

Abstract
Signals that drive interstitial fibrogenesis after renal ischemia reperfusion injury remain undefined. Sympathetic activation is manifest even in the early clinical stages of chronic kidney disease and is directly related to disease severity. A role for renal nerves in renal interstitial fibrogenesis in the setting of ischemia reperfusion injury has not been studied. In male 129S1/SvImJ mice, ischemia reperfusion injury induced tubulointerstitial fibrosis as indicated by collagen deposition and profibrotic protein expression 4 to 16 days after the injury.. Leukocyte influx, proinflammatory protein expression, oxidative stress, apoptosis, and cell cycle arrest at G2/M phase were enhanced after ischemia reperfusion injury. Renal denervation at the time of injury or up to 1 day post-injury improved histology, decreased proinflammatory/profibrotic responses and apoptosis, and prevented G2/M cell cycle arrest in the kidney. Treatment with afferent nerve-derived calcitonin gene-related peptide (CGRP) or efferent nerve-derived norepinephrine in denervated and ischemia reperfusion injury-induced kidneys mimicked innervation, restored inflammation and fibrosis, induced G2/M arrest, and enhanced TGF-β1 activation. Blocking norepinephrine or CGRP function using respective receptor blockers prevented these effects. Consistent with the in vivo study, treatment with either norepinephrine or CGRP induced G2/M cell cycle arrest in HK-2 proximal tubule cells, whereas antagonists against their respective receptors prevented G2/M arrest. Thus, renal nerve stimulation is a primary mechanism and renal nerve-derived factors drive epithelial cell cycle arrest and the inflammatory cascade causing interstitial fibrogenesis after ischemia reperfusion injury.
INTRODUCTION
Patients surviving an episode of acute kidney injury (AKI) have a significant risk of progression to chronic kidney disease (CKD) and even end-stage renal disease.1 However, the mechanisms that link AKI to CKD in humans remain poorly defined. Animal studies have shown that persistent medullary hypoxia, capillary rarefaction, inflammation, failed differentiation of epithelial cells and apoptosis following renal ischemia reperfusion injury (IRI) are possible mechanisms that may drive tubulointerstitial fibrosis.2–4 Several molecules induced after IRI are implicated in injury and inflammation as well as in repair, regeneration and in the progression of renal fibrosis. These molecules include various cytokines (IL-13, IL-21), chemokines (KC, MIP-2, MCP-1), angiogenic factors (VEGF), growth factors (EGF, TGF-β1, CTGF, PDGF), and the renin–angiotensin system.5–7 The interplay of these molecules and their downstream signaling pathways in the injured or regenerating tubular epithelium, capillary and interstitial cells could evoke inflammation, fibroblast differentiation and proliferation and matrix deposition. However, the primary stimuli that induce the myriad of signaling events that lead to the inflammatory response and fibrosis after an initial insult to the kidney remains undefined.8 Therefore, identification of the primary signal or the core signaling pathway that instigate renal fibrogenesis after an initial stimulus is essential for the elucidation of the pathophysiological mechanisms of the syndrome and in developing effective therapeutic strategies for preventing, reversing or limiting progression of fibrogenesis.9
The kidney is innervated by efferent sympathetic nerves as well as peptidergic sensory afferent nerves in which several neuroactive substances have been identified.10–12 Sympathetic nerve activity (SNA) is increased in both patients and experimental animals with chronic renal failure.13, 14 Renal denervation shows protective effects against renal failure in both animals and humans. Although the mechanisms remain to be fully elucidated, it may include decrease in blood pressure, renal efferent SNA, central SNA and sympathetic outflow, and downregulation of the renin-angiotensin system.12, 15 Given the pronounced effect of the renal nerves on CKD, we sought to determine whether afferent and efferent nerve-derived neuropeptides/neurotransmitters and their signaling pathways may be responsible for the functional, fibrotic and inflammatory responses in IRI-induced long-term sequelae. Here we report that renal nerve-derived norepinephrine and CGRP signaling is required for tubular epithelial cell injury and production of inflammatory factors and profibrogenic factors to trigger renal interstitial fibrogenesis.
RESULTS
Renal denervation attenuates interstitial fibrosis induced by IRI
To test whether kidney nerve contributes to interstitial fibrosis induced by IRI, we performed renal denervation before IRI. Collagen deposition assessed by Sirius red staining and hydroxyproline measurement increased after IRI in intact kidneys in a time-dependent manner, whereas renal denervation markedly lessened the collagen deposition (Figure 1, A and B; Supplementary Figure 1A). Renal denervation also reduced the myofibroblast marker α-smooth muscle actin (α-SMA) expression, Smad3 phosphorylation (p-Smad3) and TGF-β1 production during interstitial fibrosis induced by IRI (Supplementary Figure 1, B and C). Since inflammation plays as an important role in fibrosis, we next examined the recruitment of neutrophils and macrophages. Polymorphonuclear neutrophil (PMN)-positive neutrophils and F4/80-positive macrophages were persistently recruited into intact kidneys for at least 16 days after IRI, whereas renal denervation inhibited the recruitment of both cell populations during interstitial fibrosis (Figure 1, C–E). Additionally, we determined that postconditioning of renal denervation (up to 1 d post-injury) significantly reduced interstitial fibrosis and inflammation as demonstrated by decreased collagen deposition, profibrotic protein expression, and recruitment of neutrophils and macrophages (Supplementary Figure 2).
Figure 1. Renal denervation inhibits collagen deposition, and neutrophil and macrophage influx during a period of interstitial fibrosis after IRI.

Two days after denervation in left kidneys of mice, IRI or sham operation (S) in the left kidneys was carried out. (A) Collagen deposition using Sirius red stain on denervated or intact kidney sections at 16 days after IRI. Scale bars indicate 50 μm. (B) Percentage of Sirius red-positive area on kidney sections. (C and D) Neutrophil infiltration represented by the number of PMN-positive cells on immunohistochemically stained kidney sections. (C and E) Macrophage infiltration represented by the percentage of F4/80-positive area on immunohistochemically stained kidney sections. Error bars represent SD (n = 5). #P<0.05 versus intact. Scale bars indicate 50 μm.
Norepinephrine and CGRP signaling contribute to interstitial fibrosis after IRI
We confirmed the effectiveness of renal denervation as demonstrated by a decrease of tyrosine hydroxylase expression in IRI-subjected kidneys as well as in sham-subjected kidneys (Figure 2A and Supplementary Figure 3A). Levels of both kidney norepinephrine and CGRP remained above sham levels for at least 16 days after IRI, whereas renal denervation significantly decreased levels of both in IRI and sham kidneys (Figure 2B). Furthermore, immunohistochemistry for CGRP demonstrated faint expression in tubules and adventitia of interlobar arteries at 2 days after IRI in denervated compared to intact kidneys (Figure 2C). These results indicate successful loss of sympathetic and sensory nerve fibers by renal denervation. To determine whether norepinephrine and CGRP contribute to fibrosis, we treated denervated kidneys with exogenous norepinephrine or CGRP. Chronic administration of norepinephrine or CGRP for 16 d following IRI induced excessive collagen deposition as demonstrated by Sirius red-positive area and hydroxyproline level, and increased expression of α-SMA and p-Smad3 in denervated kidneys (Figure 2D; and Supplementary Figure 3, B and C). The exogenous norepinephrine and CGRP also increased the recruitment of neutrophils and macrophages in the denervated kidneys after IRI (Figure 2, E and F). Next, we investigated the roles of adrenergic receptors (ARs) and CGRP receptor in interstitial fibrosis using their antagonists. Sixteen days after IRI, mice treated with α2-AR antagonist (atipamezole), but not α1-AR antagonist (doxazosin) or β-AR antagonist (pronethalol), showed significant reductions of collagen level, and neutrophil and macrophage recruitment (Figure 3, A–C). Treatment with the CGRP receptor antagonist (CGRP(8-37)) also reduced collagen level, and neutrophil and macrophage recruitment in IRI kidneys, but not in sham kidneys (Figure 3, D–F). Taken together, these data suggest that norepinephrine and CGRP induce interstitial fibrosis and inflammation after IRI though α2-AR and CGRP receptors, respectively. Our study, however, did not determine whether the α2-AR antagonist, atipamezole may act presynaptically to decrease dopamine neurotransmission or dopamine overflow.
Figure 2. Exogenous norepinephrine or calcitonin gene-related peptide enhances interstitial fibrosis and inflammation induced by IRI in denervated kidneys.

(A–C) Two days after denervation in left kidneys of mice, IRI or sham operation (S) in the left kidneys was carried out. (A) Kidney expression of tyrosine hydroxylase using Western blot analysis at 16 days after IRI or sham. Anti-β-actin antibody served as a loading control. (B) Levels of norepinephrine and CGRP in the kidneys using ELISA kits. (C) Immunohistochemical staining for CGRP on the kidney sections at 2 days after IRI. Scale bars indicate 50 μm. (D–F) Denervation in left kidneys of mice was carried out; 2 days after the onset, norepinephrine, CGRP or vehicle was continuously infused into the denervated kidneys via mini-osmotic pump; and the kidneys were subjected to 30 min of ischemia followed by 16 days of reperfusion. (D) Percentage of Sirius red-positive area in kidneys sections. (E) Neutrophil infiltration represented by the number of PMN-positive cells on immunohistochemically stained kidney sections. (F) Macrophage infiltration represented by percentage of F4/80-positive area on immunohistochemically stained kidney sections. Error bars represent SD (n = 4). *P<0.05 versus sham, #P<0.05 versus intact, and $P<0.05 versus vehicle.
Figure 3. α2-AR antagonist and CGRP receptor antagonist diminish interstitial fibrosis and inflammation induced by IRI.

(A–C) Mice were continuously treated with doxazosin (α1-AR antagonist, 12 mg/kg/d), atipamezole (α2-AR antagonist, 2.4 mg/kg/d), pronethalol (β-AR antagonist, 2.4 mg/kg/d), or 10% DMSO in PBS (vehicle) via an intraperitoneal implantation of mini-osmotic pump immediately before 30 min of left kidney ischemia and 16 days of reperfusion. (A) Percentage of Sirius red-positive area on the kidney sections. (B) Neutrophil infiltration represented by counting PMN-positive cells on immunohistochemically stained kidney sections. (C) Macrophage infiltration represented by percentage of F4/80-positive area on immunohistochemically stained kidney sections. (D–F) Mice were continuously treated with the CGRP receptor antagonist (CGRP(8-37), 120 μg/kg/d) or 0.9% saline (vehicle) via an intraperitoneal implantation of mini-osmotic pump immediately before 30 min of left kidney ischemia and 16 days of reperfusion. (D) Percentage of Sirius red-positive area on the kidney sections. (E) Neutrophil infiltration represented by the number of PMN-positive cells on immunohistochemically stained kidney sections. (F) Macrophage infiltration represented by percentage of F4/80-positive area on immunohistochemically stained kidney sections. Error bars represent SD (n = 5). $P<0.05 versus vehicle.
Norepinephrine and CGRP signaling are involved in kidney injury after IRI
To determine whether norepinephrine and CGRP are involved in tubular cell death induced by IRI, we assessed histological damage and apoptosis in tubular cells. IRI consistently increased tubular injury score based on PAS stain and tubular apoptosis based on TUNEL assay for at least 16 days after the onset, whereas renal denervation diminished apoptosis and tubular injury score (Figure 4, A and B; and Supplementary Figure 4, A and B). Renal denervation also lessened increases in the expression of full-length poly(ADP-ribose) polymerase 1 (PARP1), an indicator of necrosis, and expression levels of cleaved PARP1, cleaved caspase-3, and active bax, proapoptotic markers (Supplementary Figure 4C). When denervated kidneys were treated with either norepinephrine or CGRP, tubular injury score and apoptosis were evoked at 16 days after IRI (Figure 4C and Supplementary Figure 4D). Additionally, treatment with the α2-AR antagonist or CGRP receptor antagonist significantly reduced apoptosis and tubular injury score at 16 days after IRI in innervated mice kidneys (Supplementary Figure 4, E and F). These data suggest that norepinephrine and CGRP induce tubular cell death though their respective receptors in IRI kidneys.
Figure 4. Norepinephrine and CGRP signaling contribute to kidney injury during a period of interstitial fibrosis after IRI.

(A and D) Two days after denervation in left kidneys of mice, IRI or sham in the left kidneys was carried out (n = 5). (C, E and F) Denervation or intact in left kidneys of mice was carried out; 2 days after the denervation, norepinephrine, CGRP or vehicle was continuously infused into the denervated kidneys via mini-osmotic pump; and the kidneys were subjected to 30 min of ischemia followed by 16 days of reperfusion (n = 4). (A) Tubular damage represented by PAS stain on the kidney sections at 16 days after IRI. Scale bars indicate 50 μm. (B and C) Tubular injury score measured on PAS-stained kidney sections. (D and E) Glomerular filtration rate (GFR, creatinine clearance) was measured in mice housed in metabolic cages. (F) Kidney weight obtained from mice at 16 days after IRI or sham. Error bars represent SD. #P<0.05 versus intact and $P<0.05 versus vehicle.
To examine the renal function of IRI kidneys plus/minus denervation, we measured glomerular filtration rate (GFR) after contralateral nephrectomy. As shown in Figure 4D, renal denervation did not significantly alter GFR at early post-ischemic periods 1, 2 and 4 days after IRI, compared to that in intact kidneys. However, at 8 and 16 days after IRI, GFR in denervated kidneys was significantly higher than that in intact kidneys. In denervated kidneys, treatment with either norepinephrine or CGRP significantly decreased GFR at 16 days after IRI (Figure 4E). Because unilateral IRI causes a severe reduction of kidney weight at 2–3 weeks after the onset,16, 17 we determined whether loss of renal mass would be prevented by renal denervation. As shown in Figure 5F, IRI reduced kidney weights at 16 days after the onset, compared with the weight of sham kidneys. Renal denervation inhibited the postischemic reduction of kidney weights, but treatment with either norepinephrine or CGRP decreased the weight of denervated IRI kidneys. These data suggest that norepinephrine and CGRP contribute to kidney injury during development of interstitial fibrosis induced by IRI.
Figure 5. Renal denervation reduces oxidative stress in a norepinephrine and CGRP-dependent manner in kidneys after IRI.
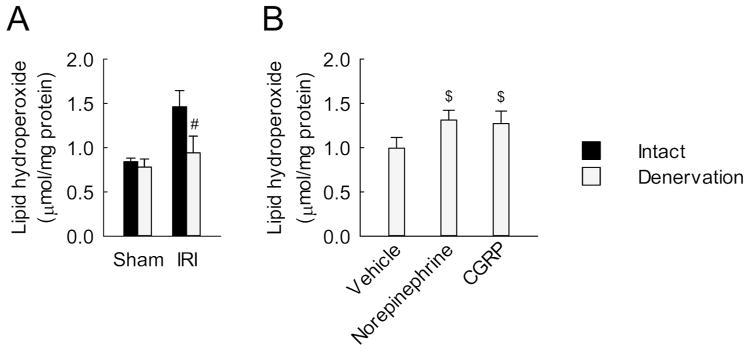
(A) Two days after denervation in left kidneys of mice, IRI or sham in the left kidneys was carried out; then the kidneys were harvested at 16 d after reperfusion (n = 5). (B) Denervation in left kidneys of mice was carried out; 2 d after the onset, norepinephrine, CGRP or vehicle was continuously infused into the denervated kidneys via mini-osmotic pump; and the kidneys were subjected to 30 min of ischemia followed by 16 d of reperfusion (n = 4). (A and B) Lipid peroxidation represented by level of lipid hydroperoxide in kidneys. Error bars represent SD. #P<0.05 versus intact and $P<0.05 versus vehicle.
To determine the effect of renal denervation on renin-angiotensin system, we measured the renin activity in plasma and kidneys. Consistent with the previous study,18 renal denervation reduced the kidney and plasma levels of renin activity; however, the renin activity was not significantly changed at 16 days after IRI (Supplementary Figure 5, A and B). Next, to determine whether the protective effects of denervation are related to systemic blood pressure, we measured systolic blood pressure. Systolic blood pressure was not altered as a function of IRI, IRI with denervation or after administration of vehicle, NE or CGRP (Supplementary Figure 5C). These data suggest that the renal nerve has no significant effect on systemic blood pressure in the setting of IRI.
Renal denervation reduces oxidative stress after IRI
Since oxidative stress contributes to tubular injury and fibrosis after IRI,4 we analyzed redox system. Sixteen days after IRI, the intact kidneys showed increases in lipid hydroperoxide level and nitrotyrosine expression as oxidative stress indicators, whereas the denervated kidneys revealed a reduced oxidative stress (Figure 5A and Supplementary Figure 6A). Conversely, IRI decreased the expression of manganese superoxide dismutase (MnSOD) and the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) at 16 days after the onset, but renal denervation inhibited the decrease of both of these antioxidative indicators (Supplementary Figure 6, A and B). In the denervated kidneys, exogenous norepinephrine and CGRP increased the level of lipid hydroperoxide and decreased the ratio of GSH to GSSG at 16 days after IRI (Figure 5B and Supplementary Figure 6C). In intact kidneys, treatment with the α2-AR antagonist or the CGRP receptor antagonist significantly decreased the lipid hydroperoxide level and the ratio of GSH to GSSG at 16 days after IRI (Supplementary Figure 6, D and E). These data suggest that norepinephrine and CGRP signaling contributes to the alteration of redox status during development of interstitial fibrosis induced by IRI.
Norepinephrine and CGRP signaling induces tubular cell cycle arrest
Tubular cell cycle arrest mediates interstitial fibrosis after IRI.19 Because of that, we tested whether norepinephrine and CGRP influence tubular cell cycle arrest after IRI. The number of tubular cells positive for phosphorylated Histone H3 (p-H3), a marker of G2/M phase in cell cycle, was increased after IRI, whereas the denervated kidneys showed a significant reduction of the number of p-H3-positive tubular cells (Figure 6, A and B). Consistent with the result of the number of p-H3-positive tubular cells, renal denervation decreased the ratio of cyclin B1 to cyclin D1, as a maker of G2/M arrest, based on Western blot analysis, from 4 days after IRI (Supplementary Figure 7A). In denervated kidneys, treatment with either norepinephrine or CGRP induced the number of p-H3-positive tubular cells and the ratio of cyclin B1 to cyclin D1 at 16 days after IRI (Figure 6C and Supplementary Figure 7C). In intact kidneys, treatments with either the α2-AR antagonist or the CGRP receptor antagonist significantly reduced the markers of G2/M arrest at 16 days after IRI (Supplementary Figure 7, D and E). Additionally, to determine whether norepinephrine and CGRP signaling directly contributes to tubular cell cycle arrest, we evaluated the effects of norepinephrine and CGRP in a HK-2 human proximal tubular cell line. Norepinephrine and CGRP increased the percentage of cells in G2/M phase (Supplementary Figure 8A). Consistent with the data from in vivo studies, treatments with either α2-AR antagonist or CGRP receptor antagonist significantly reduced the percentage of cells in G2/M phase (Supplementary Figure 8, B and C). After demonstrating that norepinephrine and CGRP induce tubular cell cycle arrest in G2/M phase, we investigated its impact on cell viability in HK-2 cells, assuming that G2/M phase arrest is induced by DNA damage and hence loss of cell viability. Using the 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, treatment with either norepinephrine or CGRP decreased the percentage of cell viability (Supplementary Figure 8, D and E), consistent with the data on the in vitro G2/M phase arrest. The α2-AR antagonist and CGRP receptor antagonist significantly increased the percentage of cell viability (Supplementary Figure 8, D and E).
Figure 6. Renal denervation inhibits tubular cell cycle arrest in a norepinephrine and CGRP-dependent manner in kidneys after IRI.
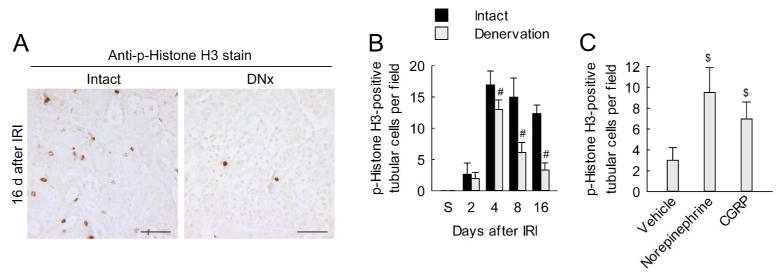
(A and B) Two days after denervation in left kidneys of mice, IRI or sham in the left kidneys was carried out (n = 5). (C) Two days after denervation in left kidneys of mice, norepinephrine, CGRP or vehicle was continuously infused into the denervated kidneys via mini-osmotic pump and the kidneys were subjected to 30 min of ischemia followed by 16 d of reperfusion (n = 4). (A) Immunohistochemial staining for phosphorylated histone H3 (p-H3) on kidney sections. Scale bars indicate 50 μm. (B and C) The number of p-H3-positive tubular cells on immunohistochemically stained kidney sections. Error bars represent SD. #P<0.05 versus intact and $P<0.05 versus vehicle.
DISCUSSION
Renal sympathetic hyperactivity is common in CKD and has been shown to contribute to glomerulonephritis and induce proteinuria both through and beyond its effect on blood pressure.20 In a model of glomerulonephritis, bilateral renal denervation significantly reduced albuminuria, mesangiolysis, microaneurysm formation and deposition of glomerular collagen IV.14 We previously reported that renal denervation can prevent UUO-induced inflammation and interstitial fibrosis and renal nerves derived signaling molecules norepinephrine and CGRP mediate the inflammatory and fibrotic response.21 Increased renal sympathetic nerve activity and norepinephrine overflow are implicated in the pathophysiology of IRI.22 Renal denervation or chemical ganglionic blockade ameliorated IRI.23 However, the effect of renal denervation or nerve-derived factors in the setting of IRI on the long-term development of renal inflammation and interstitial fibrogenesis has not been investigated.
In this study, we used a mouse model of unilateral IRI to determine whether renal nerves play a central role in activating the signaling events that lead to renal fibrogenesis. Our data demonstrate that renal denervation prior to or up to 1 day post-IRI prevents tubular injury, apoptosis, tubulointerstitial inflammation and fibrosis from 4–16 days post-IRI without altering blood pressure. Unlike prior studies,23, 24 renal denervation in our IRI model did not lessen kidney dysfunction during the early post-ischemic period (1–4 days after IRI). The discrepancy may be caused by different surgical approaches (bilateral vs. unilateral ischemia), different susceptibility to IRI (mild/moderate vs. severe ischemia), and different species (rat vs. mouse). These data suggest that the effect of renal denervation on the long term sequlae is not due to mitigation of the ischemic renal injury during the early post-ischemic period. The expression levels of profibrotic markers and the influx of neutrophils/macrophages were significantly decreased in denervated IRI-subjected kidneys at 8–16 days post-IRI. Furthermore, oxidative stress and apoptotic markers were significantly reduced by denervation. The levels of norepinephrine released from efferent renal sympathetic nerves and CGRP released from afferent renal sensory nerves were significantly increased; and both norepinephrine and CGRP initiated tubular cell apoptosis, inflammatory cascade and tubulointerstitial fibrosis after IRI. Inhibition of either CGRP receptor or α2-AR prevented the adverse effects of innervation after IRI. Cell cycle arrest in G2/M phase observed after IRI was prevented by denervation, or inhibition of either the CGRP receptor or α2-AR. Administration of either norepinephrine or CGRP induced cell cycle arrest at G2/M phase in IRI-subjected and denervated kidneys. These results suggest that the renal nerve stimulation in IRI is a primary mechanism that drives the myriad of downstream signaling events to elicit inflammation and renal fibrogenesis.
Elevated plasma norepinephrine is observed in patients with CKD and ESRD and is strongly linked with the onset and development of renal injury.25 In the present study, IRI significantly increased whole kidney norepinephrine levels from 1–16 d post-injury in mice. Further, in denervated IRI kidneys, chronic administration of norepinephrine mimicked the effect of innervation and induced inflammation and fibrosis. Norepinephrine can induce signaling via α1-AR, α2-AR and β-AR. Our data indicate that inhibition of only α2-AR prevented renal fibrogenesis after IRI. Previous reports have indicated that α1-AR or β-AR blockade is renoprotective in the 5/6-nephrectomy model and a combination of α1-AR and β-AR blockade was more effective in preventing glomerular, tubulointerstitial, and vascular injury than α1-AR blockade alone without altering blood pressure.26, 27 Our data implicating norepinephrine signaling through α2-AR to induce fibrogenesis in IRI kidneys are intriguing as presynaptic α2A-AR and α2C-AR subtypes in vasa deferentia, isolated brain and atrial tissue;28, 29 and α2A-AR subtype in kidney30 play a predominant role for regulating synaptic norepinephrine release. Deletion of α2A-AR and α2C-AR subtypes increased the susceptibility to develop heart failure following chronic pressure overload in vivo.31, 32 Intriguingly, activation of α2-AR using agonist clonidine33 and moxonidine,34 or blocking β-AR using antagonist propranolol35 protected against IRI. In contrast, our data indicate that activation of the α2-AR is detrimental in the progression of renal injury, inflammation and fibrogenesis after IRI. Thus, activation of the α2-AR in the IRI kidney may trigger signaling events other than sympathoinhibition, such as activation of profibrotic and inflammatory signaling pathways to instigate fibrogenesis. This notion is supported by our finding that administration of norepinephrine in denervated kidneys can enhance neutrophil and macrophage infiltration, activate TGF-β1 signaling as indicated by increased p-Smad-3 expression as well as α-SMA expression. Further, administration of norepinephrine can induce cell cycle arrest at the G2/M stage, as indicated by increased number of p-H3-positive epithelial cells. An increased number of tubular epithelial cells positive for p-H3 have been reported after ischemic, toxic, and obstructive injury suggesting cell cycle arrest at the G2/M stage.19 These cells contribute to renal fibrosis by releasing profibrogenic cytokines such as TGF-β1 and CTGF.19
Our data revealed that renal CGRP levels were increased soon after IRI, and persisted through 16 d of study, with maximal levels observed during the 1–4 d post-IRI. CGRP protein was evident at apical or basolateral membranes of epithelial cells as well as in sensory nerve fibers. Since CGRP has potent vasodilator activity, it has long been considered to be involved in aggravation of inflammation by increasing the blood flow, the number of circulating cells and chemotactic factors.36, 37 Consistent with this finding, our data demonstrate that inhibition of CGRP receptor prevented the expression of inflammatory molecules as well as infiltration of leukocytes into the renal parenchyma, while chronic administration of CGRP in IRI-subjected and denervated kidneys induced tubular cell apoptosis, inflammatory cascade as well as fibrogenesis without altering blood pressure. Administration of CGRP also induced cell cycle arrest in G2/M phase which may contribute to TGF-β1 activation and fibrogenesis. Blocking the CGRP receptor released the cell cycle arrest and attenuated fibrosis in the UUO-induced kidneys.21
Epithelial cell apoptosis is considered as a critical initial response to various forms of renal injury including UUO38 and IRI.39 Increased apoptosis lead to renal tubular atrophy with loss of nephrons and accompanied matrix deposition into the atrophied region resulting in interstitial fibrosis. Histological damage and apoptosis was diminished after denervation as well as after blocking α2-AR or CGRP receptor in IRI kidneys. On the other hand, administration of either norepinephrine or CGRP induced apoptosis. These data are consistent with our previous report that norepinephrine and CGRP can induce apoptosis in the UUO-induced kidneys as well as in in vitro proximal tubular cell culture model.21 Previous reports have also linked mechanical stretch induced apoptosis in in vitro models to increased oxidative stress in a catalase dependent manner.40 In catalase deficient mice, hydrogen peroxide accumulation occurs after UUO injury and contributes to apoptosis.41 Reactive oxygen species are significantly increased in the chronically obstructed kidney.42 Consistently, our data indicate that, lipid hydroperoxide and ratio of reduced to oxidized glutathione are increased after IRI but not in denervated kidneys. Further, denervation increased MnSOD levels and decreased nitrotyrosine levels in the IRI kidney. Administration of norepinephrine or CGRP in denervated mouse kidneys restored the redox status to that in innervated mice kidneys. These data indicate that one of the mechanisms by which norepinephrine or CGRP induces apoptosis is by increasing the oxidative stress in the IRI kidneys. A second mechanism by which apoptosis may be induced by is the heightened inflammatory milieu in the IRI kidney. The number of neutrophils and macrophages are increased in the IRI kidney and may further increase the oxidative stress, inflammatory milieu and cell death signals.
Interstitial fibrosis is characterized by macrophage accumulation. The influx of macrophages and the macrophage-secreted cytokines including monocyte chemoattractant protein-1 and TGF-β promote renal fibrosis.43–45 The influx of neutrophils is related with lung fibrosis,46 and the role of neutrophils has been well known in the early post-ischemic period.47 However, the role of neutrophils in the progression of renal fibrosis has not been demonstrated previously. Norepinephrine and CGRP induce the production of proinflammatory cytokines in cultured macrophages,48, 49 and activate neutrophils in vitro.50, 51 Previous reports suggest that norepinephrine and CGRP can be linked to macrophage and neutrophil functions. In our data, IRI increased the influx of macrophages and neutrophils, but renal denervation reduced the influx of those cells during the late but not the early post-ischemic period. The administration of either norepinephrine or CGRP revived the increase of the influx of those cells. It is likely that the reduction of the macrophage and neutrophil influxes in denervated kidneys may affect the reduction of damaged tubular cells and the prevention of renal fibrosis post-ischemic injury.52
In the last three years, renal denervation has been tried in 15 to 20,000 patients in Europe and showed beneficial effects on blood pressure in patients with resistant hypertension. However, the well-designed US trial—Symplicity HTN-3—showed only modest blood pressure benefit. The possible efficacy of renal denervation in 15 patients with hypertension in the setting of CKD, was carried out by Hering et al.53 The office systolic and diastolic blood pressures were significantly reduced at 3- and 6-month follow-up, although ambulatory blood pressure monitoring over 24 hours did not reveal significant reductions. Our study in mice demonstrates that renal denervation has no effect on systemic blood pressure. However, renal denervation at the time of the injury or even 1 day after the injury significantly reduces inflammation and interstitial fibrosis. Furthermore, renal denervation prevents loss of renal mass. The possibility that the prevention of renal mass may be due to improved renal blood flow and hence reduced tissue hypoxia and injury is not investigated in this study. The pharmacological intervention of norepinephrine or CGRP signaling pathways after the injury significantly reduced progression of fibrosis. These data suggest that early renal nerve ablation or blocking renal nerve-derived factors after the initialing injury may be an option to mitigate CKD complications.
In conclusion, the studies presented provide unswerving evidence that renal nerve activation is a primary mechanism instigating fibrogenesis in the IRI kidney. Our data implicate both renal afferent nerve-derived CGRP and efferent nerve-derived norepinephrine in triggering tubular atrophy, the inflammatory cascade, cell cycle arrest at the G2/M stage and fibrogenesis. Blockade of either CGRP receptor or α2-adrenergic receptor prevented activation of apoptogenic factors, inflammatory molecules and infiltration of leukocytes and profibrogenic factors, including TGF-β1 and its downstream signaling pathways. Inhibiting the actions of CGRP and norepinephrine might represent a novel effective therapeutic strategy to prevent or limit progression of renal fibrogenesis at its onset in IRI-induced CKD.
METHODS
Mice and surgical preparation, Collagen deposition, Histology, Western blot, ELISA, Cell culture and treatment, Renal function, and Blood Pressure Measurement
Please see Supplementary Methods.
Statistical analyses
Analysis of variance was used to compare data among groups. Differences between 2 groups were assessed by 2-tailed unpaired Student’s t test. P values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Youngsu Cho for technical assistance with Western blot and immunohistochemistry, and Kelly E. Long and Sherry N. Westphal for mouse care.
B.J.P. is supported by National Institutes of Health grants DK-083291 and DK-090332, and a Nebraska Research Initiative fund.
Footnotes
DISCLOSURE
All the authors declared no completing interests.
References
- 1.Ishani A, Xue JL, Himmelfarb J, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20:223–228. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venkatachalam MA, Griffin KA, Lan R, et al. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010;298:F1078–1094. doi: 10.1152/ajprenal.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basile DP, Donohoe D, Roethe K, et al. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281:F887–899. doi: 10.1152/ajprenal.2001.281.5.F887. [DOI] [PubMed] [Google Scholar]
- 4.Kim J, Seok YM, Jung KJ, et al. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am J Physiol Renal Physiol. 2009;297:F461–470. doi: 10.1152/ajprenal.90735.2008. [DOI] [PubMed] [Google Scholar]
- 5.Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819–1834. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 6.Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol. 2010;6:643–656. doi: 10.1038/nrneph.2010.120. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehal WZ, Iredale J, Friedman SL. Scraping fibrosis: Expressway to the core of fibrosis. Nat Med. 2011;17:552–553. doi: 10.1038/nm0511-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferguson M, Ryan GB, Bell C. Localization of sympathetic and sensory neurons innervating the rat kidney. Journal of the autonomic nervous system. 1986;16:279–288. doi: 10.1016/0165-1838(86)90034-2. [DOI] [PubMed] [Google Scholar]
- 11.Barajas L, Liu L, Powers K. Anatomy of the renal innervation: intrarenal aspects and ganglia of origin. Canadian journal of physiology and pharmacology. 1992;70:735–749. doi: 10.1139/y92-098. [DOI] [PubMed] [Google Scholar]
- 12.DiBona GF. Physiology in perspective: The Wisdom of the Body. Neural control of the kidney. Am J Physiol Regul Integr Comp Physiol. 2005;289:R633–641. doi: 10.1152/ajpregu.00258.2005. [DOI] [PubMed] [Google Scholar]
- 13.Converse RL, Jr, Jacobsen TN, Toto RD, et al. Sympathetic overactivity in patients with chronic renal failure. The New England journal of medicine. 1992;327:1912–1918. doi: 10.1056/NEJM199212313272704. [DOI] [PubMed] [Google Scholar]
- 14.Veelken R, Vogel EM, Hilgers K, et al. Autonomic renal denervation ameliorates experimental glomerulonephritis. J Am Soc Nephrol. 2008;19:1371–1378. doi: 10.1681/ASN.2007050552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clayton SC, Haack KKV, Zucker IH. Renal denervation modulates angiotensin receptor expression in the renal cortex of rabbits with chronic heart failure. American Journal of Physiology - Renal Physiology. 2011;300:F31–F39. doi: 10.1152/ajprenal.00088.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zager RA, Johnson AC, Andress D, et al. Progressive endothelin-1 gene activation initiates chronic/end-stage renal disease following experimental ischemic/reperfusion injury. Kidney Int. 2013;84:703–712. doi: 10.1038/ki.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zager RA, Johnson AC, Becker K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am J Physiol Renal Physiol. 2011;301:F1334–1345. doi: 10.1152/ajprenal.00431.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Draper ML, Wang J, Valego N, et al. Effect of renal denervation on renin gene expression, concentration, and secretion in mature ovine fetus. Am J Physiol Regul Integr Comp Physiol. 2000;279:R263–270. doi: 10.1152/ajpregu.2000.279.1.R263. [DOI] [PubMed] [Google Scholar]
- 19.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. 531p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schlaich MP, Socratous F, Hennebry S, et al. Sympathetic activation in chronic renal failure. J Am Soc Nephrol. 2009;20:933–939. doi: 10.1681/ASN.2008040402. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Padanilam BJ. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J Am Soc Nephrol. 2013;24:229–242. doi: 10.1681/ASN.2012070678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iaina A, Eliahou HE. The Sympathetic Nervous System in the Pathogenesis of Acute Renal Failure. Renal Failure. 1983;7:115–125. doi: 10.3109/08860228309076043. [DOI] [PubMed] [Google Scholar]
- 23.Fujii T, Kurata H, Takaoka M, et al. The role of renal sympathetic nervous system in the pathogenesis of ischemic acute renal failure. European journal of pharmacology. 2003;481:241–248. doi: 10.1016/j.ejphar.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 24.Iaina A, Eliahou HE. The sympathetic nervous system in the pathogenesis of acute renal failure. Clinical and experimental dialysis and apheresis. 1983;7:115–125. doi: 10.3109/08860228309076043. [DOI] [PubMed] [Google Scholar]
- 25.Zoccali C, Mallamaci F, Tripepi G, et al. Norepinephrine and concentric hypertrophy in patients with end-stage renal disease. Hypertension. 2002;40:41–46. doi: 10.1161/01.hyp.0000022063.50739.60. [DOI] [PubMed] [Google Scholar]
- 26.Amann K, Koch A, Hofstetter J, et al. Glomerulosclerosis and progression: Effect of subantihypertensive doses of [agr] and [bgr] blockers. Kidney Int. 2001;60:1309–1323. doi: 10.1046/j.1523-1755.2001.00936.x. [DOI] [PubMed] [Google Scholar]
- 27.Amann K, Rump LC, Simonaviciene A, et al. Effects of low dose sympathetic inhibition on glomerulosclerosis and albuminuria in subtotally nephrectomized rats. J Am Soc Nephrol. 2000;11:1469–1478. doi: 10.1681/ASN.V1181469. [DOI] [PubMed] [Google Scholar]
- 28.Trendelenburg AU, Klebroff W, Hein L, et al. A study of presynaptic alpha2-autoreceptors in alpha2A/D-, alpha2B- and alpha2C-adrenoceptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. 2001;364:117–130. doi: 10.1007/s002100100423. [DOI] [PubMed] [Google Scholar]
- 29.Trendelenburg AU, Philipp M, Meyer A, et al. All three alpha2-adrenoceptor types serve as autoreceptors in postganglionic sympathetic neurons. Naunyn Schmiedebergs Arch Pharmacol. 2003;368:504–512. doi: 10.1007/s00210-003-0829-x. [DOI] [PubMed] [Google Scholar]
- 30.Vonend O, Habbel S, Stegbauer J, et al. Alpha(2A)-adrenoceptors regulate sympathetic transmitter release in mice kidneys. Br J Pharmacol. 2007;150:121–127. doi: 10.1038/sj.bjp.0706961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brede M, Philipp M, Knaus A, et al. alpha2-adrenergic receptor subtypes - novel functions uncovered in gene-targeted mouse models. Biol Cell. 2004;96:343–348. doi: 10.1016/j.biolcel.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Brede M, Wiesmann F, Jahns R, et al. Feedback Inhibition of Catecholamine Release by Two Different α2-Adrenoceptor Subtypes Prevents Progression of Heart Failure. Circulation. 2002;106:2491–2496. doi: 10.1161/01.cir.0000036600.39600.66. [DOI] [PubMed] [Google Scholar]
- 33.Solez K, Ideura T, Silvia CB, et al. Clonidine after renal ischemia to lessen acute renal failure and microvascular damage. Kidney Int. 1980;18:309–322. doi: 10.1038/ki.1980.141. [DOI] [PubMed] [Google Scholar]
- 34.Tsutsui H, Sugiura T, Hayashi K, et al. Moxonidine prevents ischemia/reperfusion-induced renal injury in rats. Eur J Pharmacol. 2009;603:73–78. doi: 10.1016/j.ejphar.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 35.Solez K, D’Agostini RJ, Stawowy L, et al. Beneficial effect of propranolol in a histologically appropriate model of postischemic acute renal failure. The American journal of pathology. 1977;88:163–192. [PMC free article] [PubMed] [Google Scholar]
- 36.Brain SD, Williams TJ, Tippins JR, et al. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:54–56. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- 37.Girgis SI, Macdonald DW, Stevenson JC, et al. Calcitonin gene-related peptide: potent vasodilator and major product of calcitonin gene. Lancet. 1985;2:14–16. doi: 10.1016/s0140-6736(85)90059-5. [DOI] [PubMed] [Google Scholar]
- 38.Savill J, Mooney A, Hughes J. What role does apoptosis play in progression of renal disease? Curr Opin Nephrol Hypertens. 1996;5:369–374. doi: 10.1097/00041552-199607000-00014. [DOI] [PubMed] [Google Scholar]
- 39.Padanilam BJ. Cell death induced by acute renal injury: a perspective on the contributions of apoptosis and necrosis. Am J Physiol Renal Physiol. 2003;284:F608–627. doi: 10.1152/ajprenal.00284.2002. [DOI] [PubMed] [Google Scholar]
- 40.Ricardo SD, Ding G, Eufemio M, et al. Antioxidant expression in experimental hydronephrosis: role of mechanical stretch and growth factors. Am J Physiol. 1997;272:F789–798. doi: 10.1152/ajprenal.1997.272.6.F789. [DOI] [PubMed] [Google Scholar]
- 41.Sunami R, Sugiyama H, Wang D-H, et al. Acatalasemia sensitizes renal tubular epithelial cells to apoptosis and exacerbates renal fibrosis after unilateral ureteral obstruction. American Journal of Physiology - Renal Physiology. 2004;286:F1030–F1038. doi: 10.1152/ajprenal.00266.2003. [DOI] [PubMed] [Google Scholar]
- 42.Kawada N, Moriyama T, Ando A, et al. Increased oxidative stress in mouse kidneys with unilateral ureteral obstruction. Kidney Int. 1999;56:1004–1013. doi: 10.1046/j.1523-1755.1999.00612.x. [DOI] [PubMed] [Google Scholar]
- 43.Anders HJ, Ryu M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011;80:915–925. doi: 10.1038/ki.2011.217. [DOI] [PubMed] [Google Scholar]
- 44.Chow FY, Nikolic-Paterson DJ, Ozols E, et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 45.Ko GJ, Boo CS, Jo SK, et al. Macrophages contribute to the development of renal fibrosis following ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant. 2008;23:842–852. doi: 10.1093/ndt/gfm694. [DOI] [PubMed] [Google Scholar]
- 46.Pardo A, Barrios R, Gaxiola M, et al. Increase of lung neutrophils in hypersensitivity pneumonitis is associated with lung fibrosis. American journal of respiratory and critical care medicine. 2000;161:1698–1704. doi: 10.1164/ajrccm.161.5.9907065. [DOI] [PubMed] [Google Scholar]
- 47.Lauriat S, Linas SL. The role of neutrophils in acute renal failure. Seminars in nephrology. 1998;18:498–504. [PubMed] [Google Scholar]
- 48.Yaraee R, Ebtekar M, Ahmadiani A, et al. Neuropeptides (SP and CGRP) augment pro-inflammatory cytokine production in HSV-infected macrophages. International immunopharmacology. 2003;3:1883–1887. doi: 10.1016/S1567-5769(03)00201-7. [DOI] [PubMed] [Google Scholar]
- 49.Spengler RN, Allen RM, Remick DG, et al. Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol. 1990;145:1430–1434. [PubMed] [Google Scholar]
- 50.Ortega E, Marchena JM, Garcia JJ, et al. Norepinephrine as mediator in the stimulation of phagocytosis induced by moderate exercise. European journal of applied physiology. 2005;93:714–718. doi: 10.1007/s00421-004-1245-8. [DOI] [PubMed] [Google Scholar]
- 51.Richter J, Andersson R, Edvinsson L, et al. Calcitonin gene-related peptide (CGRP) activates human neutrophils--inhibition by chemotactic peptide antagonist BOC-MLP. Immunology. 1992;77:416–421. [PMC free article] [PubMed] [Google Scholar]
- 52.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. The Journal of clinical investigation. 2008;118:3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hering D, Mahfoud F, Walton AS, et al. Renal denervation in moderate to severe CKD. J Am Soc Nephrol. 2012;23:1250–1257. doi: 10.1681/ASN.2011111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.