

Summary
Colorectal cancer primarily metastasizes to the liver and kills over 600,000 people annually. By functionally screening 661 miRNAs in parallel during liver colonization, we have identified miR-551a and miR-483 as robust endogenous suppressors of liver colonization and metastasis. These miRNAs convergently target creatine kinase, brain-type (CKB), which phosphorylates the metabolite creatine, to generate phosphocreatine. CKB is released into the extracellular space by metastatic cells encountering hepatic hypoxia and catalyzes production of extracellular phosphocreatine, which is imported through the SLC6A8 transporter and used to generate ATP—fueling metastatic survival. Combinatorial therapeutic viral delivery of miR-551a and miR-483-5p through single-dose adeno-associated viral (AAV) delivery significantly suppressed colon cancer metastatic colonization, as did CKB inhibition with a small-molecule inhibitor. Importantly, human liver metastases express higher CKB and SLC6A8 levels and reduced miR-551a/miR-483 levels relative to primary tumors. We identify the extracellular space as an important compartment for malignant energetic catalysis and therapeutic targeting.
Introduction
Colorectal cancer is the third leading cause of mortality in the United States and a major cause of death globally (Davis and Schlessinger, 2012; Jemal et al., 2011; Siegel et al., 2014). Death from colorectal cancer is primarily due to the metastatic progression, with the liver being the organ of metastatic colonization in over 70% of patients. To date, efforts aimed at increasing cure rates after surgery have focused on combination chemotherapy administration as a means of preventing metastasis. Such therapy reduces metastatic relapse by roughly 7% (Meyerhardt and Mayer, 2005). The high prevalence of this disease and the lack of effective adjuvant therapeutics demand a greater understanding of the biology of its progression (Markowitz and Bertagnolli, 2009).
In recent years, post-transcriptional deregulation has emerged as a key feature of metastatic cells. In particular, specific miRNAs, which are small non-coding RNAs, have been identified that are silenced or over-expressed and act to suppress or promote metastatic progression by diverse cancer types (Lujambio and Lowe, 2012; Ma et al., 2007; Pencheva and Tavazoie, 2013; Pencheva et al., 2012; Tavazoie et al., 2008). While the use of these miRNAs as molecular probes for the identification of metastasis regulators has proved fruitful, their therapeutic utility has been limited given the inefficient delivery of miRNAs into various metastatic tissues. Interestingly, the liver represents an exception to this rule, since miRNAs tend to accumulate in hepatic tissue and since vectors such as adeno-associated viruses and nanoparticles have shown promising efficacy in enhancing hepatic delivery in non-human primates and humans (Kota et al., 2009; Mingozzi and High, 2011). Given this unique feature of the liver as well as the great need for targeted therapies that can suppress liver metastatic colonization by colon cancer, the identification of miRNAs that can suppress liver metastasis would be of great clinical value.
By screening 661 human miRNAs in parallel for their ability to suppress the colonization of the liver by multiple colon cancer cell lines representing diverse mutational subtypes, we have identified miR-551 and miR-483 as endogenous suppressors of colon cancer metastasis. We find that these miRNAs both target Creatine kinase Brain (CKB). Disseminated metastatic cells release this enzyme into the extracellular space, where it catalyzes the phosphorylation of the metabolite creatine by using extracellular ATP as the phosphate source. Phosphocreatine is then imported into disseminated colorectal cancer cells where its high-energy phosphate is used to generate intracellular ATP that sustains the energetic requirements of colon cancer cells encountering hepatic hypoxia, allowing them to survive this barrier to metastatic progression. Therapeutic viral delivery of these miRNAs to the liver and disseminated colon cancer cells via adeno-associated viral delivery strongly suppresses metastatic colonization by colon cancer cells. Moreover, small-molecule therapeutic inhibition of CKB activity also suppresses metastatic growth. Our findings delineate a druggable molecular network that governs both the metabolic state and the metastatic progression capacity of disseminated colon cancer cells. More importantly, we implicate the extracellular space as a previously unrecognized environment for malignant catalysis and identify CKB as a secreted metabolic kinase that drives cancer progression.
Results
Endogenous miR-483-5p and miR-551a Suppress Human Colorectal Cancer Metastasis
In vivo selection has been used by many investigators to identify candidate genes that regulate metastatic progression of diverse cancer types. This approach allows one to derive highly metastatic sub-populations with enhanced metastatic activity for a given organ (Fidler, 1973). The comparison of transcriptomic profiles of metastatic derivatives to the parental lines from which that they were derived has revealed numerous candidate genes for functional testing (Bruns et al., 1999; Kang et al., 2003; Minn et al., 2005; Pencheva et al., 2012; Png et al., 2012; Tavazoie et al., 2008). As a first step to identify the molecular regulators of liver colonization by colon cancer cells, we performed in vivo selection on the LS-174T (K-Ras mutant) human colon cancer line for enhanced liver colonization activity through iterative intra-hepatic injection of cancer cells into immunodeficient mice followed by surgical resection of liver colonies and dissociation of cells. Independently derived third-generation liver colonizers LS-LvM3a and LS-LvM3b displayed significantly enhanced (>50 fold) capacity for liver colonization upon intrahepatic injection relative to their parental line (Fig. 1A). Importantly, these derivatives also displayed dramatically enhanced (>150 fold) liver metastatic capacity upon portal circulation injection in metastasis assays (Fig. S1A)—revealing the acquisition of liver colonization capacity to be sufficient for imparting enhanced liver metastasis activity. As an orthogonal approach, we transduced a library of lentiviral particles, each encoding one of 661 human miRNAs, into two independent colon cancer cell lines—the WiDR (K-Ras wild-type) and SW620 (K-Ras mutant) human lines. These cancer populations, containing cancer cells expressing each of 661 miRNAs, were then intra-hepatically injected into mice to allow for selection of cells capable of colonizing the liver. Genomic PCR amplification of lentiviral-derived miRNA sequences and miRNA profiling of miRNA inserts allowed for the quantification of miRNA insert representation (Fig. 1B, Supplemental Table 1). We identified miRNAs that displayed reduced representation in the context of liver colonization in both colon cancer cell-lines on the basis that over-expression of these miRNAs suppressed liver colonization by colon cancer cells. We next asked whether endogenous forms of these miRNAs exhibited silencing in highly metastatic derivatives relative to isogenic poorly metastatic parental cells. Indeed, two of the miRNAs, miR-483-5p and miR-551a, were found to be silenced in highly metastatic LS-LVM3a and LS-LVM3b liver colonizers relative to their parental line (Fig. S1B; Supplemental Table 2). Consistent with a suppressive role for these miRNAs in liver colonization, over-expression of miR-483-5p or miR-551a robustly suppressed metastatic colonization by LS-LvM3b cells introduced into the portal circulation (Fig. 1C, Fig S1C), while inhibition of endogenous miR-483-5p or miR-551a in poorly metastatic parental lines SW480 and LS-174T significantly enhanced liver metastatic colonization (Fig. 1D, Fig. S1D). The effects of these miRNAs on metastatic progression were not secondary to modulation of proliferative capacity since miR-551a inhibition did not affect in vitro proliferation, while miR-483-5p inhibition minimally increased proliferation (10%)—an order of magnitude less than its effect on metastasis (Fig. S1E). Importantly, over-expression of either miRNA did not suppress primary tumor growth (Fig. S1F).
Figure 1. miR-483-5p and miR-551a are endogenous miRNAs that suppress liver metastasis.

A, Bioluminescence plot of liver colonization by 5 × 105 LS-Parental, LvM3a and LvM3b cells after direct intrahepatic injection (n>5). Mice were imaged at day 21 after injection and livers extracted for ex vivo imaging and gross morphological examination. Photon flux ratio is the ratio of bioluminescence signal at day 21 normalized to signal on day 0. B, Schematic for the identification of miR-483-5p and miR-551a as suppressors of metastasis. C, Liver metastasis of mice injected with 5 × 105 LvM3b cells over-expressing either a control hairpin, miR-483-5p or miR-551a (n>5). D, Liver metastasis in mice injected with 5 × 105 SW480 cells, whose endogenous miR-483-5p or miR-551a was inhibited (n>5). E, F, Organotypic slice culture imaging of SW480 cells (n=8) whose endogenous miR-483-5p (E) or miR-551a (F) were inhibited by LNAs. 5 × 105 cells were labeled with cell-tracker green (control LNA) or cell-tracker red (miRNA specific LNA) and introduced into the livers prior to slice culture. Dye-swap experiments were performed to compensate for dye bias. Representative images at day 0 and day 3 are shown. Total area of each cell population at indicated time points are measured and normalized to start of experiment. Scale bar is equal to 50um. G, Bioluminescent metastatic signal from mice (n=5) injected with 5 × 105 SW480 cells whose endogenous miR-483-5p or miR-551a activities were inhibited. Images and measurements were taken 24hr after tumor cells inoculation. H, Relative in vivo caspase activity of SW480 cells whose endogenous miR-483-5p or miR-551a was inhibited (n=3). Caspase activity was monitored using a caspase-3/7 activated DEVD-luciferin and normalized with bioluminescent signal from regular luciferin. Error bars, s.e.m; all P values are based on one-sided Student’s t-tests, or where appropriate, Mann-Whitney test for non-Gaussian distribution. *p<0.05; **p<0.01; ***p<0.001. See also Figure S1.
To better investigate the mechanism(s) by which these miRNAs exert their anti-metastatic effects, we employed an in vitro liver organotypic slice culture system to study early events during liver metastasis subsequent to single-cell dissemination of colon cancer cells in the liver microenvironment (Fig. S1G). Consistent with prior studies, which revealed a significant selection on cell survival during metastatic colonization (Gupta and Massagué, 2006; Talmadge and Fidler, 2010), we noted that highly metastatic LvM3b cells were significantly better at persisting in the liver microenvironment than their poorly metastatic parental line; consistent with a key role for intrahepatic persistence in metastatic progression (Fig. S1H). We examined whether the enhanced capacity of metastatic cells to persist in the hepatic microenvironment is regulated by miR-483-5p or miR-551a. Indeed, over-expression of miR-483-5p and miR-551a in LS-LvM3b cells suppressed (Fig. S1I, J), while inhibition of these miRNAs enhanced (Fig 1E, F) colon cancer persistence in the hepatic microenvironment. In agreement with our organotypic slice culture findings, we noted that as early as 24 hours after injection of cells into the portal circulation, cells whose endogenous miRNAs were inhibited out-competed control cells (Fig. 1G). As neither of these miRNAs significantly affected proliferation, we asked if they elicited their effects by suppressing cancer cell survival during metastatic progression. To quantify cell death in vivo, we utilized a bioluminescence-based luciferin reporter of caspase-3/7 activity (Hickson et al., 2010). miRNA inhibition significantly reduced in vivo caspase activity in colon cancer cells during the early phase of hepatic colonization (Fig. 1H), revealing cancer survival to be the phenotype suppressed by these miRNAs. These in vivo findings provide corroboration and a mechanistic basis for the organotypic slice culture observations. Our findings reveal miR-483 and miR-551a to suppress liver metastatic colonization through suppression of metastatic cell survival in the liver—a phenotype exhibited by highly metastatic colon cancer cells.
miR-483-5p and miR-551a Suppress Colorectal Cancer Cell Survival and Metastasis in the Liver Through Targeting of CKB
We next sought to identify the downstream effectors of these miRNAs. Through transcriptomic profiling, we identified transcripts that were down-regulated by over-expression of each miRNA and that contained 3′-UTR or coding-sequence elements complementary to the miRNAs. Interestingly, CKB was identified as a putative target of both miRNAs, suggesting that these miRNAs, which exhibit common in vivo and organotypic phenotypes, might mediate their effects through a common target gene (Supplemental Table S3). Importantly, endogenous miR-483 and miR-551a were found to suppress CKB protein levels (Fig. 2A). Quantitative RT-PCR validation also revealed suppression or up-regulation of CKB transcript levels upon over-expression or inhibition of the miRNAs, respectively (Fig. S2A, B). Mutagenesis and luciferase-based reporter assays revealed miR-483 to directly target the 3′UTR and miR-551a to directly target the coding region of CKB (Fig. S2C, D). Over-expression of CKB in poorly metastatic SW480 cells was sufficient to significantly enhance liver metastasis (>3-fold; Fig. 2B), while CKB knockdown in metastatic LS-LvM3b cells and SW480 cells, through the use of two independent hairpins for each line, robustly suppressed liver metastatic colonization (>5 fold Fig. 2C, Fig. S2E). Importantly, metastases that grew out in knockdown experiments had ‘escaped’ shRNA knockdown and displayed restored CKB expression (Fig S2F). Consistent with the effects of its regulatory miRNAs, CKB over-expression was sufficient to significantly enhance the ability of colon cancer cells to persist in the liver micro-environment and enhanced their representation in the liver (Fig. 2D), while CKB knockdown substantially reduced intra-hepatic persistence (Fig. 2E). Consistent with this, CKB over-expression reduced (Fig. 2F), while CKB knockdown significantly enhanced (Fig. 2G), in vivo caspase-3/7 activity in colon cancer cells during the initial phase of hepatic colonization. To investigate whether CKB acts directly downstream of miR-483-5p and miR-551a, we performed gain-of-function, loss-of-function, and epistasis studies. Knockdown of CKB in cells inhibited for miR-483-5p or miR-551a prevented the enhanced metastasis effect seen with miR-483-5p or miR-551a silencing (Fig. 2H). Conversely over-expression of CKB was sufficient to rescue the suppressed liver metastatic phenotypes of cells over-expressing miR-483-5p or miR-551a (Fig. S2G). The results of the above mutational, gain-of-function, loss-of-function, and epistasis analyses reveal CKB to be a direct target of miR-483-5p and miR-551a, to act as a downstream effector of these miRNAs in the regulation of colon cancer metastatic progression, and be a promoter of colon cancer survival during hepatic metastatic colonization.
Figure 2. miR-483-5p and miR-551a suppress colorectal cancer cells survival and metastasis in the liver through regulation of CKB.
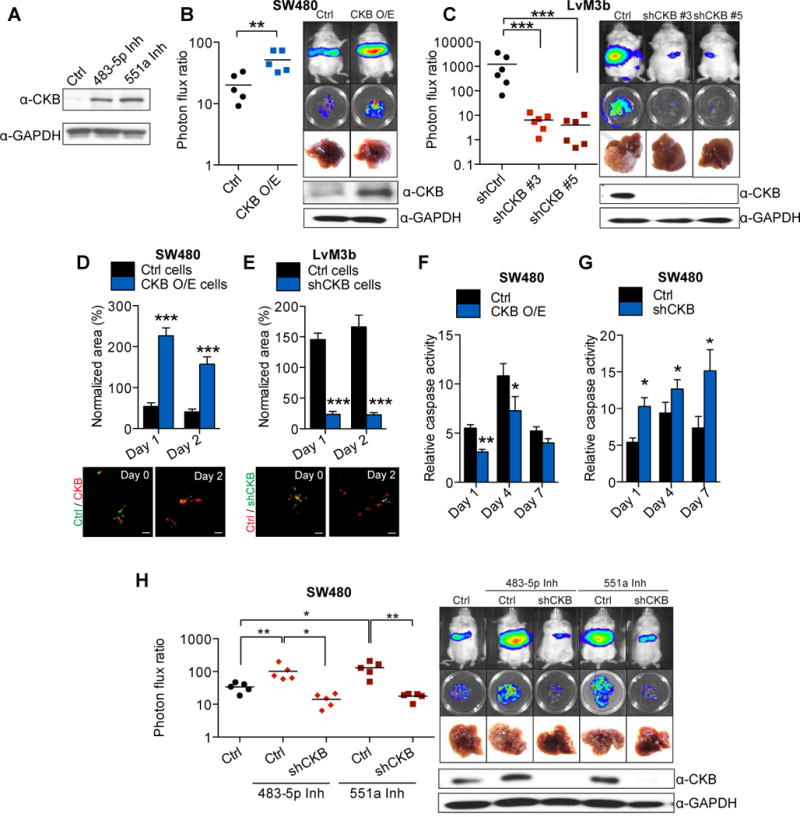
A, Expression of CKB in SW480 cells whose endogenous miR-483-5p or miR-551a was inhibited with LNAs. B, Liver metastasis in mice injected intrasplenically with 5 × 105 control SW480 cells and CKB over-expressing SW480 cells (n=5). Mice were euthanized at 28 days after injection. C, Liver metastasis in mice injected intrasplenically with 5 × 105 LvM3b expressing a control hairpin or two independent shRNA hairpins targeting CKB (n=6). Mice were euthanized 21 days after injection. D, Survival of control SW480 and CKB over-expressing SW480 cells in organotypic liver slices (n=8). E, Survival of LvM3b cells expressing a control hairpin or hairpin targeting CKB in organotypic slice cultures (n=8). Representative images at day 0 and day 2 are shown. Scale bar represent 50um. F, Relative in vivo caspase activity of control or CKB over-expressing SW480 cells Caspase activity was measured on day 1, 4 and 7 after injection (n=3). G, Relative in vivo caspase-3 activity of SW480 cells expressing a control shRNA or shRNA targeting CKB. Caspase activity was measured on day 1, 4 and 7 after injection (n=3). H, Liver metastasis in mice injected with 5 × 105 SW480 cells whose endogenous miR-483-5p or miR-551a was inhibited by LNA, with and without CKB knockdown. Error bars, s.e.m; all P values are based on one-sided Student’s t-tests, or where appropriate, Mann-Whitney test for non-Gaussian distribution. *p<0.05; **p<0.01; ***p<0.001. See also Figure S2.
CKB Promotes Colorectal Cancer Cell Survival during Acute Intrahepatic Hypoxia through Modulation of the Phosphocreatine/ATP shuttle
CKB is known to regulate the reservoir of rapidly mobilized high-energy phosphates in tissues such as the brain by catalyzing the transfer of a high-energy phosphate group from ATP to the metabolite creatine, yielding phosphocreatine (Wyss and Kaddurah-Daouk, 2000). Recent studies have implicated the involvement of metabolic pathways in tumorigenesis and cancer progression (Cairns et al., 2011; Christofk et al., 2008; Dang et al., 2009; Kaelin and McKnight, 2013). The maintenance of intracellular ATP levels is also critical for cancer cell survival under metabolic stress (Jeon et al., 2012). Highly metabolic cells maintain phosphocreatine stores in order to buffer against low ATP states, since phosphocreatine’s high-energy phosphate can be transferred to ADP to generate ATP (Wallimann et al., 1992; Wyss and Kaddurah-Daouk, 2000). Consistent with this, over-expression and knockdown of CKB in colon cancer cells increased and decreased, respectively, intracellular phosphocreatine levels (Fig. 3A) and CKB depletion resulted in decreased ATP levels that could be rescued by phosphocreatine supplementation (Fig. 3B). Consistent with our findings that miR-483 and miR-551a regulate CKB expression, modulation of either of these miRNAs also modulated intracellular phosphocreatine (Fig. S3A, B) and ATP levels (Fig. S3C, D). What purpose could CKB-generated phosphocreatine and ATP play during colon cancer metastatic progression? The liver microenvironment is known to contain hypoxic regions, with metabolically active hepatocytes at the periportal region displaying high rates of oxygen consumption and hepatocytes at the perivenous region actively undergoing glycolysis (Arteel et al., 1995; Jungermann and Kietzmann, 2000). Additionally, colon cancer cells metastasize to the liver via the portal circulation, which is relatively hypoxemic. We hypothesized that colorectal cancer cells experience acute hypoxia and intense competition for glycolytic substrates during initial dissemination to the liver and could be poorly adapted to the liver microenvironment prior to HIF activated responses (Semenza, 2011). ATP generated from rapid utilization of intracellular phosphocreatine reservoirs might enable colon cancer cells to survive acute hepatic hypoxia. To determine if colon cancer cells experience hypoxia during early metastatic colonization, we utilized a Hif-1 alpha transcriptional luciferase-reporter (HRE-Luc) as an in vivo sensor and reporter of cellular hypoxia (Fig. S3E) and observed that colon cancer cells experience hypoxia early after hepatic dissemination (Fig. S3F). We found that CKB depletion increased caspase-mediated cell death in HRE-Luc expressing cells experiencing hypoxia in vivo (Fig. 3C). Conversely, inhibition of either miR-483-5p or miR-551a protected HRE-Luc expressing cells experiencing hypoxia in vivo (Fig. S3G). Consistent with a role for CKB and phosphocreatine in promoting cancer-cell survival during hypoxia, cells depleted of CKB through RNAi displayed reduced survival while experiencing hypoxia in vitro—an effect that was abrogated upon phosphocreatine supplementation (Fig. 3D). In agreement with our in vitro findings, pre-incubation of colon cancer cells depleted of CKB with phosphocreatine enhanced their ability to metastasize to the liver (Fig 3E). Conversely, liver metastasis was inhibited when we pre-incubated colon cancer cells with cyclocreatine (Lillie et al., 1993), an inhibitor of CKB that depletes phosphocreatine levels (Fig. 3F). Our findings suggest that hepatic hypoxia poses a barrier for colon cancer cells during early metastatic colonization and that cells endure this phase through the generation of ATP from phosphocreatine reserves. Indeed, The ability of phosphocreatine pre-loading to enhance metastasis in vivo supports the importance of the acute initial hypoxic barrier and energetic demands in shaping metastatic colonization by cancer cells as they enter the liver microenvironment.
Figure 3. CKB modulates colorectal cancer cell survival during acute intrahepatic hypoxia through modulation of the phosphocreatine/ATP shuttle.
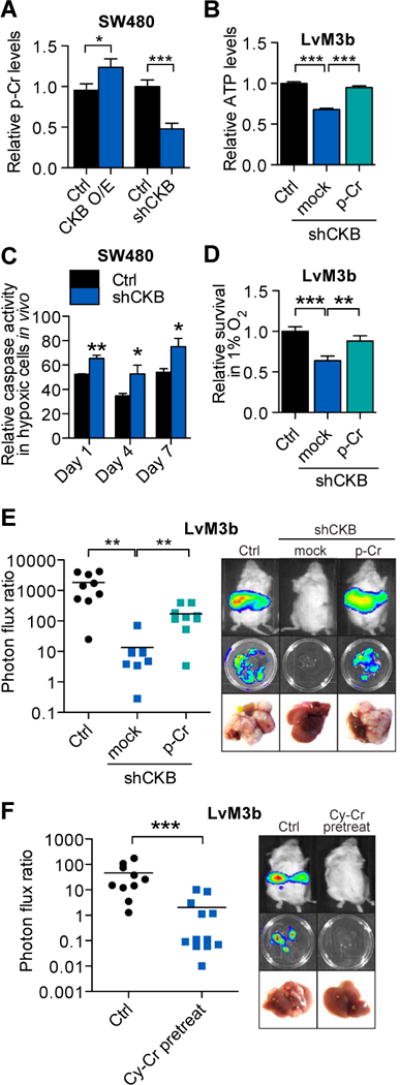
A, Relative intracellular phosphocreatine levels in SW480 cells over-expressing CKB or depleted for CKB (n=5). B, Relative intracellular ATP levels in LvM3b cells depleted for CKB, with and without exogenous 10um phosphocreatine supplementation (n=5). C, In vivo caspase activity of control and CKB knockdown SW480 cells experiencing hypoxia within the livers of mice (n=3). D, Survival of colorectal cancer cells in hypoxia in vitro with and without CKB knockdown, and 10um phosphocreatine supplementation (n=3). E, Liver metastasis by CKB depleted LvM3b cells pre-incubated overnight with 10um phosphocreatine. 5 × 105 cells were then inoculated into the liver of mice through intrasplenic injection. F, Liver metastasis in mice injected with 5 × 105 LvM3b cells with and without pre-treatment with 10mM cyclocreatine for 48hrs. Error bars, s.e.m; all p values are based on one-sided Student’s t-tests, or where appropriate, Mann-Whitney test for non-Gaussian distribution. *p<0.05; **p<0.01; ***p<0.001. See also Figure S3.
CKB is Secreted by Colorectal Cancer Cells and Promotes Malignant Conversion of Extracellular ATP and Creatine to Phosphocreatine
While considering CKB’s role in the setting of the hypoxic hepatic microenvironment that colorectal cancer cells arrive into as they disseminate from the gut to the liver, we faced a conundrum: how can colon cancer cells arriving and residing in a hypoxic hepatic microenvironment replenish phosphocreatine if they are depleted of ATP, especially during the acute phase, prior to any hypoxia-response (Bertout et al., 2008; Semenza, 2013; Wheaton and Chandel, 2011)? Earlier clinical studies have described the detection of CKB proteins and CKB activity in the sera of patients with various forms of malignancies and physiological insults (Huddleston et al., 2005; Rubery et al., 1982; Wyss and Kaddurah-Daouk, 2000). The presence of extracellular ATP in the microenvironment of macro-metastases was also reported (Pellegatti et al., 2008; Stagg and Smyth, 2010). Interestingly, the liver is the main synthetic organ for creatine synthesis in the body. We hypothesized that colorectal cancer cells may release CKB into the extracellular space, which can then convert extracellular ATP and liver-produced creatine into phosphocreatine that is then taken up by cancer cells, thereby exerting a protective effect on hypoxic colorectal cancer cells prior to their adaptation to the hypoxic liver microenvironment. We thus investigated the possibility of metastatic colorectal cancer cells releasing CKB extracellularly. Indeed, extracellular CKB was released from metastatic LvM3b cells, but not LvM3b cells expressing a CKB targeting shRNA (Fig. 4A). To determine if extracellular CKB was released from live or dying cells, we expressed CKB tagged with a FLAG-epitope through a linker containing a caspase-3/7 recognition DEVD motif. Caspase activation in apoptotic cells would result in caspase recognition and cleavage of the DEVD motif between the FLAG-epitope tag and CKB, causing loss of the FLAG-epitope from the expressed CKB (Fig. 4B). We observed that extracellular CKB was released primarily by live cells, as the FLAG-epitope was not lost (Fig. 4C). As generation of extracellular phosphocreatine requires exogenous ATP and creatine, we confirmed the presence of extracellular ATP in the microenvironment of incipient micrometastases using a plasma membrane-anchored luciferase reporter for detecting extracellular ATP (pME-Luciferase; Fig 4D)(Pellegatti et al., 2005). If the pro-metastatic effects of CKB result from utilization of extracellular ATP as a substrate, then depleting extracellular ATP should suppress the pro-metastatic activity of CKB. Indeed, expressing CD39, a plasma membrane anchored ATP hydrolase in SW480 cells significantly precluded the ability of CKB over-expression to promote metastasis without affecting CKB levels (Fig. 4E). Consistent with CKB consumption of extracellular ATP, cells over-expressing CKB or cells whose endogenous miR-483-5p or miR-551a were inhibited displayed significantly lower extracellular ATP levels in vivo relative to the control cells (Fig 4F, Fig. S4A). Conversely, the microenvironment surrounding CKB knockdown cells displayed higher extracellular ATP levels (Fig S4B). If extracellular CKB catalysis can enhance metastasis, we reasoned that supplying the product of CKB-mediated catalysis, phosphocreatine, in the extracellular space should rescue the effect of CKB loss-of-function. In order to test this, we implanted a mini-osmotic pump that continuously released phosphocreatine into the peritoneal cavity of immunodeficient mice, which is eventually drained into the portal circulation. Remarkably, exogenous phosphocreatine was sufficient to significantly enhance metastasis (>10 fold) by CKB-depleted cells in vivo (Fig. 4G). We next used a boyden chamber co-culture system to determine if colorectal cancer cells over-expressing CKB could promote the survival of CKB knockdown cells under hypoxia (Fig. 4H). CKB over-expressing cells were indeed able to compensate for the survival of CKB knockdown cells across the trans-well, while addition of a CKB-activity neutralizing antibody abrogated this effect (Fig. 4I). We extended our findings to an in vivo system using colorectal cancer cells depleted of intracellular CKB but expressing a secreted form of CKB fused to the IgK secretory signal sequence. Remarkably, over-expressing secreted CKB was sufficient to enhance colorectal cancer metastasis (Fig. 4J). We further examined the levels of serum CKB in mice injected with CKB knockdown cells. Interestingly, we found that mice with escaped tumors invariably had increased serum CKB levels (Fig. S4C).
Figure 4. CKB is secreted by colorectal cancer cells and promotes malignant conversion of extracellular ATP and liver creatine to phosphocreatine to enhance metastasis.
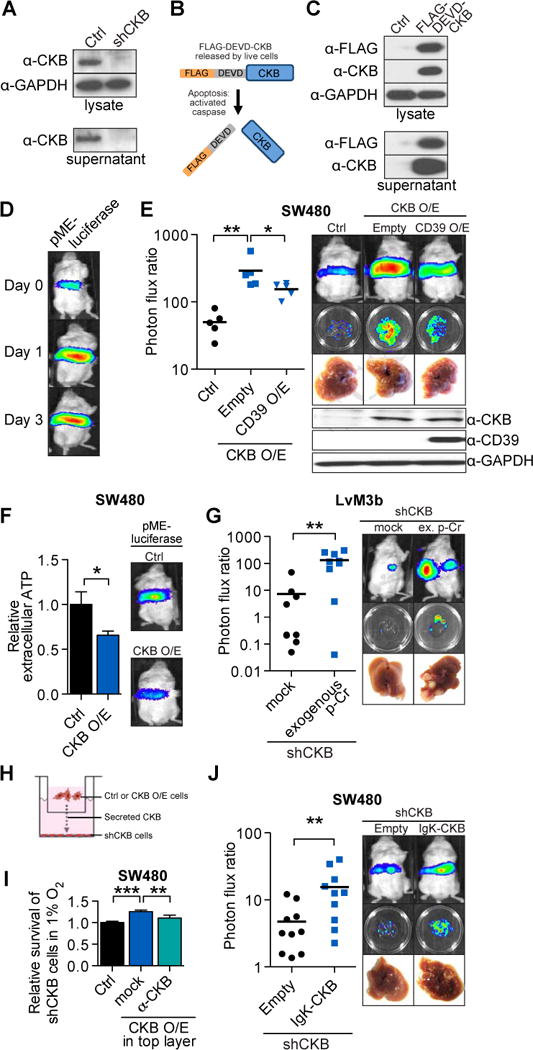
A, Extracellular and intracellular CKB protein levels in control and LvM3b cells depleted of CKB through RNAi. B, FLAG-tagged CKB with a caspase 3/7 recognition site linker. The FLAG-DEVD-CKB has a FLAG-tag linked to the N-terminal of CKB by a linker containing a caspase 3/7 recognition motif (DEVD-amino sequence). Caspase activation in apoptotic cells will result in cleavage of linker and release of FLAG-tag. C, Western-blot of FLAG-DEVD-CKB over-expressing cells demonstrate release of CKB by live cells into the extracellular space. D, Bioluminescent imaging of immunodeficient mice injected with SW480 cells expressing pME-Luc for detection of extracellular ATP (n=5). E, Liver metastasis by 5 × 105 SW480 cells over-expressing CKB with concomitant over-expression of CD39. F, Relative extracellular ATP levels in CKB over-expressing cells. Control and CKB over-expressing pME-Luc SW480 cells were injected into mice (n=5). G, Liver metastasis by CKB-depleted LvM3b cells in mice implanted with an osmotic pump releasing phosphocreatine into the portal circulation. H, Scheme for co-culture experiment. 5 × 104 CKB-knockdown cells were cultured on the bottom of 24-well plates, while control or CKB-over-expressing cells were plated onto boyden chambers above CKB-knockdown cells with pores for exchange of metabolites and proteins. Cells were counted after 4 days in hypoxia. I, Relative survival of CKB-knockdown cells in 1% oxygen when co-cultured with control, CKB-over-expressing cells or with CKB-over-expressing cells in the presence of a neutralizing antibody (n=4). J, Liver metastasis by endogenous CKB-knockdown SW480 cells over-expressing a secreted form of CKB. Error bars, s.e.m; all P values are based on one-sided Student’s t-tests, or where appropriate, Mann-Whitney test for non-Gaussian distribution. *p<0.05; **p<0.01; ***p<0.001. See also Figure S4.
The Creatine Transporter, SLC6a8 modulates CKB-mediated Colorectal Cancer Metastasis
Having implicated exogenous creatine/phosphocreatine metabolism in colorectal cancer metastasis, we next investigated the regulation of creatine/phosphocreatine metabolism in colon cancer metastatic progression. Depletion of guanidinoacetate methyltransferase (GAMT), the enzyme required for the final step of creatine synthesis, in colon cancer cells did not affect metastasis (Fig. S5A), consistent with a model wherein extracellular (the liver is the primary site of creatine biosynthesis) rather than intracellular creatine drives colorectal cancer metastasis. Next, we asked if SLC6a8, a transporter of creatine compounds (Salomons et al., 2001) modulates phosphocreatine levels in colon cancer cells. Indeed, SLC6a8 knockdown reduced intracellular phosphocreatine and ATP levels (Fig. 5A). If extracellular phosphocreatine uptake promotes metastasis, such impairment in phosphocreatine levels should reduce metastasis. Indeed, multiple colon cancer cell lines depleted of SLC6a8 displayed substantially reduced (10 to 100-fold) metastatic activity (Fig. 5B, Fig. S5B), whereas metastatic tumors that eventually grew out from SLC6a8 knockdown cells were escapers and displayed restored SLC6a8 expression (Fig. S5C). Importantly SLC6a8 knockdown, which depleted cellular uptake of extracellular phosphocreatine, also abrogated the effect of CKB over-expression on colorectal cancer metastasis (Fig. 5C), revealing extracellular phosphocreatine uptake to be downstream of CKB catalysis. Additionally, depleting SLC6a8 in CKB knockdown cells abrogated the protective effect of phosphocreatine during hypoxic stress (Fig. 5D). Finally exogenously added phosphocreatine was not able to promote liver metastasis by SLC6a8 knockdown cells (Fig. S5D). These findings reveal SLC6a8 to be downstream of CKB and phosphocreatine and to mediate their metastasis promoting effects.
Figure 5. SLC6a8 modulates CKB-mediated colon cancer metastasis through modulation of intracellular phosphocreatine levels.
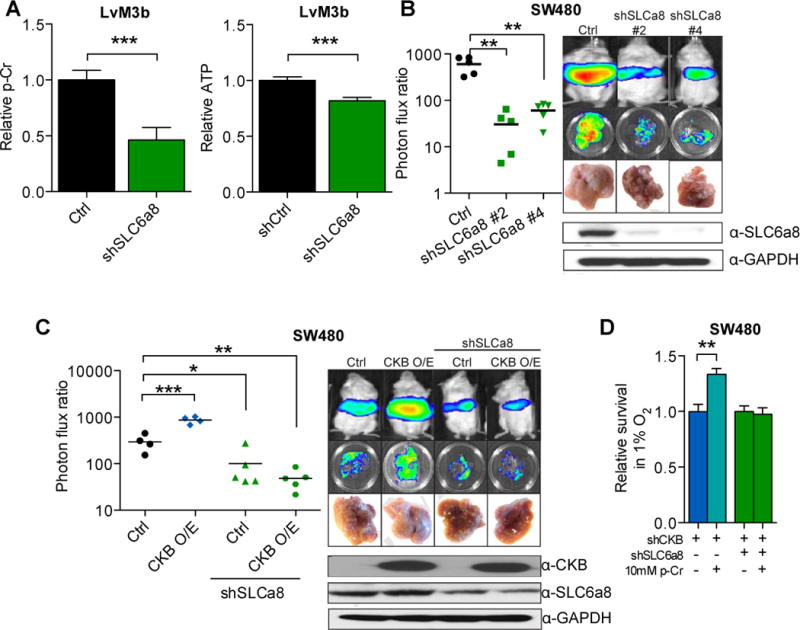
A, Relative intracellular phosphocreatine and ATP levels in LvM3b cells expressing a control shRNA or shRNA targeting SLC6a8 (n=4). B, Liver metastasis by 5 × 105 LvM3b cells expressing two independent short hairpins targeting SLC6a8 (n=5). C, Liver metastasis by SW480 cells over-expressing CKB with and without SLC6a8 depletion (n>4). D, In vitro survival of LvM3b cells depleted of CKB, SLC6a8 with and without phosphocreatine supplementation in hypoxia (n=3). Error bars, s.e.m; all P values are based on one-sided Student’s t-tests, or where appropriate, Mann-Whitney test for non-Gaussian distribution. *p<0.05; **p<0.001; ***p<0.0001. See also Figure S5.
miR-483-5p, miR-551a, CKB and SLC6a8 associate with human progression stage
We next analyzed the expression levels of miR-483 and miR-551a in a set of 66 surgically resected human primary colon cancer and liver metastases from MSKCC. Consistent with a metastasis-suppressive role for these miRNAs during cancer progression, miR-483 and miR-551a both independently displayed significantly reduced expression levels in human liver metastases relative to primary colon cancers (Fig. 6A,B; p=0.02 for miR-483 and p=0.001 for miR-551a; N=66). CKB (Fig 6C; p=0.05, N=233) and SLC6a8 (Fig. 6D; p<0.001, N=233) expression levels were significantly higher in liver metastases relative to primary tumors in an independent publicly available gene expression dataset, suggesting a selection for enhanced expression of CKB and SLC6a8 during progression. We also constructed a tissue microarray from a collection of primary colorectal tumors and liver metastases that were surgically resected from patients at Weill-Cornell Medical Center and immunohistochemically stained for CKB and SLC6a8 expression. CKB (Fig. 6E, p<0.05, N=92) and SLC6a8 (Fig. 6F, p<0.001, N=88) protein expression levels were found to be elevated in liver metastases relative to primary tumors of patients. These findings are consistent with, and suggest the pathophysiological basis for, previous studies revealing elevated expression levels of CKB in advanced stage cancer (Wallimann and Hemmer, 1994) and reveal significant association between the components of this multi-miRNA network and colon cancer progression.
Figure 6. miR-483-5p, miR-551a, CKB and SLC6a8 are clinically relevant in independent cohorts of patients and can be therapeutically targeted.
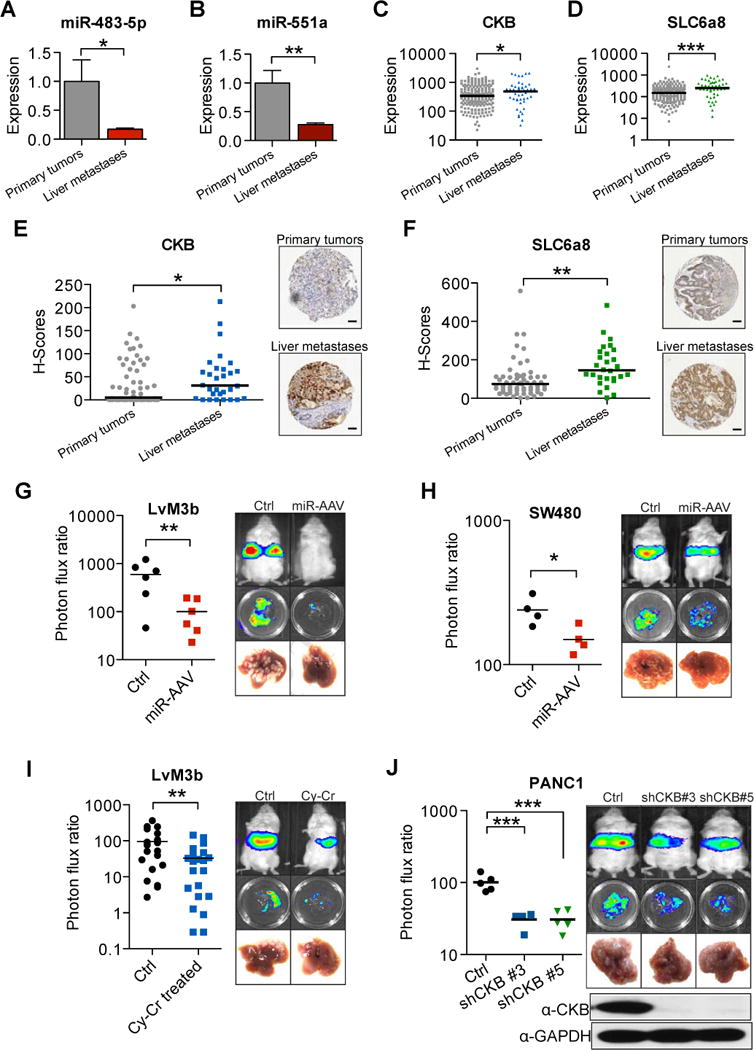
A, B, miR-483-5p and miR-551a levels in 36 primary colorectal adenocarcinomas and 30 liver metastases were quantified by quantitative real-time PCR. C, D, CKB and SLC6a8 expression from a public microarray dataset (GSE41258) comparing primary tumors and liver metastases (N=233). E, CKB expression in primary tumors compared to liver metastases examined through immunohistochemical staining of a tissue microarray (N=92). Scale bar represents 50um. F, SLC6a8 expression in primary tumors compared to liver metastases examined through immunohistochemical staining of above mentioned tissue microarray (N=88). Scale bar represents 50um. G, Liver metastasis in mice injected with 5 × 105 LvM3b cells and treated with a single dose of AAV doubly expressing miR-483-5p and miR-551a one day after injection cells (n=6). H, Liver metastasis in mice injected with 5 × 105 SW480 cells and treated with a single dose of AAV doubly expressing miR-483-5p and miR-551a one day after injection cells (n=4). I, Liver metastasis in mice injected with 5 × 105 LvM3b cells and treated with cyclocreatine daily for two weeks (n>15). J, Liver metastasis by pancreatic cancer cells, PANC1, with knockdown of CKB with two independent shRNA hairpins (n=5). Error bars, s.e.m; all P values are based on one-sided Student’s t-tests, or where appropriate, Mann-Whitney test for non-Gaussian distribution. *p<0.05; **p<0.01; ***p<0.001.
miR-483-5p, miR-551a and CKB modulation provides clinical benefit
We sought to investigate the therapeutic potential of targeting this clinically relevant miRNA regulatory network. We first tested the ability of adeno-associated virus to transduce colon cancer cells in vitro and detected viral genomic DNA (gDNA) in colon cancer cells even with low multiplicity of infection (Fig. S6A). Injection of mice bearing macroscopic hepatic metastases with adeno-associated virus revealed that adeno-associated virus was able to infect colon cancer metastases in vivo (Fig. S6B). We next injected mice with 5×105 highly metastatic LvM3b cells and 24 hours later administered a single intra-venous dose of adenoviral-associated virus (AAV) encoding miR-483-5p and miR-551a from a single transcript. Surprisingly, a single therapeutic dose of AAV delivering both miRNAs dramatically and significantly reduced metastatic colonization (>5 fold; Fig. 6G). Therapeutic efficacy was also seen in mice injected with SW480 cells (Fig. 6H). Importantly, this treatment did not cause any adverse phenotypic outcomes or pathological abnormalities in mice. Moreover, autopsied mice treated with this AAV therapy did not harbor spontaneous tumors. Even mice injected with BEAS-2B immortalized lung epithelial cells that are prone to oncogenic transformation did not develop tumors (Fig. S6C). We next investigated the therapeutic potential of targeting this metabolic network by determining the impact of small-molecule inhibition of CKB on colon cancer metastasis using cyclocreatine. Therapeutic treatment of mice with cyclocreatine after colorectal cancer cell inoculation also significantly reduced metastatic colonization, demonstrating proof-of-principle for targeting this kinase as a means of metastasis suppression (Fig. 6I). Given that the liver is a common site of metastasis for other gastrointestinal cancers such as pancreatic cancer, we sought to determine if knockdown of components in this pathway could also inhibit metastasis by pancreatic cancer cells. Knockdown of CKB and SLC6a8 in PANC1 cells (a KRAS mutant human pancreatic line) with multiple shRNAs, strongly suppressed their ability to metastasize (Fig. 6J, Fig. S6G >10 fold). This finding suggests that CKB and SLC6a8, and their associated metabolic pathway, may more broadly govern liver metastasis by other gastrointestinal cancers.
Discussion
Colorectal cancer is diagnosed in over a million patients a year globally, with the majority of these patients (over 600,000) experiencing liver metastatic progression (Jemal et al., 2011). Using a systematic approach, we have identified two miRNAs that act as suppressors of liver metastatic colonization by colon cancer cells. MiR-483-5p had been recently reported to be oncogenic in lung adenocarcinoma by enhancing invasion and progression, and tumor growth in IGF2-dependent sarcoma (Song et al., 2014), while miR-551a has been recently implicated in suppressing gastric cancer (Li et al., 2012b). Given that miRNAs are widely known to act in a context-specific manner, we had experimentally validated the role of these miRNAs in suppressing liver metastasis by colon cancer cells and we find that these miRNAs suppress a metabolic axis that drives liver colonization by convergent targeting of CKB—a key gene that allows colon cancer cells to expand their phosphocreatine reserves during periods of positive cellular energy balance (Wyss and Kaddurah-Daouk, 2000). Moreover, enhanced phosphocreatine reserves, which can fuel ATP generation provide great utility during periods of intense energetic requirement such as during metastatic colonization of the liver microenvironment—a hypoxic microenvironment that also contains perivenous hepatocytes that compete with cancer cells for glycolytic substrates (Jungermann and Kietzmann, 1996). We find that colon cancer cells, which enter the liver via the hypoxemic portal circulation, undergo substantial cell death. Cells capable of generating sufficient phosphocreatine reserves are better able to survive the initial phase of hypoxic hepatic colonization. Colon cancer cells that survive the initial selective pressure of hypoxic hepatic dissemination can then activate pathways involved in energy homeostasis and generation (DeBerardinis et al., 2008; Hardie et al., 2012; Inoki et al., 2012; Jeon et al., 2012; Kaelin and McKnight, 2013; Semenza, 2011) and harness additional pro-metastatic programs for successful competition and further colonization of the liver (Chiang and Massagué, 2008).
As incipient metastatic cells are dependent on CKB-mediated intracellular phosphocreatine generation, which arises from miR-483-5p/miR-551a silencing and CKB over-expression, a selection for sub-populations that exhibit silencing of these miRNAs and consequent induction of CKB would occur. We demonstrate that cells, which over-express the metabolic kinase CKB, are able to survive and progress within the hepatic parenchyma. However, in the context of energetic stress and ATP limitation, a paradoxical situation would arise wherein intracellular phosphocreatine generation from ATP would comprise a futile cycle. Interestingly, we find that colon cancer cells secrete CKB into the extracellular space, where it catalyzes the ATP-dependent phosphorylation of creatine—yielding phosphocreatine (Fig. 7). Extracellular phosphocreatine had been shown to be protective against hypoxic, ischemic and other energetic insults in neurons and myocardium, with increased phosphocreatine uptake observed in ischemic myocardium (Brustovetsky et al., 2001; Li et al., 2012a; Sharov et al., 1987). Our findings reveal that colon cancer cells can actually generate extracellular phosphocreatine and import it as a means of enhancing energy stores. Interestingly, we find that colon cancer cells have developed a remarkable adaptive mechanism— secretion of CKB—that enables them to catalytically enhance extracellular phosphocreatine levels from exogenous precursors. The ‘harvesting’ of extracellular metabolites through secretion of CKB by colon cancer cells thus represents a powerful mechanism for survival when malignant cells are highly vulnerable.
Figure 7. Model for the miR-483-5p, miR-551a, CKB and SLC6a8 axis.
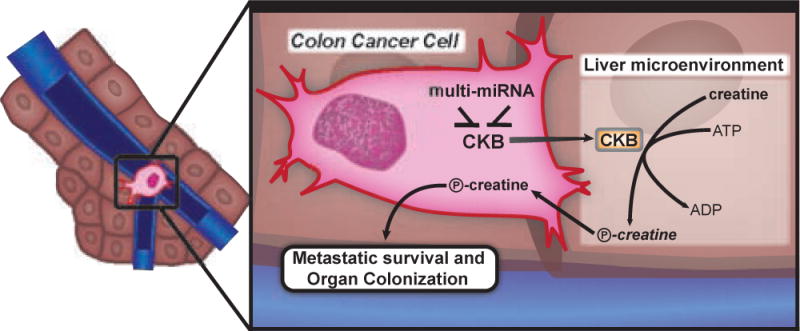
Disseminated colon cancer cells arrive in the liver microenvironment through the hypoxemic portal circulation. Within the liver microenvironment, they experience hypoxic stress and ATP depletion. Cells that up-regulate CKB through loss of miRNAs, release CKB into the extracellular matrix where it converts available creatine and ATP into phosphocreatine that is then taken up by the cell to fuel metastatic survival and subsequent organ colonization. Colon cancer cells with higher levels of CKB also build up a larger pool of intracellular phosphocreatine that acts as a buffer against energetic stress.
While our findings reveal an important pro-metastatic role for extracellular CKB, we do not rule out the possibility that intracellular CKB could also play a role in cancer progression. One potential intracellular role for CKB might occur during contexts when cancer cells have adequate levels of ATP, such as during primary tumor growth in the colonic epithelium, or subsequent to metastatic tumor expansion and recruitment of functional blood vessels, which could provide adequate oxygenation and glucose for fueling ATP generation. In such contexts, intracellular CKB could allow cancer cells to expand their intracellular buffer of phosphocreatine to be drawn upon during subsequent periods of reduced nutrients. While we have identified one pathway that sub-serves cancer progression, it is possible that malignant cells conduct other metabolic reactions in the extracellular space that could allow for the extraction of extracellular energy and substrates, or the degradation of toxic metabolic wastes.
Our findings from human primary and metastatic tissue specimens support our model (Fig 7), revealing enhanced expression of CKB and reduced expression of its repressive miRNAs in liver metastases—consistent with a selection for cells with molecular activation of this pathway in the hepatic parenchyma. While our findings have revealed a key pathway that governs colon cancer metastatic colonization of the liver, the predominant site of metastasis by colorectal cancer, and suggest that the in vivo-selected metastatic sublines we have derived display the same organotropism for the liver that the majority of human colorectal cancers display, we do not know if this pathway also regulates the colonization of other organs such as the lungs by these cells.
We speculate that part of this organotropism may arise from the production of creatine by the liver—the primary organ for creatine biosynthesis. Our findings that CKB and SLC6a8 depletion suppress pancreatic cancer metastasis highlight the possibility that additional gastrointestinal cancer types, which exhibit tropism for the liver, may also utilize this pathway and as such, may be also vulnerable to CKB inhibition therapy.
While the promise of miRNA therapeutics has been great, its actual clinical implementation has been more limited given inadequate stability and delivery of small RNA therapeutics to target tissues. The liver, however, is an exceptional organ in this respect, since miRNAs and RNAi molecules accumulate to higher degrees in the hepatic parenchyma relative to other organs and we have demonstrated adeno-associated virus to be efficient in infecting colon cancer metastases. Additionally, adeno-associated approaches of gene delivery have demonstrated proof-of-concept in human trials (Nathwani et al., 2011). Given these features of the liver, our identification of metastasis suppressor miRNAs in colorectal cancer and our proof-of-principle demonstration of their therapeutic activity have important clinical implications since both nanoparticle-mediated and adeno-associated viral delivery of these metastasis suppressors could be viable paths clinically. A more conventional path towards targeting this pathway could be the development of highly potent inhibitors of CKB that would act in a similar manner as cyclocreatine, given that highly specific kinase inhibitors can be designed (Dar and Shokat, 2011; Davis and Schlessinger, 2012). Importantly, such inhibitors do not have to be cell-permeable, since they would be targeting extracellular CKB. This could substantially increase the therapeutic index of such compounds. The poor overall efficacy of the current standard-of-care chemotherapy regimen FOLFOX in reducing metastatic relapse rates in high-risk patients necessitates the development and testing of such targeted therapeutic approaches in this prevalent disease.
Experimental Procedures
Animal Studies
All animal work was conducted in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at The Rockefeller University. 5–6 weeks old age-matched male NOD-SCID mice were used for organotypic slice culture, intrahepatic colonization and liver metastasis assays involving LS174T, SW620, WiDR, LvM3a and LvM3b cell-lines. 5–6 weeks old age-matched male NOD/SCID gamma male mice were used for liver metastasis assays for the SW480 and PANC1 cell-lines.
Lenti-miR library screening
Cells were transduced with a lentiviral Lenti-miR library (System Biosciences) at a low multiplicity of infection (MOI<1) such that each cell over-expressed a single miRNA. The transduced population was then injected intra-hepatically into NOD-SCID mice for in vivo selection of miRNAs that when over-expressed, either promoted or suppressed metastatic liver colonization. Genomic DNA PCR amplification and recovery of lenti-viral miRNA inserts was performed on cells prior to injection, and from liver nodules, according to the manufacturer’s protocol. miRNA array profiling allowed for miRNA insert quantification prior to and subsequent to in vivo selection.
Organotypic slice culture system
Cells to be injected were labeled with cell-tracker red or green (Invitrogen) and inoculated into the livers of NOD-SCID or NOD-SCID gamma mice through intrasplenic injection. The livers were then extracted and cut into 150um slices using a McIlwain tissue chopper (Ted Pella) and plated onto organotypic tissue culture inserts (Millipore) and cultured in William’s E Medium supplemented with Hepatocyte Maintenance Supplement Pack (Invitrogen). After indicated time periods, the liver slices were fixed in paraformaldehyde and imaged. Extended protocol can be found in supplemental materials.
Adeno-associated viral therapy
miR-483-5p and miR-551a were cloned as a polycistron consisting of both miRNA precursor with flanking genomic sequences (Supplementary Table S5) in tandem into the BglII and NotI site of scAAV.GFP (Plasmid 21893, Addgene). Adeno-associated virus was packaged, purified and titered by Vector Biolabs (PA). One day after mice were inoculated with colorectal cancer cells, 1 × 1012 AAV viral particles were injected into each mouse through intravenous injection.
Cyclocreatine treatment of mice
One day after inoculation of colon cancer cells, mice were injected with 10mg of cyclocreatine in 300ul PBS. Treatment was continued daily for 2 weeks until the mice were euthanized.
shRNA and Primer Sequences
shRNA, primers and cloning sequences are listed in supplementary tables 4–6.
Supplementary Material
Acknowledgments
We thank the members of the Tavazoie Laboratory and Dr. Siavash Kurdistani for helpful comments and discussion. We thank Dr. Victor Brodsky, Yifang Liu and Liza Rivera of Weill Cornell Medical College for invaluable help with the tissue microarray and image acquisition. The work was supported by the Irma T. Hirschl/Monique Weill-Caulier Trust, the Starr Cancer Consortium, the Leona M. and Harry B. Helmsley Charitable Trust and by the National Institutes of Health Director’s New Innovator Award under award number 1DP2OD006506-01. J.M.L is an A*STAR National Science Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional experimental procedures can be found in supplemental materials.
Author Contributions
S.F.T. conceived the project and supervised all research. J.M.L. and S.F.T. wrote the manuscript. J.M.L., A.S., A.N., F.Y.M. and E.W. performed the experiments. P.B.P., Z.Z., and L.S. obtained, curated and provided access to the clinical samples.
Accession Numbers
The data from the miRNA screen experiments are deposited at GEO under the accession number GSE56320
References
- Arteel GE, Thurman RG, Yates JM, Raleigh JA. Evidence that hypoxia markers detect oxygen gradients in liver: pimonidazole and retrograde perfusion of rat liver. Br J Cancer. 1995;72:889–895. doi: 10.1038/bjc.1995.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Dubinsky JM. On the mechanisms of neuroprotection by creatine and phosphocreatine. J Neurochem. 2001;76:425–434. doi: 10.1046/j.1471-4159.2001.00052.x. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Chiang AC, Massagué J. Molecular basis of metastasis. N Engl J Med. 2008;359:2814–2823. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu Rev Biochem. 2011;80:769–795. doi: 10.1146/annurev-biochem-090308-173656. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Schlessinger J. The genesis of Zelboraf: targeting mutant B-Raf in melanoma. J Cell Biol. 2012;199:15–19. doi: 10.1083/jcb.201205167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Selection of successive tumour lines for metastasis. Nat New Biol. 1973;242:148–149. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson J, Ackler S, Klaubert D, Bouska J, Ellis P, Foster K, Oleksijew A, Rodriguez L, Schlessinger S, Wang B, et al. Noninvasive molecular imaging of apoptosis in vivo using a modified firefly luciferase substrate, Z-DEVD-aminoluciferin. Cell Death Differ. 2010;17:1003–1010. doi: 10.1038/cdd.2009.205. [DOI] [PubMed] [Google Scholar]
- Huddleston HG, Wong KK, Welch WR, Berkowitz RS, Mok SC. Clinical applications of microarray technology: creatine kinase B is an up-regulated gene in epithelial ovarian cancer and shows promise as a serum marker. Gynecol Oncol. 2005;96:77–83. doi: 10.1016/j.ygyno.2004.08.047. [DOI] [PubMed] [Google Scholar]
- Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400. doi: 10.1146/annurev-pharmtox-010611-134537. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- Jungermann K, Kietzmann T. Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology. 2000;31:255–260. doi: 10.1002/hep.510310201. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Wang N, Zhao M. Neuroprotective effect of phosphocreatine on focal cerebral ischemia-reperfusion injury. J Biomed Biotechnol. 2012a;2012:168756. doi: 10.1155/2012/168756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cao Y, Jie Z, Liu Y, Li Y, Li J, Zhu G, Liu Z, Tu Y, Peng G, et al. miR-495 and miR-551a inhibit the migration and invasion of human gastric cancer cells by directly interacting with PRL-3. Cancer Lett. 2012b;323:41–47. doi: 10.1016/j.canlet.2012.03.029. [DOI] [PubMed] [Google Scholar]
- Lillie JW, O’Keefe M, Valinski H, Hamlin HA, Varban ML, Kaddurah-Daouk R. Cyclocreatine (1-carboxymethyl-2-iminoimidazolidine) inhibits growth of a broad spectrum of cancer cells derived from solid tumors. Cancer Res. 1993;53:3172–3178. [PubMed] [Google Scholar]
- Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482:347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med. 2005;352:476–487. doi: 10.1056/NEJMra040958. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegatti P, Falzoni S, Pinton P, Rizzuto R, Di Virgilio F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol Biol Cell. 2005;16:3659–3665. doi: 10.1091/mbc.E05-03-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One. 2008;3:e2599. doi: 10.1371/journal.pone.0002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencheva N, Tavazoie SF. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol. 2013;15:546–554. doi: 10.1038/ncb2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencheva N, Tran H, Buss C, Huh D, Drobnjak M, Busam K, Tavazoie SF. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell. 2012;151:1068–1082. doi: 10.1016/j.cell.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Png KJ, Halberg N, Yoshida M, Tavazoie SF. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature. 2012;481:190–194. doi: 10.1038/nature10661. [DOI] [PubMed] [Google Scholar]
- Rubery ED, Doran JF, Thompson RJ. Brain-type creatine kinase BB as a potential tumour marker–serum levels measured by radioimmunoassay in 1015 patients with histologically confirmed malignancies. Eur J Cancer Clin Oncol. 1982;18:951–956. doi: 10.1016/0277-5379(82)90243-7. [DOI] [PubMed] [Google Scholar]
- Salomons GS, van Dooren SJ, Verhoeven NM, Cecil KM, Ball WS, Degrauw TJ, Jakobs C. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet. 2001;68:1497–1500. doi: 10.1086/320595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347–353. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov VG, Saks VA, Kupriyanov VV, Lakomkin VL, Kapelko VI, Steinschneider AYa, Javadov SA. Protection of ischemic myocardium by exogenous phosphocreatine. I. Morphologic and phosphorus 31-nuclear magnetic resonance studies. J Thorac Cardiovasc Surg. 1987;94:749–761. [PubMed] [Google Scholar]
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- Song Q, Xu Y, Yang C, Chen Z, Jia C, Chen J, Zhang Y, Lai P, Fan X, Zhou X, et al. miR-483-5p promotes invasion and metastasis of lung adenocarcinoma by targeting RhoGDI1 and ALCAM. Cancer Res. 2014;74:3031–3042. doi: 10.1158/0008-5472.CAN-13-2193. [DOI] [PubMed] [Google Scholar]
- Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29:5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massagué J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallimann T, Hemmer W. Creatine kinase in non-muscle tissues and cells. Mol Cell Biochem. 1994:133–134. doi: 10.1007/BF01267955. [DOI] [PubMed] [Google Scholar]
- Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281(Pt 1):21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton WW, Chandel NS. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol. 2011;300:C385–393. doi: 10.1152/ajpcell.00485.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.