

Abstract
Rationale
Trimethylamine-N-oxide (TMAO), a gut microbial-dependent metabolite of dietary choline, phosphatidylcholine (lecithin) and L-carnitine, is elevated in chronic kidney diseases (CKD) and associated with coronary artery disease pathogenesis.
Objective
To both investigate the clinical prognostic value of TMAO in subjects with versus without CKD, and to test the hypothesis that TMAO plays a direct contributory role in the development and progression of renal dysfunction.
Methods and Results
We first examined the relationship between fasting plasma TMAO and all-cause mortality over 5-year follow-up in 521 stable subjects with CKD (estimated glomerular filtration rate [eGFR] <60 ml/min/1.73m2). Median TMAO level among CKD subjects was 7.9 μM (interquartile range 5.2–12.4μM), which was markedly higher (P<0.001) than in non-CKD subjects (n=3,166). Within CKD subjects, higher (4th vs. 1st quartile) plasma TMAO level was associated with a 2.8-fold increased mortality risk. Following adjustments for traditional risk factors, hsCRP and eGFR, elevated TMAO levels remained predictive of 5-year mortality risk (HR 1.93 [95%CI 1.13–3.29], p<0.05). TMAO provided significant incremental prognostic value (net reclassification index 17.26%, p<0.001; and differences in area under Receiver Operator Characteristic curve, 63.26% vs. 65.95 %, p=0.036). Among non-CKD subjects, elevated TMAO levels portend poorer prognosis within cohorts of high and low cystatin C. In animal models, elevated dietary choline or TMAO directly led to progressive renal tubulointerstitial fibrosis and dysfunction.
Conclusion
Plasma TMAO levels are both elevated in patients with CKD and portend poorer long-term survival. Chronic dietary exposures that increase TMAO appear to directly contribute to progressive renal fibrosis and dysfunction.
Keywords: Trimethylamine N-oxide, renal insufficiency, fibrosis, cardiovascular disease, gut microbiota
INTRODUCTION
Patients with chronic kidney disease (CKD) are at increased risk for development of cardiovascular disease (CVD) beyond traditional risk factors. Yet the mechanism(s) through which CKD is linked to enhanced atherosclerotic heart disease is not fully elucidated. While a role for uremic toxins in the pathogenesis of cardiovascular and renal disease progression in CKD has been suggested for some time, the precise participants involved, and their mechanisms of action remain unclear. Recent studies have suggested involvement of gut microbiota in the generation of metabolites that display uremic toxicity1. Further, perturbations of the composition of the gut microbial community in both human and experimental CKD are associated with significant elevations of gut-derived uremic toxins2. Such alterations have also been associated with increased systemic inflammatory burden, and thus are suspected to potentially play a role in the pathogenesis of cardiovascular and renal disease progression in subjects with CKD. Bacterial structural components such as lipopolysaccharide3, and metabolites such as indoxyl sulfate, p-cresyl sulfate, amines, and ammonia, have been identified as potential microbial byproducts capable of initiating pro-inflammatory cytokine/chemokine cascades seen in the setting of CKD and end stage renal disease4. Thus, it has been hypothesized that gut-microbiota derived uremic toxins may serve as both therapeutic targets and/or assessment tools for renal diseases in this vulnerable population1, 5–7. However, demonstration of a role for these uremic toxins in CVD pathogenesis has been indirect.
Our group recently identified a novel mechanistic link between gut microbiota metabolism of dietary trimethylamine-containing nutrients, and CVD pathogenesis8–13 Specifically, gut microbiota-mediated metabolism of phosphatidylcholine, choline or L-carnitine each have been shown to ultimately produced trimethylamine-N-oxide (TMAO), and in multiple clinical studies, TMAO levels have been shown to be associated with cardiovascular risks8–10, 13. Further, animal model studies have revealed TMAO is mechanistically linked to atherosclerosis development through multiple distinct pathways8, 14. Animal model and human clinical studies demonstrate an obligatory role for gut microbiota in TMAO formation from TMA-containing nutrients including choline and phosphatidylcholine9, 10, L-carnitine8, and more recently, γ-butyrobetaine15, and to a lesser degree, the choline oxidation metabolite betaine11.
TMAO is cleared by the kidney, and prior studies have reported TMAO is elevated in subjects with impaired renal function16–18. It is thus an attractive hypothesis that TMAO may represent an excellent mechanism-based marker of CVD risk in subjects with impaired renal function. In one large study of subjects with predominantly preserved renal function, we reported the prognostic value of TMAO for predicting three year risk for major adverse cardiovascular events (MACE = myocardial infarction, stroke or death) remained significant following adjustments for renal function 9. However, no studies to date have directly looked at the long-term mortality risk of TMAO among CKD subjects. Interestingly, plasma TMAO levels in apparently healthy donors was identified in a prospective multicenter study aimed at identifying donor factors associated with delayed graft function in renal transplant recipients19. A direct role for TMAO in impacting renal functional impairment has not been reported. Herein, we sought to further examine both the prognostic value of TMAO among CKD subjects and the potential contribution of dietary-induced, gut microbiota-associated TMAO formation in the development and progression of CKD.
METHODS
Human studies
This is a single-center, prospective cohort study approved by the Cleveland Clinic Institutional Review Board. We included adult subjects (ages 18 years and above) who underwent elective diagnostic coronary angiography for cardiac evaluation at our institution from 2001–2007 as previously described9. We excluded subjects with known acute coronary syndromes or revascularization procedures within 30 days of enrollment, or history of congenital heart disease. After informed consent, fasting plasma blood samples were collected using ethylenediaminetetraacetic acid tubes prior to any drug administration via the arterial sheath, and immediately processed and frozen in −80ºC until analysis. Estimated glomerular filtration rate (eGFR) was calculated according to the CKD-EPI creatinine and cystatin C formula20, with CKD defined as eGFR <60 ml/min/1.73m2 (CKD stage 3 or beyond). Ascertainment of all-cause mortality at 5-years was performed by prospective telephone contact and chart review plus interrogation of the Social Security Death Index (up to 2011).
Plasma analysis
Trimethylamine-N-oxide (TMAO) levels were determined by stable isotope dilution high-performance liquid chromatography with online electrospray ionization tandem mass spectrometry (LC/MS/MS) on an AB SCIEX 5500 triple quadrupole mass spectrometer (AB SCIEX, Framingham MA) using d9-(trimethyl)-labeled internal standards as previously described10, 21. High-sensitivity C-reactive protein (hsCRP), fasting lipid panel, cystatin C, and serum creatinine were measured using the Architect ci8200 platform (Abbott Laboratories, Abbott Park, IL).
Animal study
To directly test for a potential contribution of dietary choline or TMAO to promotion of renal dysfunction, C57BL6J mice were fed with the following diets for 6 weeks: i) a chemically defined diet comparable in composition to standard chow diet (Teklad 2018, Harland Laboratories) that contains 0.08% (gm/gm) total choline; ii) the same diet supplemented with choline (1.0% total); and iii) the same diet supplemented with TMAO (0.12%). A separate study included C57BL6J mice with ApoE−/− background were fed with the same diet groups for comparing their cystatin C levels at 14 weeks of follow-up using a commercially available mouse enzyme-linked immunosorbent assay (R&D systems, Minneapolis MN). This study has been approved by the Cleveland Clinic Institutional Animal Care and Use Committee.
Quantitative histologic techniques
Mason’s trichome staining was performed on deparafinized 5 μm serial kidney sections. The kidney sections were mounted under a Leica DM 2500 microscope and digitized with a QImaging MicroPublisher 5.0 RTV camera for wide field microscopy. Quantitative morphometric analysis was performed on cortical fields (at least 8 from each animal) lacking major blood vessels and the collagen volume was determined using automated (for batch analysis) and customized algorithms/scripts (ImageIQ Inc., Cleveland, OH) written for Image Pro Plus 7.0. Briefly, a set of representative images are chosen that demonstrated a wide range of staining intensities and prevalence. In an automated script, these “training” images were loaded one after another prompting the user to delineate “blue” pixels representing positive collagen staining using an interactive color picking tool. An iterative color profile or classifier was generated and subsequently applied to all images in a given directory using a fully automated algorithm. Positive pixels, as defined by the color profile, were segmented and summed to provide positive staining area. Total tissue area was determined by extracting the “saturation” channel, applying a lo-pass filter, and thresholding the result. Any area within the general tissue boundary that was empty (i.e. white) was removed by converting the original image to grayscale and applying a fixed threshold for non background pixels on adequately white-balanced images. Finally, total tissue area and total stained area were exported to Excel. For post-processing verification, segmented regions were superimposed onto the original image (green outlines) and saved for each image analyzed.
Preparation of tissue homogenates and immunoblotting
Equal amounts of protein were prepared using standard biochemical methods and subjected to SDS-PAGE and electrotransfer of proteins from gels to Immobilon-P membranes (Millipore). Membranes were incubated with the following antibodies: SMAD3 and phospho-SMAD-3(Ser423/425) (Cell Signaling Technology); tubulin (Santa Cruz Biotechnology); KIM-1 (Novus Biologicals). Detection for all immunoblots was performed with the SNAP i.d.™ Protein Detection System (Millipore) and Super Signal Chemiluminescent Substrate Products (Pierce), and band intensity was analyzed by densitometry (ImageQuant™, GE Healthcare).
Statistical analyses
Continuous variables were summarized as mean ± standard deviation if normally distributed and median (interquartile ranges [IQR]) if non-normally distributed. The student’s t-test or Wilcoxon-Rank sum test for continuous variables and chi-square test for categorical variables were used to examine the difference between groups. Spearman's correlation was used to examine the associations between TMAO and other laboratory measurements. Kaplan-Meier survival plots and Cox proportional hazards analysis were used to determine Hazard ratio (HR) and 95% confidence intervals (95%CI) for all-cause mortality stratified according to TMAO in quartiles. Adjustments were made for individual traditional risk factors including age, sex, systolic blood pressure, low-density lipoprotein cholesterol (LDLc), high-density lipoprotein cholesterol (HDLc), smoking, diabetes mellitus, as well as log-transformed hsCRP to predict all-cause mortality risks. Additional adjustment for log-transformed eGFR was also performed. Net reclassification and area under Receiver Operator Characteristic curve were calculated according to mortality risk estimated using Cox models adjusted for above mentioned traditional risk factors with versus without TMAO as previously described22. All analyses performed used R 2.15.1 (Vienna, Austria). P<0.05 was considered statistically significant.
RESULTS
Elevated TMAO in patients with renal insufficiency portend poorer survival
Baseline clinical and laboratory characteristics of the cohort are reported in Table 1. A total of 3,687 subjects were included in this analysis, among which 521 subjects fulfilled criteria for CKD and 3,166 subjects for non-CKD. Compared to non-CKD subjects, TMAO levels were elevated in patients with CKD (median TMAO: 7.9 [IQR 5.2–12.4]μM versus 3.4 [IQR 2.3–5.3]μM, p<0.001; Figure 1A). Overall in the CKD cohort, TMAO modestly correlated with eGFR (r= −0.48, p<0.001) and cystatin C (r=0.46, p<0.001), but did not correlate with hsCRP (r=0.04, p=0.332).
Table 1.
Baseline Characteristics
| Characteristic | Non-CKD Cohort eGFR ≥60 (n=3,166) | CKD Cohort eGFR<60 (n=521) | p-value |
|---|---|---|---|
| Age (years) | 62±11 | 70±10 | <0.001 |
| Male (%) | 66 | 48 | <0.001 |
| Diabetes (%) | 27 | 53 | <0.001 |
| Hypertension (%) | 69 | 88 | <0.001 |
| Smoking (%) | 66 | 61 | 0.047 |
| History of MI (%) | 40 | 53 | <0.001 |
| History of stroke (%) | 5 | 13 | <0.001 |
| History of CABG (%) | 28 | 42 | <0.001 |
| History of PCI (%) | 31 | 30 | 0.728 |
| LDL (mg/dL) | 97 (79–118) | 93 (72–114) | <0.001 |
| HDL (mg/dL) | 34 (28–41) | 32 (26–40) | <0.001 |
| hsCRP (mg/L) | 2.2 (0.9–5.0) | 4.1 (1.8–9.6) | <0.001 |
| eGFR (ml/min/1.73m2) | 89 (78–101) | 49 (38–55) | <0.001 |
| Cystatin C (mg/L) | 0.9 (0.8–1) | 1.5 (1.3–1.8) | <0.001 |
| ACE inhibitor/ARB (%) | 48 | 66 | <0.001 |
| Statins (%) | 61 | 59 | 0.437 |
| Beta blockers (%) | 63 | 68 | 0.04 |
| Aspirin (%) | 75 | 67 | <0.001 |
|
| |||
| TMAO (μM) | 3.4 (2.3–5.3) | 7.9 (5.2–12.4) | <0.001 |
Figure 1. Prognostic Value of Plasma trimethylamine N-oxide (TMAO) Levels in the CKD Cohort.
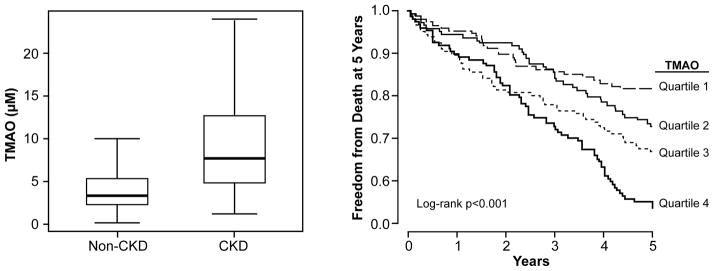
In a cohort of stable cardiac patients undergoing elective diagnostic coronary evaluation, subjects with underlying Stage 3+ chronic kidney disease demonstrated higher levels of fasting plasma TMAO than those with no CKD (p<0.01, Panel A). Increasing quartiles fasting plasma TMAO levels portend increased risk for all-cause mortality at 5 years in patients with CKD (n=521, Panel B).
In the CKD cohort, higher TMAO levels (quartiles 4 versus 1) were associated with a 2.8-fold increase in risk for all-cause mortality at 5 years (unadjusted HR 2.76, 95%CI 1.74–4.37, p<0.001). After adjusting for traditional CVD risk factors, log-transformed hsCRP, and log-transformed eGFR, higher TMAO levels still were associated with a 1.9-fold poorer 5-year survival (adjusted HR 1.93, 95%CI 1.13–3.29, p<0.05; Table 2, Kaplan-Meier curve shown in Figure 1B). When stratified according to median levels (7.9 μM), higher TMAO conferred a 1.7-fold increase in risk for all-cause mortality (HR 1.70, 95%CI 1.25–2.30, p<0.001), and remained significant after adjusting for traditional risk factors and log-transformed hsCRP (adjusted HR 1.72, 95%CI 1.16–2.34, p<0.001), as well as with addition of cystatin C to the model (adjusted HR 1.45, 95%CI 1.05–2.02, p<0.05). Using median cohort cut-offs with low cystatin C (<1.4 mg/dL) and low TMAO (<7.9 μM) as reference, those with concomitant high cystatin C and high TMAO had a 3-fold increase in mortality risk (HR 3.01, 95%CI 1.97–4.59, p<0.001). These findings are consistent with the notion that elevated TMAO is associated with poor prognosis in patients with established CKD.
Table 2.
Cox Proportional Hazards Analysis of Plasma TMAO Levels Stratified at quartile Levels in Predicting Risk of All-cause Mortality at 5 Years Stratified by CKD and Non-CKD Cohorts
| CKD Cohort (n=521) | TMAO (range) | |||
|---|---|---|---|---|
| Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | |
| Range (μM) | <5.2 | 5.2–7.9 | 7.9–12.4 | ≥12.4 |
| Events | 26/129 (20.2%) | 42/131 (32.1%) | 43/130 (33.1%) | 63/131 (48.1%) |
| Unadjusted HR | 1 | 1.70 (1.04–2.79)* | 1.75 (1.07–2.87)* | 2.76 (1.74–4.37)** |
| Adjusted HR | 1 | 1.42 (0.85–2.35) | 1.51 (0.90–2.51) | 1.93 (1.13–3.29)* |
| Non-CKD Cohort (n=3,166) | TMAO (range) | |||
|---|---|---|---|---|
| Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | |
| Range (mM) | <2.3 | 2.3–3.4 | 3.4–5.3 | ≥5.3 |
| Events | 48/787 (6.1%) | 59/793 (7.4%) | 81/791 (10.2%) | 104/795 (13.1%) |
| Unadjusted HR | 1 | 1.22 (0.83–1.79) | 1.70 (1.19–2.43)** | 2.21 (1.57–3.12)** |
| Adjusted HR | 1 | 1.08 (0.74–1.58) | 1.23 (0.84–1.78) | 1.47 (1.02–2.12)* |
Adjusted model: adjusted for traditional risk factors (age, gender, systolic blood pressure, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, smoking, diabetes mellitus), log(hsCRP), and log(eGFR).
p<0.05;
p<0.001
Increased TMAO levels in non-CKD patients with elevated Cystatin C
Within the non-CKD cohort (n=3,166), the prognostic value of elevated TMAO (quartile 4 vs 1) remained predictive of 5-year mortality risk (HR 2.21, 95%CI 1.57–3.12, p<0.001), as well as after adjusting for traditional risk factors, log-transformed hsCRP, and log-transformed eGFR (adjusted HR 1.47, 95%CI 1.02–2.12, p<0.05, Table 2B). These findings were similar when restricted to subjects with preserved eGFR (≥60 ml/min/1.73m2) plus normal cystatin C (<1.4mg/dL, n=3,151). Elevated TMAO levels are associated with higher 5-year mortality risk among subjects with either normal or elevated cystatin C levels (Figure 2). Using median cohort cut-offs with low cystatin C (<0.9 mg/dL) and low TMAO (<3.4μM) as reference, those with concomitant high cystatin C and high TMAO had a 3.7-fold increase in mortality risk (HR 3.67, 95%CI 2.57–5.23, p<0.001).
Figure 2. Comparative Prognostic Value of Plasma TMAO and Cystatin C in the Non-CKD Cohort.

Subjects with elevated cystatin C (>0.9 mg/dL) and TMAO (>3.4 μM) had the highest 5-year mortality risk in this non-CKD cohort (n=3,188).
Dietary choline and dietary TMAO both promote renal fibrosis and dysfunction in animal models
To directly test the hypotheses that either dietary TMAO itself, or dietary nutrients that contribute to gut microbiota dependent production of TMAO, can impact development and progression of CKD, we performed animal model studies. Conventionally housed 8-week old male mice (C57BL/6J background) were fed ad libitum a chemically defined diet comparable to normal chow (0.08gm% choline), or the same diet supplemented with either choline (1.0% final) or TMAO (0.12%), as described under Methods. After 6 weeks we observed significant (p<0.01) increases in TMAO levels in both the TMAO-supplemented and choline-supplemented groups of mice (Figure 3A), with TMAO levels observed within the range of values detected among CKD subjects studied (97.5 percentile 77.6μM, 99 percentile 96.3μM). Importantly, elevated TMAO levels were associated with corresponding increases in tubulointerstitital fibrosis and collagen deposition (Figure 3B–C) and phosphorylation of Smad3, an important regulator of the pro-fibrotic TGF-β/Smad3 signaling pathway during fibrotic kidney disease23 (Figure 3E). Furthermore, TMAO-fed and choline-fed mice experienced increased kidney injury marker-1 (KIM-1, Figure 4A). Extending the TMAO/choline feeding to 16 weeks was associated with increased serum cystatin C levels compared to chow-fed mice (Figure 4C). Upon further examination, striking dose-dependent relationships were noted between plasma TMAO levels and monitored indices of renal histopathological (Figure 3D, 3F) and functional impairment (Figure 4B).
Figure 3. Dietary choline/TMAO Exposure Contributes to Progressive Renal Fibrosis in Murine Model.

Plasma TMAO (A) levels are increased after 6 week feeding TMAO (0.12%), or Choline (1.0%) diets vs chow (0.08% choline) fed mice. Representative Mason trichrome histology (B) quantitative morphometry (C) and its relationship with TMAO levels (D), SMAD3 activation by phosphorylation at serine 423/425 (E) and its relationship with TMAO levels (F) in mouse kidneys after 6 week feeding of chow (0.08% choline), TMAO (0.12%), and Choline (1.0%) diets. Scale bar represents 100 um. **P<0.01 vs. chow fed, n≥5 mice per group.
Figure 4. Dietary choline/TMAO Exposure Contributes to Progressive Renal Injury and Dysfunction in Murine Model.
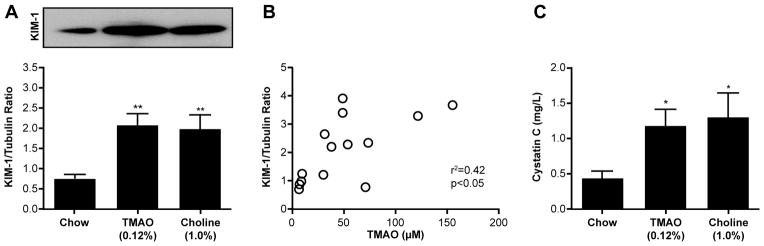
Immunoblot of KIM-1 expression (A) and its relationship with TMAO levels (B) in mouse kidneys after 6 week feeding of chow (0.08% choline), TMAO (0.12%), and Choline (1.0%) diets. Also shown are plasma cystatin C levels (C) after 16 week feeding of chow, TMAO (0.12%), and Choline (1.0%) diets. **P<0.01 vs. chow fed, n≥5 mice per group.
DISCUSSION
There are several key findings in this report. First, we observed in subjects with CKD, that TMAO levels are not only elevated compared to non-CKD subjects, but importantly, portend poorer overall survival. Second, we observed that within the non-CKD cohort, higher levels of TMAO portend poorer survival both within the cohort of low levels as well as high levels of cystatin C (stratified at median levels). Interestingly, the prognostic value for the highest TMAO quartile in predicting future mortality risk in this cohort remained robust even after adjustment for traditional risk factors. Third, extending to animal models studies, dietary exposure of either choline or TMAO lead to the development of renal tubulointerstitial fibrosis and early measures of dysfunction (elevated cystatin C). These studies thus suggest both a causal relationship and clinical relevance of dietary choline-induced, gut microbiota-mediated, TMAO formation in CKD development and progression.
Trimethylamine N-oxide is a low molecular weight compound that is easily filtered by the kidney, and effectively removed by hemodialysis16. Considered a nitrogenous waste product whose levels rise with diminished renal function, elevated TMAO levels have been reported in small cohorts (n<20) of subjects with either end-stage renal disease, or CKD, where levels were shown to correlate with both serum urea and creatinine18. Detailed animal and human experiments on the renal clearance of methylamines such as TMA and TMAO have been performed, and confirming the kidneys as the primary elimination route24. Interestingly, the urinary clearances of both TMA and TMAO are higher than the glomerular filtration rate, and TMAO clearance also decreases with increasing dose, which suggests that saturable renal tubular secretion occurs25. The majority of a dose of TMA is also excreted in the urine, with varying proportions in the forms of TMA and TMAO being dependent on the dose level26. Urinary TMAO levels are reported to rise with episodes of kidney graft dysfunction in renal transplant recipients, suggesting an intrinsic accumulation of TMAO (presumably as an osmolyte like urea) that is released during damage of the renal medullary cells27–30. The mechanistic link between elevated TMAO and adverse prognosis in CKD and even in the setting of subclinical renal insufficiency (elevated cystatin C) in non-CKD patients observed in our study is therefore consistent with the heightened risk of developing CVD in the CKD population.
Our results from animal studies showed for the first time a direct mechanistic link between dietary choline, or dietary TMAO, and progressive renal dysfunction, even in the C57BL/6J mouse model that is known to be relatively resistant to renal injury31. Indeed, exposure to either a high choline diet or a diet supplemented directly with TMAO both led to increased levels of the early kidney injury marker KIM-1, and enhanced phosphorylation of Smad3, an important regulator of renal fibrosis32. A more prolonged exposure to either the high choline diet or the TMAO supplemented diet both led to increased plasma levels of cystatin C, a sensitive indicator of renal functional impairment. Importantly, the plasma TMAO levels observed within the animal models on either choline or TMAO supplemented diets were well within the range of TMAO levels observed among subjects with CKD. Further, examining TMAO levels as a continuous variable, a dose-dependent relationship between plasma TMAO levels and mortality risk was observed (log[TMAO]: adjusted HR=1.41, 95%CI 1.23–1.61, p<0.001 for CKD cohort; adjusted HR=1.21, 95%CI 1.07–1.36, p<0.01 for non-CKD cohort). While there is the appearance of a threshold level of TMAO that is associated with increased risk with the predicted outcomes using quartile-based analyses in the current sized study, the biological data collected in the animal model studies (scatter plots comparing TMAO levels versus the CKD-related phenotypes) shows a continuous dose dependent relationship between TMAO levels and renal tubulointerstitial fibrosis (pSMAD3, and various measures of collagen or fibrosis), and renal functional impairment (cystatin C). Interestingly, a recent untargeted metabolomic study from the Framingham Heart Study among subjects with normal renal function identified elevated choline and TMAO levels at baseline were each associated with an increased future risk of developing CKD 33. Our animal model findings therefore provide a potential mechanistic rationale for the Framingham observational data, and collectively, further link elevated systemic TMAO levels with increased susceptibility for development of CKD.
The prospects that exposure to specific dietary nutrients like choline, phosphatidylcholine (lecithin) and L-carnitine via gut microbiota may impact susceptibility to the development and progression of both CKD and CVD has important potential public health implications. Randomized nutritional intervention studies in CKD patients to date have not explored a potential role for choline, phosphatidylcholine, L-carnitine or TMAO (which can be abundant in certain types of fish) in disease progression. Similarly, epidemiological studies are rather limited on the topic of diet and CKD risks, even though a recommended renal diet is typically low in protein intake. Dietary management of CKD patients represents a challenge, and much less is known about nutritional factors that might predispose to enhanced risk for development of CKD or its progression. Interestingly, in a sub-study (n=3,296) amongst women who had urine microalbumin levels available from the Nurses Health Study, two or more servings of red meat (primary dietary source of L-carnitine) per week were directly associated with enhanced risk for development of microalbuminuria (OR: 1.51; 95% CI: 1.01 to 2.26)34. Further investigations in dietary predisposition to CKD development and progression are warranted. Based upon the present studies, a diet monitored by following TMAO levels and designed to limit TMAO precursors (low in red meat, meats, liver, egg yolk, and high fat dairy products) and TMAO itself (certain fish) would be an attractive diet to test to see if it limited rate of CKD progression. However, it is important to note that choline is an essential nutrient; therefore, its total elimination from the diet is ill advised, and could result in development of a deficiency state35.
Collectively, the present data indicate a dietary-induced, intestinal microbiota-dependent mechanism may contribute to both progressive renal fibrosis and dysfunction, and mortality risks, among subjects with CKD. They also build upon the recent body of evidence demonstrating a mechanistic link between gut microbiota-associated metabolic dysregulation and cardiovascular risk in humans8–12. The discovery of the metaorganismal pathway involved in TMAO generation thus affords a unique opportunity to systematically investigate the potential contributions of discrete participants in the overall diet → microbe → host enzyme pathways for TMAO formation and development and progression of cardio-renal dysfunction, thereby offering insights into modulation of such pathways. It is interesting that in both animal models and patients with established CKD, pre- and probiotic intervention studies have been performed, with reports of changes in gut microbiota composition and activity. For example, lactobacillus acidophilus or bifidobacterium have been reported to reduce inflammatory signaling associated with the microbiota-derived metabolites that accumulate in CKD36–39, in addition to modestly improving renal function40, 41. Similarly, prebiotic compound use to interrupt pathways that lead to gut microbiota derived uremic toxins such as indoxyl sulfate and p-cresyl sulfate has shown some efficacy in both human and animal trials of CKD5, 42. Further studies are warranted to see whether dietary interventions, or disruption of the meta-organismal pathway involving TMAO production, may retard development of CKD, progression of renal functional impairment among subjects with CKD, and adverse CVD event risks in subjects with CKD.
Conclusion
The gut microbiota-dependent product, TMAO, is associated with both higher risk of progressive renal fibrosis and functional impairment, and poorer long-term survival.
Supplementary Material
Novelty and Significance.
What Is Known?
Gut microbe-dependent metabolism of dietary choline, phosphatidylcholine and L-carnitine contributes to the development and progression of atherosclerotic cardiovascular diseases (CVD) via formation of trimethylamine-N-oxide (TMAO).
Patients with chronic kidney disease (CKD) have heightened risk of developing CVD.
What New Information Does This Article Contribute?
Plasma TMAO levels are both increased in subjects with CKD, and portend poorer overall survival, independent of traditional risk factors.
In subjects without CKD, higher levels of TMAO portend poorer survival, both among those with high or low levels of cystatin C. Interestingly, the prognostic value for the highest TMAO quartile in predicting future MACE (MI, stroke or death) in this cohort remained robust even after adjustment for traditional risk factors and renal function.
In animal model studies, dietary exposure to either choline or TMAO each lead to the development of renal tubulointerstitial fibrosis and early measures of dysfunction (elevated cystatin C).
Previous studies have shown that intestinal microbiota-associated choline metabolism and TMAO generation contribute to the development and progression of atherosclerotic CVD. Hence, we examined the potential contribution of dietary choline and gut microbiota-dependent TMAO production in the development and progression of CKD. These studies indicate both a causal relationship and clinical relevance of dietary choline-induced, gut microbiota-mediated, TMAO formation in CKD development and progression. Our findings support a mechanistic link between TMAO and CVD risk in this vulnerable population. The significance of a dietary-induced CKD susceptibility strongly implies the need to focus preventive efforts on dietary modulation and therapeutic targeting of the gut microbiota-dependent TMAO pathway to potentially retard CKD development, progression, and CVD risks.
Acknowledgments
SOURCES OF FUNDING
This research was supported by grants from the National Institutes of Health and the Office of Dietary Supplements (R01HL103866, P20HL113452). The GeneBank study has been supported by NIH grants P01HL076491, P01HL098055, R01HL103931, and the Cleveland Clinic Clinical Research Unit of the Case Western Reserve University CTSA (UL1TR 000439). Dr. Wang was partially supported by an American Heart Association Scientist Development Grant 12SDG12050473. Dr. Kennedy was partially supported by an American Heart Association Scientist Development Grant 14SDG18650010. Dr. Hazen is also partially supported by a gift from the Leonard Krieger endowment. Mass spectrometry studies were performed on instruments housed in a facility supported in part by a Center of Innovations Award by AB SCIEX.
Nonstandard Abbreviations and Acronyms
- CKD
chronic kidney disease
- CVD
cardiovascular disease
- eGFR
estimated glomerular filtration rate
- HDL-c
high-density lipoprotein cholesterol
- HR
hazard ratio
- hsCRP
high-sensitivity C-reactive protein
- IQR
interquartile range
- KIM1
kidney injury molecular 1
- LDL-c
low-density lipoprotein cholesterol
- Smad3
Mothers against decapentaplegic homolog 3
- TMAO
trimethylamine n-oxide
Footnotes
DISCLOSURES
Drs.Wang, Levison and Hazen are named as co-inventor on pending patents held by the Cleveland Clinic relating to cardiovascular diagnostics and/or therapeutics. Dr. Hazen reports having been paid as a consultant for the following companies: Cleveland Heart Lab, Esperion, Liposcience Inc., and P&G. Dr. Hazen reports receiving research funds from Cleveland Heart Lab, Liposcience Inc., P&G and Takeda. Drs. Wang, Levison and Hazen report having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics and/or therapeutics from Cleveland Heart Lab, and Dr. Hazen also from the companies shown below: Siemens, Esperion, Frantz Biomarkers, LLC. All other authors have no relationships to disclose.
References
- 1.Mafra D, Lobo JC, Barros AF, Koppe L, Vaziri ND, Fouque D. Role of altered intestinal microbiota in systemic inflammation and cardiovascular disease in chronic kidney disease. Future Microbiol. 2014;9:399–410. doi: 10.2217/fmb.13.165. [DOI] [PubMed] [Google Scholar]
- 2.Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, DeSantis TZ, Ni Z, Nguyen TH, Andersen GL. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83:308–315. doi: 10.1038/ki.2012.345. [DOI] [PubMed] [Google Scholar]
- 3.Stenvinkel P, Alvestrand A. Inflammation in end-stage renal disease: Sources, consequences, and therapy. Seminars in dialysis. 2002;15:329–337. doi: 10.1046/j.1525-139x.2002.00083.x. [DOI] [PubMed] [Google Scholar]
- 4.Anders HJ, Andersen K, Stecher B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013;83:1010–1016. doi: 10.1038/ki.2012.440. [DOI] [PubMed] [Google Scholar]
- 5.Lee CT, Hsu CY, Tain YL, Ng HY, Cheng BC, Yang CC, Wu CH, Chiou TT, Lee YT, Liao SC. Effects of ast-120 on blood concentrations of protein-bound uremic toxins and biomarkers of cardiovascular risk in chronic dialysis patients. Blood Purif. 2014;37:76–83. doi: 10.1159/000357641. [DOI] [PubMed] [Google Scholar]
- 6.Lekawanvijit S, Kompa AR, Wang BH, Kelly DJ, Krum H. Cardiorenal syndrome: The emerging role of protein-bound uremic toxins. Circ Res. 2012;111:1470–1483. doi: 10.1161/CIRCRESAHA.112.278457. [DOI] [PubMed] [Google Scholar]
- 7.Ramezani A, Raj DS. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol. 2014;25:657–670. doi: 10.1681/ASN.2013080905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-n-oxide. Eur Heart J. 2014;35:904–910. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. 2014;124:4204–4211. doi: 10.1172/JCI72331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang WH, Wang Z, Fan Y, Levison BS, Hazen JE, Donahue L, Wu Y, Hazen SL. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-n-oxide in patients with heart failure: Refining the gut hypothesis. J Am Coll Cardiol. 2014;2014:1908–1914. doi: 10.1016/j.jacc.2014.02.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL, Lusis AJ. Trimethylamine-n-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. doi: 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koeth RA, Levison BS, Culley MK, Buffa JA, Wang Z, Gregory JC, Org E, Wu Y, Li L, Smith JD, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. G-butyrobetaine is a pro-atherogenic intermediate in gut microbial metabolism of l-carnitine to tmao. Cell Metab. 2014 doi: 10.1016/j.cmet.2014.10.006. Published on November 4, 2014 at http://dx.doi.org/2010.1016/j.cmet.2014.2010.2006. [DOI] [PMC free article] [PubMed]
- 16.Bain MA, Faull R, Fornasini G, Milne RW, Evans AM. Accumulation of trimethylamine and trimethylamine-n-oxide in end-stage renal disease patients undergoing haemodialysis. Nephrol Dial Transplant. 2006;21:1300–1304. doi: 10.1093/ndt/gfk056. [DOI] [PubMed] [Google Scholar]
- 17.Bain MA, Faull R, Milne RW, Evans AM. Oral l-carnitine: Metabolite formation and hemodialysis. Curr Drug Metab. 2006;7:811–816. doi: 10.2174/138920006778520561. [DOI] [PubMed] [Google Scholar]
- 18.Bell JD, Lee JA, Lee HA, Sadler PJ, Wilkie DR, Woodham RH. Nuclear magnetic resonance studies of blood plasma and urine from subjects with chronic renal failure: Identification of trimethylamine-n-oxide. Biochim Biophys Acta. 1991;1096:101–107. doi: 10.1016/0925-4439(91)90046-c. [DOI] [PubMed] [Google Scholar]
- 19.Robert R, Guilhot J, Pinsard M, Longeard PL, Jacob JP, Gissot V, Hauet T, Seguin F. A pair analysis of the delayed graft function in kidney recipient: The critical role of the donor. J Crit Care. 2010;25:582–590. doi: 10.1016/j.jcrc.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, Kusek JW, Manzi J, Van Lente F, Zhang YL, Coresh J, Levey AS. Estimating glomerular filtration rate from serum creatinine and cystatin c. N Engl J Med. 2012;367:20–29. doi: 10.1056/NEJMoa1114248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Levison BS, Hazen JE, Donahue L, Li XM, Hazen SL. Measurement of trimethylamine-n-oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal Biochem. 2014;455:35–40. doi: 10.1016/j.ab.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pencina MJ, D'Agostino RB, Sr, D'Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: From area under the roc curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- 23.Qu X, Li X, Zheng Y, Ren Y, Puelles VG, Caruana G, Nikolic-Paterson DJ, Li J. Regulation of renal fibrosis by smad3 thr388 phosphorylation. Am J Pathol. 2014;184:944–952. doi: 10.1016/j.ajpath.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Al-Waiz M, Mitchell SC, Idle JR, Smith RL. The metabolism of 14c-labelled trimethylamine and its n-oxide in man. Xenobiotica. 1987;17:551–558. doi: 10.3109/00498258709043962. [DOI] [PubMed] [Google Scholar]
- 25.Smith JL, Wishnok JS, Deen WM. Metabolism and excretion of methylamines in rats. Toxicol Appl Pharmacol. 1994;125:296–308. doi: 10.1006/taap.1994.1076. [DOI] [PubMed] [Google Scholar]
- 26.Zeisel SH, daCosta KA, Youssef M, Hensey S. Conversion of dietary choline to trimethylamine and dimethylamine in rats: Dose-response relationship. J Nutr. 1989;119:800–804. doi: 10.1093/jn/119.5.800. [DOI] [PubMed] [Google Scholar]
- 27.Hauet T, Mothes D, Bon D, Baumert H, Le Moyec L, Goujon JM, Robert R, Caritez JC, Tallineau C, Carretier M, Eugene M. Proton nmr spectroscopy as a novel approach to the monitoring of citrate and trimethylamine-n-oxide excretion after kidney preservation. Transplant Proc. 1997;29:2323–2325. doi: 10.1016/s0041-1345(97)00385-0. [DOI] [PubMed] [Google Scholar]
- 28.Le Moyec L, Pruna A, Eugene M, Bedrossian J, Idatte JM, Huneau JF, Tome D. Proton nuclear magnetic resonance spectroscopy of urine and plasma in renal transplantation follow-up. Nephron. 1993;65:433–439. doi: 10.1159/000187525. [DOI] [PubMed] [Google Scholar]
- 29.Serkova N, Fuller TF, Klawitter J, Freise CE, Niemann CU. H-nmr-based metabolic signatures of mild and severe ischemia/reperfusion injury in rat kidney transplants. Kidney Int. 2005;67:1142–1151. doi: 10.1111/j.1523-1755.2005.00181.x. [DOI] [PubMed] [Google Scholar]
- 30.Foxall PJ, Mellotte GJ, Bending MR, Lindon JC, Nicholson JK. Nmr spectroscopy as a novel approach to the monitoring of renal transplant function. Kidney Int. 1993;43:234–245. doi: 10.1038/ki.1993.37. [DOI] [PubMed] [Google Scholar]
- 31.Walkin L, Herrick SE, Summers A, Brenchley PE, Hoff CM, Korstanje R, Margetts PJ. The role of mouse strain differences in the susceptibility to fibrosis: A systematic review. Fibrogenesis Tissue Repair. 2013;6:18. doi: 10.1186/1755-1536-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Runyan CE, Schnaper HW, Poncelet AC. Smad3 and pkcdelta mediate tgf-beta1-induced collagen i expression in human mesangial cells. Am J Physiol Renal Physiol. 2003;285:F413–422. doi: 10.1152/ajprenal.00082.2003. [DOI] [PubMed] [Google Scholar]
- 33.Rhee EP, Clish CB, Ghorbani A, Larson MG, Elmariah S, McCabe E, Yang Q, Cheng S, Pierce K, Deik A, Souza AL, Farrell L, Domos C, Yeh RW, Palacios I, Rosenfield K, Vasan RS, Florez JC, Wang TJ, Fox CS, Gerszten RE. A combined epidemiologic and metabolomic approach improves ckd prediction. J Am Soc Nephrol. 2013;24:1330–1338. doi: 10.1681/ASN.2012101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin J, Hu FB, Curhan GC. Associations of diet with albuminuria and kidney function decline. Clin J Am Soc Nephrol. 2010;5:836–843. doi: 10.2215/CJN.08001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeisel SH. Choline: Clinical nutrigenetic/nutrigenomic approaches for identification of functions and dietary requirements. J Nutrigenet Nutrigenomics. 2010;3:209–219. doi: 10.1159/000324357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hegazy SK, El-Bedewy MM. Effect of probiotics on pro-inflammatory cytokines and nf-kappab activation in ulcerative colitis. World J Gastroenterol. 2010;16:4145–4151. doi: 10.3748/wjg.v16.i33.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seth A, Yan F, Polk DB, Rao RK. Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a pkc- and map kinase-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1060–1069. doi: 10.1152/ajpgi.00202.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simenhoff ML, Dunn SR, Zollner GP, Fitzpatrick ME, Emery SM, Sandine WE, Ayres JW. Biomodulation of the toxic and nutritional effects of small bowel bacterial overgrowth in end-stage kidney disease using freeze-dried lactobacillus acidophilus. Miner Electrolyte Metab. 1996;22:92–96. [PubMed] [Google Scholar]
- 39.Takayama F, Taki K, Niwa T. Bifidobacterium in gastro-resistant seamless capsule reduces serum levels of indoxyl sulfate in patients on hemodialysis. Am J Kid Dis. 2003;41:S142–145. doi: 10.1053/ajkd.2003.50104. [DOI] [PubMed] [Google Scholar]
- 40.Ranganathan N, Patel B, Ranganathan P, Marczely J, Dheer R, Chordia T, Dunn SR, Friedman EA. Probiotic amelioration of azotemia in 5/6th nephrectomized sprague-dawley rats. ScientificWorldJournal. 2005;5:652–660. doi: 10.1100/tsw.2005.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ranganathan N, Ranganathan P, Friedman EA, Joseph A, Delano B, Goldfarb DS, Tam P, Rao AV, Anteyi E, Musso CG. Pilot study of probiotic dietary supplementation for promoting healthy kidney function in patients with chronic kidney disease. Adv Ther. 2010;27:634–647. doi: 10.1007/s12325-010-0059-9. [DOI] [PubMed] [Google Scholar]
- 42.Niwa T, Ise M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J Lab Clin Med. 1994;124:96–104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.