) was associated with a 20% increase in the risk of fibrosis progression (HR 1.2; 95% CI 1.0–1.3).
Conclusions
Liver fibrosis progression was associated with HIV rebound, but not blips, and with increasing cumulative exposure to HIV RNA, highlighting the importance of achieving and maintaining HIV suppression in the setting of HIV/HCV coinfection.
Keywords: fibrosis, hepatitis C virus, HIV, virological blips, virological rebound
Introduction
HIV combination antiretroviral therapy (cART) has led to a substantial decrease in AIDS-related morbidity and mortality in HIV-positive individuals 1. However, among those coinfected with the hepatitis C virus (HCV), liver-related diseases including cirrhosis, hepatocellular carcinoma (HCC), and end-stage liver disease (ESLD) have emerged as leading causes of comorbidity and death 2,3. It is well established that HIV infection negatively impacts the natural history of HCV by accelerating the progression of liver disease and reducing the likelihood of achieving a sustained virological response with HCV antiviral therapy 3,4. Identifying factors that can be acted on to modify liver fibrosis progression rates among patients untreated for HCV remains highly relevant despite the prospect of very effective HCV treatments on the horizon. It may be some time before patients will be able to access such therapy and, in many parts of the world, their cost will be prohibitive.
Even during effective HIV treatment, routine testing may occasionally reveal episodes of detectable HIV viraemia (i.e. virological blips) punctuating periods of virological suppression 5–9. Factors contributing to blips include: HIV replication bursts from stable reservoirs, ongoing cycles of replication, random biological fluctuation, and the consequence of increasingly sensitive detection methods 10–14. HIV RNA blips do not appear to be associated with decreased antiretroviral adherence or dose-timing irregularities 5–9. HIV RNA blips > 500 HIV-1 RNA copies/ml have been associated with subsequent virological rebound 15 and with a dampened CD4 cell count increase 16,17 and thus could affect outcomes in coinfection.
In contrast, virological rebounds have been identified as an independent predictor of loss to follow-up 18. Virological rebounds have been defined as detectable RNA levels on two occasions after HIV suppression is achieved or as HIV RNA values ≥ 1000 copies/mL 15,18. HIV RNA rebounds are generally interpreted as evidence of poor adherence. Other factors contributing to HIV viral rebounds include the presence of pre-existing resistance mutations that are expressed following the initiation of cART, poor gastrointestinal absorption, and drug−drug interactions leading to diminished antiretroviral drug levels 15,18.
While HIV viral blips and rebounds have been well evaluated in HIV monoinfection, their effects on the natural history of HCV-induced liver disease and HCV antiviral treatment outcomes have not been described in HIV/HCV coinfection. Treatment interruption has been associated with liver fibrosis progression in HIV/HCV coinfection 19, but it is not known if low levels of viraemia might also affect the natural history of liver disease. To this end, we evaluated HIV RNA blips and rebounds as predictors of liver fibrosis progression in HIV/HCV coinfection.
Methods
Study setting and population
The Canadian Co-infection Cohort Study (CCC; CTN222) is a prospective multicentre study recruiting HIV/HCV-coinfected patients from existing HIV clinic populations at 16 centres across Canada since 2003. As of July 2012, 1119 patients were enrolled. Details of the cohort design and protocol have been published previously 20. Eligible patients were adults aged over 16 years with documented HIV infection [enzyme-linked immunosorbent assay (ELISA) with western blot confirmation] and with chronic HCV infection or evidence of HCV exposure [e.g. HCV seropositive by ELISA with recombinant immunoblot assay (RIBA) II or enzyme immunoassay (EIA) confirmation, and/or HCV RNA positive]. After providing written informed consent, participants underwent an initial evaluation followed by study visits every 6 months. Sociodemographic, behavioural and clinical care information was collected using standardized questionnaires. At each visit, laboratory assessments included complete blood count, creatinine measurement, liver profile, plasma HIV RNA measurement (COBAS TaqMan HIV-1 Test, Roche Diagnostics, Laval, Quebec, Canada; limit of detection 40 copies/mL), CD4 T cell count and plasma HCV RNA measurement (COBAS Amplicor HCV Test, v2.0, Roche Diagnostics, Laval, Quebec, Canada; limit of detection 60 IU/mL). The study was approved by the community advisory committee of the Canadian HIV Trials Network (CIHR)-Canadian HIV Trials Network and the research ethics boards of all participating institutions.
The analytic sample (n = 288) included participants 16 years of age or older who were chronically infected with HCV and who were currently on HIV antiretroviral treatment with at least two consecutive visits and two consecutive undetectable HIV RNA measurements in follow-up or undetectable HIV RNA at cohort entry with prior stable antiretroviral treatment defined as > 1 year on combination antiretroviral therapy (cART). The primary outcome of interest was the progression to liver fibrosis; thus, participants with significant fibrosis or ESLD at study entry were excluded (see definitions below). Patients were censored on their last clinic visit prior to July 2012, when an outcome occurred, at death or at initiation of HCV treatment.
Definitions
The aspartate aminotransferase (AST) to platelet ratio index (APRI) was used as a noninvasive surrogate for liver fibrosis and defined as: 100 × (AST/upper limit of normal)/platelet count (109/L) 21,22. An APRI score ≥ 1.5 has been validated as a marker of significant liver fibrosis in coinfected patients (corresponding to a biopsy score > F2) 21.
HIV RNA virological ‘blips’ were defined as an HIV RNA value ≥ 50 copies/mL and < 1000 copies/mL, preceded and followed by an undetectable value (< 50 copies/mL) 23,24. HIV RNA virological ‘rebounds’ were determined as either: (i) an HIV RNA value ≥ 50 copies/mL during two consecutive serological measurements, or (ii) a single HIV RNA measurement ≥ 1000 copies/mL 15.
ESLD diagnoses included liver cirrhosis, ascites, hepatic encephalopathy, bleeding esophageal varices, spontaneous bacterial peritonitis and hepatocellular carcinoma 25. Time since HIV diagnosis was determined using the date of HIV seroconversion, where known, or the date of the first positive HIV antibody test. The duration of HCV infection was determined using the date of HCV seroconversion, if known, or the year of first injecting drug use (IDU) or blood product exposure as a proxy of HCV infection.
Alcohol abuse was defined as self-reported alcohol intake of more than two drinks per day or binge drinking (greater than six drinks at any one time).
Statistical analysis
For all analyses, baseline (time zero) corresponded to cohort entry for those on stable cART with undetectable HIV RNA or the second of two consecutive visits at which HIV RNA was observed to be undetectable for those starting cART during follow-up.
Incidence rates of liver fibrosis were estimated among those without fibrosis at baseline by dividing the number of participants developing the outcome by the number of person-years (PY) at risk, and expressed in cases per 100 PY. Poisson count models were used to calculate confidence intervals (CIs) for incidence rates.
Multivariate proportional hazards models were built to assess the effect of different states of HIV viraemia on progression to significant liver fibrosis (APRI ≥ 1.5) using variables that were determined a priori to be clinically important. We used a discrete time version of the Cox model (with a complementary log log link function, an offset to allow for variation in the time between visits and robust standard errors) 26 because liver fibrosis could only be assessed at each cohort visit. Final models were adjusted for both time-independent covariates fixed at baseline [age, gender, ethnicity, alcohol abuse and ln(APRI)] and time-dependent exposures. The natural logarithm of the APRI [ln(APRI)], which nearly normalizes the distribution 22, was used in all analyses. Time-dependent exposures were: CD4 cell count, virological blip and virological rebound status. Once a blip or rebound had occurred, this status was kept ‘on’ for future risk intervals. In a secondary analysis, viral blip and rebound categories were replaced with cumulative log copies of HIV RNA.
In a sensitivity analysis, we estimated the association between significant fibrosis progression and cumulative log copies of HIV RNA where the latter was represented by a linear spline with two knots selected a priori at 50 and 1000 copies/mL 26. The second knot corresponds to the threshold used to differentiate viral blips from rebounds.
All analyses were performed using stata software version 11 (StatCorp LP, College Station, TX, USA).
Results
At the time of analysis, 1119 HIV/HCV-coinfected participants were enrolled in the CTN222 cohort (Fig. 1). Data were analysed for 288 participants who met study inclusion criteria. The mean age of the patients was 45 years, 26% were female, 14% were aboriginal, 82% reported a history of IDU, and 51% currently used alcohol. The median baseline CD4 T-lymphocyte count was 440 cells/μL [interquartile range (IQR) 298, 609 cells/μL]. Baseline characteristics comparing those who did and did not progress to advanced fibrosis, as determined by APRI > 1.5, are shown in Table 1. Most characteristics were similar. However, those progressing to fibrosis were more likely to be on protease inhibitor-based cART at baseline than those not progressing to fibrosis [n = 47 (82%) vs. 151 (65%), respectively].
Fig 1.
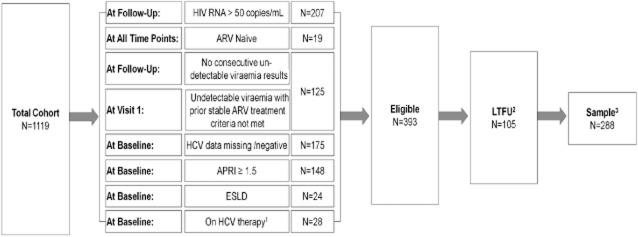
Participant selection flow chart. 1Hepatitis C virus (HCV) treatment censoring was carried out during the follow-up period. Antiretroviral (ARV) interruption was not censored. 2Participants not attending visits beyond study enrolment. 3The analysed data set is comprised of ‘valid risk-sets’. If one or multiple visits were skipped then that risk-set was excluded from analysis. APRI, aspartate aminotransferase to platelet ratio index; ESLD, end-stage liver disease; LTFU, lost to follow-up.
Table 1.
Sociodemographic and clinical characteristics of HIV/hepatitis C virus (HCV)-coinfected patients at baseline according to whether they progressed to significant liver fibrosis [aspartate aminotransferase to platelet ratio index (APRI) ≥ 1.5] in follow-up
| Total (n = 288) | Progressors (n = 57) | Nonprogressors (n = 231) | |
|---|---|---|---|
| Follow-up time (years) [median (IQR)] | 1.5 (0.7, 2.9) | 1.0 (0.5, 1.9) | 1.8 (0.9, 3.0) |
| Age (years) [median (IQR)] | 45 (40, 50) | 46 (42, 51) | 45 (40, 50) |
| Female [n (%)] | 75 (26) | 15 (26) | 60 (26) |
| Aboriginal [n (%)] | 40 (14) | 6 (11) | 34 (15) |
| IDU ever [n (%)] | 236 (82) | 51 (89) | 185 (80) |
| IDU in previous 6 months [n (%)] | 90 (31) | 16 (28) | 74 (32) |
| Current alcohol use [n (%)] | 148 (51) | 32 (56) | 116 (50) |
| Current alcohol abuse* [n (%)] | 41 (28) | 13 (42) | 28 (24) |
| Time since HIV diagnosis (years) [median (IQR)] | 12 (7, 17) | 11 (7, 15) | 12 (7.6, 18) |
| Duration of HCV infection (years) [median (IQR)] | 19 (11, 27) | 20 (12, 28) | 19 (11, 26) |
| Nadir CD4 count (cells/μL) [median (IQR)] | 150 (50, 249) | 158 (84, 264) | 150 (50, 240) |
| CD4 count (cells/μL) [median (IQR)] | 440 (298, 609) | 419 (249, 642) | 440 (305, 602) |
| HIV RNA load (copies/mL) [median (IQR)] | 49 (39, 49) | 49 (39, 49) | 49 (39, 49) |
| APRI [median (IQR)] | 0.5 (0.4, 0.8) | 0.8 (0.5, 1.1) | 0.5 (0.4, 0.7) |
| On cART [n (%)] | 277 (96) | 55 (96) | 222 (96) |
| cART regimen† [n (%)] | |||
| PI | 198 (69) | 47 (82) | 151 (65) |
| NNRTI | 82 (28) | 9 (16) | 73 (32) |
| Others | 16 (6) | 1 (2) | 15 (6) |
| HCV RNA (log10 copies/mL) [median (IQR)]‡ | 6.2 (5.4, 6.8) | 6.5 (5.3, 6.8) | 6.2 (5.4, 6.7) |
| HCV treatment naïve [n (%)] | 253 (88) | 48 (84) | 205 (89) |
cART, combination antiretroviral therapy; IDU, injecting drug use; PI, protease inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor.
Alcohol abuse was defined as self-reported alcohol intake of more than two drinks per day or binge drinking (greater than six drinks at any one time). †Sum of regimens >100% as some participants are on both PI, NNRTI and/or other cART. ‡For HCVRNA, only 114 [26/57 (46%) progressors and 88/231 (38%) non-progressors] had available quantitative HCV RNA values.
Fifty-seven participants (20%) [12.4/100 PY; 95% CI 9.2–15.7/100 PY] progressed to an APRI ≥ 1.5 over a mean 1.1 (IQR 0.6, 2.0) years of follow-up time at risk (Table 2). Multivariate discrete proportional hazards models of factors associated with progression to significant fibrosis are shown in Table 3 (also represented in Fig. 2). Viral rebounds [adjusted hazard ratio (HR) 2.3; 95% CI 1.1–4.7] but not blips (adjusted HR 0.5; 95% CI 0.2–1.1) predicted progression to APRI ≥ 1.5. Each additional 1 log10 copies/mL HIV RNA exposure (cumulative) was associated with an 20% increase in risk of progression to significant fibrosis (adjusted HR 1.2; 95% CI 1.0–1.3). Only eight of 44 participants experiencing rebound were reported to be on a treatment interruption, suggesting nonadherence in the remainder. Other factors associated with progression to significant fibrosis were baseline ln(APRI), older age and alcohol abuse.
Table 2.
Incidence of progression to significant fibrosis [defined as aspartate aminotransferase to platelet ratio index (APRI) ≥ 1.5] according to level of HIV RNA replication
| n (%) | Denominator | Person-years | Rate per 100/person-years (95% CI) | |
|---|---|---|---|---|
| Overall | 57 (20) | 288 | 456.5 | 12.4 (9.2, 15.7) |
| No virological blip or rebound ever | 42 (19) | 216 | 303.8 | 13.8 (9.6, 18.0) |
| Virological blip only | 3 (11) | 28 | 45.8 | 6.6 (−0.9, 14.0) |
| Any virological rebound | 12 (27) | 44 | 69.4 | 17.3 (7.5, 27.1) |
| Number of follow-up visits with detectable HIV RNA | ||||
| 0 | 38 (18) | 209 | 304.2 | 12.5 (10.5, 14.4) |
| 1 | 8 (19) | 42 | 82.2 | 9.7 (3.0, 16.5) |
| 2–3 | 5 (25) | 20 | 36.2 | 13.8 (1.7, 25.9) |
| > 3 | 1 (17) | 6 | 18.3 | 5.5 (−5.3, 16.2) |
| Cumulative log copies of HIV RNA up to last visit | ||||
| < 2 | 39 (17) | 224 | 330.7 | 11.8 (8.1, 15.5) |
| 2–4 | 4 (20) | 20 | 43.3 | 9.2 (0.2, 18.3) |
| > 4 | 9 (27) | 33 | 66.9 | 13.4 (4.7, 22.2) |
CI, confidence interval.
Table 3.
Multivariate discrete time proportional hazards models of factors associated with time to developing significant fibrosis (APRI ≥ 1.5) in follow-up
| Primary analyses | Secondary analyses | |||
|---|---|---|---|---|
| Adjusted HR | 95% CI | Adjusted HR | 95% CI | |
| Time-updated exposures | ||||
| Virological blip | 0.5 | 0.2, 1.1 | – | – |
| Virological rebound | 2.3 | 1.1, 4.7 | – | – |
| Total log copies HIV RNA | – | – | 1.2 | 1.0, 1.3 |
| CD4 count (per 100 cells/μL) | 1.0 | 0.9, 1.2 | 1.0 | 0.9, 1.1 |
| Time-independent baseline covariates | ||||
| Age (per 10 years) | 1.3 | 0.9, 1.8 | 1.3 | 0.9, 1.8 |
| Sex (female) | 1.2 | 0.6, 2.3 | 1.3 | 0.6, 2.6 |
| Ethnicity (Aboriginal) | 1.0 | 0.4, 2.7 | 0.8 | 0.3, 2.4 |
| Current alcohol abuse | 1.2 | 0.7, 2.1 | 1.2 | 0.7, 2.3 |
| ln (APRI) | 6.3 | 3.1, 12.6 | 5.9 | 2.9, 12.1 |
APRI, aspartate aminotrasferase to platelet ratio; CI, confidence interval; HR, hazard ratio.
Fig 2.
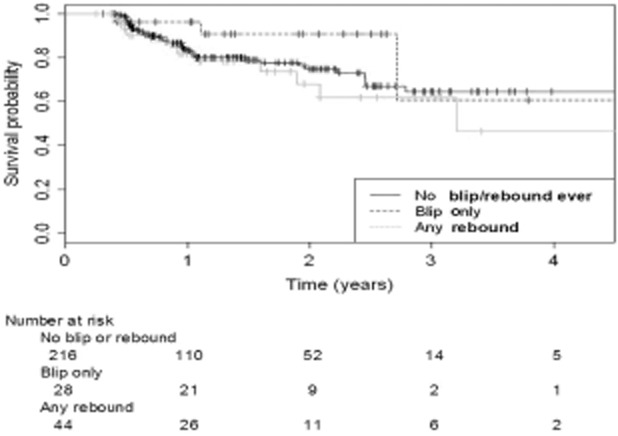
Kaplan−Meier estimated time to significant fibrosis [aspartate aminotransferase to platelet ratio index (APRI) ≥ 1.5] stratified by HIV RNA blips and rebound.
The sensitivity analysis using a linear spline was consistent with our main findings: adjusted HRs (95% CI) per 1 log copies/ml of: 0.7 (0.2–2.1) for HIV RNA < 50 copies/mL; 0.7 (0.1–6.0) for 50–1000 copies/mL and 1.4 (1.3–1.6) for > 1000 copies/mL. These findings suggest that the risk of fibrosis progression only clearly increased at HIV RNA levels > 1000 copies/mL.
Given that baseline characteristics suggested that patients with liver fibrosis progression were more likely to use protease inhibitor-based cART, we performed a post hoc sensitivity analysis that included cumulative time on protease inhibitors from baseline and total duration of cART (to account for potentially longer duration of cART among those receiving protease inhibitors) as additional covariates in the main models above. Cumulative protease inhibitor use in follow-up was associated with progression to significant fibrosis (adjusted HR 2.4; 95% CI 1.2–4.9) and its inclusion in the model attenuated the estimates for viral rebound somewhat (adjusted HR 1.9; 95% CI 0.9–4.1). However, the effect of cumulative HIV RNA exposure was unchanged (adjusted HR 1.2; 95% CI 1.0–1.3 per 1 log copies/mL).
Discussion
Lack of continuous HIV suppression negatively influences clinical outcomes in HIV infection. The Strategies for Management of Antiretroviral Therapy (SMART) study demonstrated that episodic CD4 T-lymphocyte count-guided ART interruption is associated with accelerated immunodeficiency and increased HIV replication 27. Moreover, interrupted ART increased the risk of morbidity and mortality by both opportunistic infection and nonopportunistic disease 28. The HR for major hepatic, cardiovascular and renal diseases was 1.8 (95% CI 1.2–2.9). Hepatic disease outcomes were identified based on histological (liver biopsy and autopsy), clinical (ascites, hepatic encephalopathy and varices), laboratory (prothrombin time/international normalized ratio and albumin) and radiological criteria 28.
In HIV/HCV-coinfected individuals, the interruption of ART has been associated with an increased risk of opportunistic and nonopportunistic infection outcomes 19,29. Liver biopsy studies have shown that detectable HIV RNA levels, as a consequence of inadequate HIV suppression, pose a risk for accelerated liver fibrosis 30,31. As a consequence of the inherent inconsistency and limited availability of liver biopsies, serum biochemical indexes have been used to further investigate hepatic disease. APRI studies demonstrate that interrupted ART is associated with liver fibrosis progression and ESLD 19,32.
The published literature is inconsistent as to whether virological blips predict adverse clinical outcomes such as death and end-organ disease 16,17,33–39. This is in part attributable to inconsistent definitions. Blips defined as transiently detectable viraemia within the 50–400 copies/mL range do not seem to increase the risk of virological failure 8,9. However, greater magnitude blips (i.e. 500–999 copies/mL) are associated with subsequent treatment failure 15,16,35,36.
There are several proposed mechanisms by which nonsuppressed HIV viraemia may contribute to accelerated fibrosis 40. These include impaired HCV-specific immune response 41 and cytokine deregulation leading to increased inflammatory milieu within the liver leading to activation of profibrotic processes 42 and the potential for greater drug-induced hepatotoxicity 43,44. Increased microbial translocation from the gut during periods of viral rebound may contribute to increased concentrations of inflammatory markers (i.e. tumour necrosis factor alpha and interleukin-6) within the liver parenchyma contributing to increased fibrosis progression 45.
Our study has several strengths. It was nested in a large, prospective cohort study that is broadly representative of HIV/HCV-coinfected persons in care in Canada. In previous studies, patients were only selected based on having undergone liver biopsy, which potentially introduces selection bias. Additional methods to quantify fibrosis including biopsy, transient elastography and other calculated systems (e.g. FibroTest BioPredictive, Paris, France) would ideally have been valuable to corroborate our findings using APRI. However, there is ample literature demonstrating a strong correlation between APRI and other methods of evaluating liver fibrosis 46,47.
Limitations of our analyses are noted, including a relatively short period of follow-up. The fact that viral rebound was identified as a predictor of fibrosis progression despite this short period of follow-up highlights the critical role that achieving and maintaining HIV RNA suppression can play in protecting the liver from HCV-induced injury. Our relatively small sample size and short follow-up duration also limited inference about other variables traditionally associated with fibrosis progression such as age, sex and alcohol use, which were not significantly associated with fibrosis progression in our analyses. Interestingly, protease inhibitor use appeared to be an additional strong predictor of fibrosis progression in sensitivity analyses even after accounting for time on cART. Inclusion of protease inhibitor use in our models attenuated, but did not remove the association of viral rebound with fibrosis progression. It is possible that protease inhibitor use is simply a marker of more advanced HIV disease and more frequent episodes of viral rebound prior to cohort entry which may have affected risk of fibrosis progression in this cohort. Protease inhibitor use might also be a proxy for poor adherence if, because of their higher barrier to resistance, protease inhibitors were given preferentially to patients expected not to adhere to therapy. Alternatively, there is evidence that boosted protease inhibitors may contribute to long-term hepatoxicity both directly and indirectly through metabolic changes such as insulin resistance and hepatic steatosis 48. We were unable to fully explore the association of protease inhibitor use and fibrosis progression in these analyses as data on total exposure to protease inhibitors prior to cohort entry were not available for all patients.
Liver fibrosis progression, as estimated by APRI score, was greater in patients experiencing HIV rebound, but not blips, and was associated with the degree of HIV RNA exposure over time. Optimizing antiretroviral adherence, which would reduce the frequency of virological rebound episodes, may protect the liver from HCV-induced liver fibrosis progression.
Acknowledgments
This study was funded by the Fonds de recherche en santé du Québec, Réseau SIDA/maladies infectieuses (FRQ-S), the Canadian Institutes of Health Research (CIHR MOP-79529) and the CIHR Canadian HIV Trials Network (CTN222). CC acknowledges The Ontario HIV Treatment Network (Applied HIV Research Chair) and The Ottawa Hospital Department of Medicine for funding support. MBK is supported by a ‘Chercheurs nationaux’ career award from the FRQ-S. We thank all study coordinators and nurses for their assistance with study coordination, participant recruitment and care.
Author contributions: As the corresponding author, MBK has had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. She supervised the study design, conduct and reporting and participated in revising the manuscript. CC conceived the study, revised results and drafted the manuscript. KR-K conducted the data analysis and prepared data reports for use in the manuscript. All of the other authors participated by recruiting and following patients in the cohort and critically reviewed the manuscript as part of the writing group. All authors have seen and approved the final manuscript and have participated sufficiently in the work to take public responsibility for its content.
Appendix
The Canadian Co-infection Cohort Study (CTN222) Investigators are: Dr Jeff Cohen, Windsor Regional Hospital Metropolitan Campus, Windsor, ON; Dr Brian Conway, Vancouver Infectious Diseases Research and Care Centre, Vancouver, BC; Dr Pierre Côté, Clinique du Quartier Latin, Montréal, QC; Dr Joseph Cox, Montréal General Hospital, Montréal, QC; Dr Shariq Haider, McMaster University, Hamilton, ON; Dr Marianne Harris, St Paul's Hospital, Vancouver, BC; Dr David Haase, Capital District Health Authority, Halifax, NS; Dr Mark Hull, BC Centre for Excellence in HIV/AIDS, Vancouver, BC; Dr Julio Montaner, St Paul's Hospital, Vancouver, BC; Dr Erica Moodie, McGill University, Montreal, QC; Dr Anita Rachlis, Sunnybrook & Women's College Health Sciences Centre, Toronto, ON; Dr Danielle Rouleau, Centre Hospitalier de l'Université de Montréal, Montréal, QC; Dr Roger Sandre, HAVEN Program, Sudbury, ON; Dr Marie-Louise Vachon, Centre Hospitalier Universitaire de Québec, Québec, QC; and Dr David Wong, University Health Network, Toronto, ON.
References
- Weber R, Sabin CA, Friis-Moller N, et al. Liver-related deaths in persons infected with the human immunodeficiency virus: the D:A:D study. Arch Intern Med. 2006;166:1632–1641. doi: 10.1001/archinte.166.15.1632. [DOI] [PubMed] [Google Scholar]
- Ioannou GN, Bryson CL, Weiss NS, Miller R, Scott JD, Boyko EJ. The prevalence of cirrhosis and hepatocellular carcinoma in patients with human immunodeficiency virus infection. Hepatology. 2013;57:249–257. doi: 10.1002/hep.25800. [DOI] [PubMed] [Google Scholar]
- Lacombe K, Rockstroh J. HIV and viral hepatitis coinfections: advances and challenges. Gut. 2012;61(Suppl 1):i47–i58. doi: 10.1136/gutjnl-2012-302062. [DOI] [PubMed] [Google Scholar]
- Cooper CL. Therapies for HIV and viral hepatitis coinfection. Expert Rev Anti Infect Ther. 2005;3:81–89. doi: 10.1586/14787210.3.1.81. [DOI] [PubMed] [Google Scholar]
- Dornadula G, Zhang H, VanUitert B, et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA. 1999;282:1627–1632. doi: 10.1001/jama.282.17.1627. [DOI] [PubMed] [Google Scholar]
- Di Mascio M, Markowitz M, Louie M, et al. Viral blip dynamics during highly active antiretroviral therapy. J Virol. 2003;77:12165–12172. doi: 10.1128/JVI.77.22.12165-12172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Cozzi-Lepri A, Ledergerber B, et al. Intermittent and sustained low-level HIV viral rebound in patients receiving potent antiretroviral therapy. AIDS. 2002;16:1967–1969. doi: 10.1097/00002030-200209270-00017. [DOI] [PubMed] [Google Scholar]
- Mira JA, Macias J, Nogales C, et al. Transient rebounds of low-level viraemia among HIV-infected patients under HAART are not associated with virological or immunological failure. Antivir Ther. 2002;7:251–256. doi: 10.1177/135965350200700404. [DOI] [PubMed] [Google Scholar]
- Sklar PA, Ward DJ, Baker RK, et al. Prevalence and clinical correlates of HIV viremia (‘blips’) in patients with previous suppression below the limits of quantification. AIDS. 2002;16:2035–2041. doi: 10.1097/00002030-200210180-00008. [DOI] [PubMed] [Google Scholar]
- Lima V, Harrigan R, Montaner JS. Increased reporting of detectable plasma HIV-1 RNA levels at the critical threshold of 50 copies per milliliter with the Taqman assay in comparison to the Amplicor assay. J Acquir Immune Defic Syndr. 2009;51:3–6. doi: 10.1097/QAI.0b013e31819e721b. [DOI] [PubMed] [Google Scholar]
- Nettles RE, Kieffer TL. Update on HIV-1 viral load blips. Curr Opin HIV AIDS. 2006;1:157–161. doi: 10.1097/01.COH.0000203834.24221.13. [DOI] [PubMed] [Google Scholar]
- Nettles RE, Kieffer TL, Kwon P, et al. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. JAMA. 2005;293:817–829. doi: 10.1001/jama.293.7.817. [DOI] [PubMed] [Google Scholar]
- Smit E, Bhattacharya S, Osman H, Taylor S. Increased frequency of HIV-1 viral load blip rate observed after switching from Roche Cobas Amplicor to Cobas Taqman assay. J Acquir Immune Defic Syndr. 2009;51:364–365. doi: 10.1097/QAI.0b013e3181aa13b3. [DOI] [PubMed] [Google Scholar]
- Stosor V, Palella FJ, Jr, Berzins B, et al. Transient viremia in HIV-infected patients and use of plasma preparation tubes. Clin Infect Dis. 2005;41:1671–1674. doi: 10.1086/498025. [DOI] [PubMed] [Google Scholar]
- Grennan JT, Loutfy MR, Su D, et al. Magnitude of virologic blips is associated with a higher risk for virologic rebound in HIV-infected individuals: a recurrent events analysis. J Infect Dis. 2012;205:1230–1238. doi: 10.1093/infdis/jis104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easterbrook PJ, Ives N, Waters A, et al. The natural history and clinical significance of intermittent viraemia in patients with initial viral suppression to < 400 copies/ml. AIDS. 2002;16:1521–1527. doi: 10.1097/00002030-200207260-00009. [DOI] [PubMed] [Google Scholar]
- Martinez V, Marcelin AG, Morini JP, et al. HIV-1 intermittent viraemia in patients treated by non-nucleoside reverse transcriptase inhibitor-based regimen. AIDS. 2005;19:1065–1069. doi: 10.1097/01.aids.0000174453.55627.de. [DOI] [PubMed] [Google Scholar]
- Orrell C, Kaplan R, Wood R, Bekker LG. Virological breakthrough: a risk factor for loss to followup in a large community-based cohort on antiretroviral therapy. AIDS Res Treat. 2011;2011:469127. doi: 10.1155/2011/469127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe J, Saeed S, Moodie EE, Klein MB. Antiretroviral treatment interruption leads to progression of liver fibrosis in HIV-hepatitis C virus co-infection. AIDS. 2011;25:967–975. doi: 10.1097/QAD.0b013e3283455e4b. [DOI] [PubMed] [Google Scholar]
- Klein MB, Saeed S, Yang H, et al. Cohort profile: the Canadian HIV-hepatitis C co-infection cohort study. Int J Epidemiol. 2010;39:1162–1169. doi: 10.1093/ije/dyp297. [DOI] [PubMed] [Google Scholar]
- Al-Mohri H, Cooper C, Murphy T, Klein MB. Validation of a simple model for predicting liver fibrosis in HIV/hepatitis C virus-coinfected patients. HIV Med. 2005;6:375–378. doi: 10.1111/j.1468-1293.2005.00330.x. [DOI] [PubMed] [Google Scholar]
- Al-Mohri H, Murphy T, Lu Y, Lalonde RG, Klein MB. Evaluating liver fibrosis progression and the impact of antiretroviral therapy in HIV and hepatitis C coinfection using a noninvasive marker. J Acquir Immune Defic Syndr. 2007;44:463–469. doi: 10.1097/QAI.0b013e318030ff8e. [DOI] [PubMed] [Google Scholar]
- Di Mascio M, Ribeiro RM, Markowitz M, Ho DD, Perelson AS. Modeling the long-term control of viremia in HIV-1 infected patients treated with antiretroviral therapy. Math Biosci. 2004;188:47–62. doi: 10.1016/j.mbs.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Percus JK, Percus OE, Markowitz M, Ho DD, Di Mascio M, Perelson AS. The distribution of viral blips observed in HIV-1 infected patients treated with combination antiretroviral therapy. Bull Math Biol. 2003;65:263–277. doi: 10.1016/S0092-8240(02)00095-2. [DOI] [PubMed] [Google Scholar]
- Lo ReV, 3rd, Lim JK, Goetz MB, et al. Validity of diagnostic codes and liver-related laboratory abnormalities to identify hepatic decompensation events in the Veterans Aging Cohort Study. Pharmacoepidemiol Drug Saf. 2011;20:689–699. doi: 10.1002/pds.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin JB, Wolfe R, Coffey C, Patton GC. Analysis of binary outcomes in longitudinal studies using weighted estimating equations and discrete-time survival methods: prevalence and incidence of smoking in an adolescent cohort. Stat Med. 1999;18:2655–2679. doi: 10.1002/(sici)1097-0258(19991015)18:19<2655::aid-sim202>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Lundgren JD, Babiker A, El-Sadr W, et al. Inferior clinical outcome of the CD4+ cell count-guided antiretroviral treatment interruption strategy in the SMART study: role of CD4+ Cell counts and HIV RNA levels during follow-up. J Infect Dis. 2008;197:1145–1155. doi: 10.1086/529523. [DOI] [PubMed] [Google Scholar]
- El-Sadr WM, Lundgren JD, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355:2283–2296. doi: 10.1056/NEJMoa062360. [DOI] [PubMed] [Google Scholar]
- Tedaldi E, Peters L, Neuhaus J, et al. Opportunistic disease and mortality in patients coinfected with hepatitis B or C virus in the strategic management of antiretroviral therapy (SMART) study. Clin Infect Dis. 2008;47:1468–1475. doi: 10.1086/593102. [DOI] [PubMed] [Google Scholar]
- Brau N, Salvatore M, Rios-Bedoya CF, et al. Slower fibrosis progression in HIV/HCV-coinfected patients with successful HIV suppression using antiretroviral therapy. J Hepatol. 2006;44:47–55. doi: 10.1016/j.jhep.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Schiavini M, Angeli E, Mainini A, et al. Fibrosis progression in paired liver biopsies from HIV/HCV co-infected patients. Hepat Mon. 2011;11:525–531. [PMC free article] [PubMed] [Google Scholar]
- Lopez-Dieguez M, Montes ML, Pascual-Pareja JF, et al. The natural history of liver cirrhosis in HIV-hepatitis C virus-coinfected patients. AIDS. 2011;25:899–904. doi: 10.1097/QAD.0b013e3283454174. [DOI] [PubMed] [Google Scholar]
- Garcia-Gasco P, Maida I, Blanco F, et al. Episodes of low-level viral rebound in HIV-infected patients on antiretroviral therapy: frequency, predictors and outcome. J Antimicrob Chemother. 2008;61:699–704. doi: 10.1093/jac/dkm516. [DOI] [PubMed] [Google Scholar]
- Havlir DV, Bassett R, Levitan D, et al. Prevalence and predictive value of intermittent viremia with combination hiv therapy. JAMA. 2001;286:171–179. doi: 10.1001/jama.286.2.171. [DOI] [PubMed] [Google Scholar]
- Masquelier B, Pereira E, Peytavin G, et al. Intermittent viremia during first-line, protease inhibitors-containing therapy: significance and relationship with drug resistance. J Clin Virol. 2005;33:75–78. doi: 10.1016/j.jcv.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Moore AL, Youle M, Lipman M, et al. Raised viral load in patients with viral suppression on highly active antiretroviral therapy: transient increase or treatment failure? AIDS. 2002;16:615–618. doi: 10.1097/00002030-200203080-00013. [DOI] [PubMed] [Google Scholar]
- Podsadecki TJ, Vrijens BC, Tousset EP, Rode RA, Hanna GJ. Decreased adherence to antiretroviral therapy observed prior to transient human immunodeficiency virus type 1 viremia. J Infect Dis. 2007;196:1773–1778. doi: 10.1086/523704. [DOI] [PubMed] [Google Scholar]
- Raboud JM, Rae S, Woods R, Harris M, Montaner JS. Consecutive rebounds in plasma viral load are associated with virological failure at 52 weeks among HIV-infected patients. AIDS. 2002;16:1627–1632. doi: 10.1097/00002030-200208160-00008. [DOI] [PubMed] [Google Scholar]
- Sungkanuparph S, Overton ET, Seyfried W, Groger RK, Fraser VJ, Powderly WG. Intermittent episodes of detectable HIV viremia in patients receiving nonnucleoside reverse-transcriptase inhibitor-based or protease inhibitor-based highly active antiretroviral therapy regimens are equivalent in incidence and prognosis. Clin Infect Dis. 2005;41:1326–1332. doi: 10.1086/496985. [DOI] [PubMed] [Google Scholar]
- Cooper CL. HIV antiretroviral medications and hepatotoxicity. Curr Opin HIV AIDS. 2007;2:466–473. doi: 10.1097/COH.0b013e3282f0dd0b. [DOI] [PubMed] [Google Scholar]
- Leroy V, Vigan I, Mosnier JF, et al. Phenotypic and functional characterization of intrahepatic T lymphocytes during chronic hepatitis C. Hepatology. 2003;38:829–841. doi: 10.1053/jhep.2003.50410. [DOI] [PubMed] [Google Scholar]
- Glassner A, Eisenhardt M, Kokordelis P, et al. Impaired CD4(+) T cell stimulation of NK cell anti-fibrotic activity may contribute to accelerated liver fibrosis progression in HIV/HCV patients. J Hepatol. 2013;59:427–433. doi: 10.1016/j.jhep.2013.04.029. [DOI] [PubMed] [Google Scholar]
- Renton KW. Alteration of drug biotransformation and elimination during infection and inflammation. Pharmacol Ther. 2001;92:147–163. doi: 10.1016/s0163-7258(01)00165-6. [DOI] [PubMed] [Google Scholar]
- Morgan ET, Li-Masters T, Cheng PY. Mechanisms of cytochrome P450 regulation by inflammatory mediators. Toxicology. 2002;181–182:207–210. doi: 10.1016/s0300-483x(02)00283-4. [DOI] [PubMed] [Google Scholar]
- Reus S, Portilla J, Sanchez-Paya J, et al. Low-level HIV viremia is associated with microbial translocation and inflammation. J Acquir Immune Defic Syndr. 2013;62:129–134. doi: 10.1097/QAI.0b013e3182745ab0. [DOI] [PubMed] [Google Scholar]
- Cales P, Halfon P, Batisse D, et al. Comparison of liver fibrosis blood tests developed for HCV with new specific tests in HIV/HCV co-infection. J Hepatol. 2010;53:238–244. doi: 10.1016/j.jhep.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Loko MA, Castera L, Dabis F, et al. Validation and comparison of simple noninvasive indexes for predicting liver fibrosis in HIV-HCV-coinfected patients: ANRS CO3 Aquitaine cohort. Am J Gastroenterol. 2008;103:1973–1980. doi: 10.1111/j.1572-0241.2008.01954.x. [DOI] [PubMed] [Google Scholar]
- Moodie EE, Pant Pai N, Klein MB. Is antiretroviral therapy causing long-term liver damage? A comparative analysis of HIV-mono-infected and HIV/hepatitis C co-infected cohorts. PLoS ONE. 2009;4:e4517. doi: 10.1371/journal.pone.0004517. [DOI] [PMC free article] [PubMed] [Google Scholar]