

Abstract
Eclipta prostrata L. is one of the Chinese medicinal tonics which are usually used for treating loose teeth, dizziness, tinnitus, hemoptysis, hematuria, and uterine bleeding. However, quality control of this herbal medicine has been not satisfactory. This study reported its qualitative and quantitative analyses based on LC/MS method. UHPLC-DAD-Q-TOF-MS fingerprinting and MS fragmentation cleavage pathway were investigated for qualitative analysis. Furthermore, a method for simultaneous quantitative determination of nine compounds, luteolin 7-O-β-D-glucopyranoside, ecliptasaponin C, luteolin, eclalbasaponin IV, apigenin, ecliptasaponin A, echinocystic acid 28-O-β-D-glucopyranoside, echinocystic acid, and 3-oxo-16α-hydroxy-olean-12-en-28-oic acid in E. prostrata, was established. The method was validated for samples of E. prostrata from different habitats. The results showed good linear correlation, precision, accuracy, and repeatability that could be used for contents determination of the nine compounds in E. prostrata from different habitats.
1. Introduction
Eclipta prostrata L. (Compositae) is one of the Chinese medicinal tonics, widely distributed in the tropical and subtropical regions of the world. It has been used for the treatment of loose teeth, dizziness, tinnitus, hemoptysis, hematuria, and uterine bleeding [1]. Modern pharmacological research has confirmed its biological effects such as in antiosteoporosis [2], anti-inflammatory [3], antihyperlipemia [4, 5], and antitumor [6] activities.
Literature on its phytochemical constituents and our previous study showed that it contained triterpenoid saponins, flavonoids, thiophenes, and steroids [6–10]. Several analytical methods including HPLC, UPLC, LC/MS, and GC-MS have been used for quality control analyses of E. prostrata [11–15]; however, the standards which those methods used were finite (one or two flavonoids which could be obtained on the market). Furthermore, analytical time of the reported methods was relatively long in order to obtain ideal resolution. Therefore, we intended to establish a method which is based on our previous phytochemical study to determine main constituents of E. prostrata. Although there was a report on qualitative analysis of E. prostrata through UHPLC-Q-TOF/MS [12], the results lacked to deduce fragmentation pathway since they did not contain MS/MS data.
In this paper, we demonstrated how modern analytical methods could be used for quality control on natural product medicine. We initially analyzed a UHPLC-DAD-Q-TOF-MS fingerprint for rapid profiling of chemical constituents, and eighteen compounds in the extract of E. prostrata were identified or tentatively characterized. A rapid LC-QQQ-MS method was later validated for simultaneous determination of nine major compounds in E. prostrata. The results showed good linear correlation, precision, accuracy, and repeatability that could be used for quality control analysis of E. prostrata from different habitats.
2. Experimental
2.1. Reference Substances, Reagents, and Plant Materials
The references (1, 2, 5–9, 11, 13, 14, and 16–18) (Figure 1) were isolated from the aerial part of E. prostrata, and the structures elucidated based on 1H-NMR and 13C-NMR spectral analyses as previously reported by [10]. The purities of these reference compounds were determined to be above 98% by normalization of the peak areas detected by HPLC-ELSD (Alltech Grace Evaporative Light Scattering Detector 3300 with the following acquisition parameters: temp: 40°C; gas flow: 1.8 L/min; gain: 10).
Figure 1.

The structures of compounds 1–18 identified from Eclipta prostrata L.
HPLC grade acetonitrile and methanol were acquired from Fisher Chemicals (Pittsburg, USA). Formic acid (HPLC grade) was obtained from Tedia, USA. The other chemicals and reagents used were of analytical grade and purchased from Tianjin Concord Technology Company (Tianjin, China). Purified water was obtained using a Milli-Q system (Millipore, USA).
Thirteen dried samples (S1–S13) from the aerial part of E. prostrata were collected from different habitats and identified by Professor Lijuan Zhang (College of Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, China). Voucher specimens (20120903) deposited in our laboratory.
2.2. Preparation of the Reference and Sample Solutions
A concentration of 1 mg/mL of the reference compounds 1, 2, 5–9, 11, 13, 14, and 16–18 was prepared using 50% methanol (v/v) for UHPLC-Q-TOF-MS analysis.
For LC-QQQ-MS analysis, nine of the reference compounds were accurately weighed, put into 10 mL volumetric flasks separately, and dissolved in 50% methanol (v/v) to make reference stock solution 1. Their respective concentrations were as follows: luteolin 7-O-β-D-glucoside (18.15 mg/mL), ecliptasaponin C (47.05 mg/mL), luteolin (4.64 mg/mL), eclalbasaponin IV (11.85 mg/mL), apigenin (1.56 mg/mL), ecliptasaponin A (21.50 mg/mL), echinocystic acid 28-O-β-D-glucoside (2.60 mg/mL), echinocystic acid (8.45 mg/mL), and 3-oxo-16α-hydroxy-olean-12-en-28-oic acid (6.42 mg/mL). 1 mL of the stock solution 1 was then further diluted with 50% methanol (v/v) to 10 mL to obtain reference stock solution 2. All the solutions were stored at 4°C and brought to room temperature before use. Calibrated reference working solutions were freshly prepared by appropriate dilution of the mixed stock solution 2, giving final concentration in the range of 0.028–20.17 μg/mL for 1, 0.070–52.28 μg/mL for 11, 0.012–8.59 μg/mL for 5, 0.060–43.89 μg/mL for 13, 0.008–5.78 μg/mL for 8, 0.033–23.89 μg/mL for 14, 0.013–9.63 μg/mL for 16, 0.013–9.39 μg/mL for 17, and 0.010–7.13 μg/mL for 18.
The aerial part of E. prostrata was crushed into powder and 1.00 g weighed into a 50 mL flask with 25 mL 50% methanol (v/v). It was extracted in an ultrasonic bath at room temperature for 30 min. The extract was filtered. 1 mL of the filtrate was obtained and diluted to the mark in a 10 mL volumetric flask. It was then centrifuged at 13171 ×g for 10 min for LC/MS analysis.
2.3. UHPLC-DAD-Q-TOF-MS Fingerprint
UHPLC-DAD-Q-TOF-MS fingerprint analysis was performed on an Agilent 1290 UHPLC consisting of a binary pump, a diode-array detector, an autosampler, and a column thermostat connected to an Agilent 6520 Q-TOF spectrometry system via an ESI interface (Agilent Corp., Santa Clara, CA, USA).
The peaks were separated on an ACQUITY UPLC HSS T3 column (100 mm × 2.1 mm, 1.8 μm, Waters, USA) using a mobile phase consisting of acetonitrile (A) and water (containing 0.1% HCOOH; B). The following gradient elution system was used: 0–15 min, 10–30% A; 15–28 min, 30% A; 28–30 min, 30–40% A; 30–38 min, 40% A; 38–45 min, 40–100% A; 45–50 min, 100–100% A; 50–50.5 min, 10% A; 50.5–60 min, 10% A. An injection volume of 5 μL and a flow rate of 0.3 mL/min were used, with the column temperature set at 30°C.
Full-scan analyses in negative ionization modes were conducted and the spectra were recorded in the range of m/z 100–1700. Liquid nitrogen was used as the nebulizer and drying gas. High purity nitrogen was used as the collision gas. The major parameters used were as follows: drying gas with a flow rate of 8.0 L/min, drying gas temperature 350°C, nebulizer 30 psig, capillary voltage 3500 V, fragmentor voltage 175 V, and collision energy 35, 70 V.
2.4. Simultaneous Quantification System
The mass spectrometer was an Agilent 6430 triple Quad MS (QQQ) coupled with Agilent 1200 HPLC, consisting of a binary pump, an online vacuum degasser, an autosampler, and a column oven, through an electrospray ionization (ESI) source (Agilent Corp., Santa Clara, CA, USA). All separations were carried out on an Agilent eclipse XDB-C18 column (150 mm × 4.6 mm, 5.0 μm, Agilent, USA) and the column temperature was set at 35°C. The mobile phase was composed of acetonitrile (A)-water (containing 0.1% HCOOH; B) at a gradient elution of 0–5 min, 30–100% A; 5–10 min, 100% A; 10-11 min, 100–30% A; 11–20 min, 30% A, with a flow rate of 0.5 mL/min, and 1 μL injection volume.
The ESI source parameter of drying gas was set at the flow rate of 8.0 L/min, temperature of 350°C, 45 psig nebulizer, and 4000 V capillary voltage. The MS/MS analysis was conducted on multiple reactions monitoring (MRM) mode; the MRM parameters were shown in Table 1.
Table 1.
MRM parameters for quantitative analysis.
| Compounds | Ion pairs (m/z) | Fragmentor (V) | CE (V) | Dwell time (ms) |
|---|---|---|---|---|
| 1 | 447.1 → 285.0 | 240 | 26 | 100 |
| 11 | 841.5 → 633.4 | 210 | 30 | 100 |
| 5 | 285.0 → 133.1 | 170 | 30 | 100 |
| 13 | 795.5 → 633.4 | 310 | 40 | 100 |
| 8 | 269.0 → 117.1 | 150 | 34 | 100 |
| 14 | 633.4 → 587.4 | 270 | 38 | 100 |
| 16 | 679.4 → 471.3 | 150 | 22 | 100 |
| 17 | 471.3 → 407.3 | 230 | 38 | 100 |
| 18 | 469.3 → 407.3 | 220 | 34 | 100 |
2.5. Method Validation for Simultaneous Quantification
Seven serial working solutions were prepared as described above and injected into the LC/MS system. Calibration curves were plotted based on linear regression analysis of the integrated peak areas (Y) versus concentrations (X, μg/mL). Each solution was tested in triplicate, with limits of detection (LOD) and quantification (LOQ) for each analyte defined at signal-to-noise ratios (S/N) of 3 and 10, respectively. Intraday and interday precision for each analyte at a specific concentration were performed by six replicates on the same day (intraday) and on three consecutive days (interday). Recovery tests were performed by spiking reference standards into appropriately weighed sample 9. Six different samples were spiked with the reference standards, extracted, and prepared as described above. Three replicates were performed for each analysis. To confirm the repeatability, six replicates of the same sample were extracted and analyzed. Variations were expressed in terms of relative standard deviation (RSD) in all the tests.
3. Results and Discussion
3.1. Optimization of Extraction Procedure
In order to obtain satisfactory extraction efficiency, several extraction solvents including water, 50% methanol (v/v), and methanol were examined. The 50% methanol (v/v) solvent was chosen and used as the extraction solvent due to its high yield of target compounds. Reflux and ultrasonic extraction methods were similarly effective in the extraction of the target analytes. The ultrasonic extraction method was ultimately chosen because of its flexibility. Different extraction times (30, 45, and 60 min) were compared, which showed similar percent yields and as such 30 minutes was chosen as the ideal extraction time.
3.2. Optimization of LC-QQQ-MS Chromatographic Conditions
Chromatographic conditions of the mobile phase and gradient elution system were optimized in this study. In order to achieve good resolution and symmetric peak shapes of the nine reference compounds, we chose methanol-water (with and without acid) and acetonitrile-water (with and without acid) to optimize the mobile phase. We also optimized the column temperature from 25 to 45°C with 5°C in one step. Finally, acetonitrile-water (with 0.1% formic acid) and column temperature of 35°C were chosen which showed good resolution of adjacent peaks within a short time.
3.3. UHPLC-Q-TOF-MS Fingerprint Analysis
Figure 2 shows the total ion current (TIC) chromatogram of the extract from E. prostrata (S9). A total of 18 compounds were identified and 13 of them confirmed by comparing their MS features and retention times with those of reference compounds. According to the structural characteristics, the 18 compounds can be grouped into three types, namely, flavonoids, triterpenoids, and other types.
Figure 2.
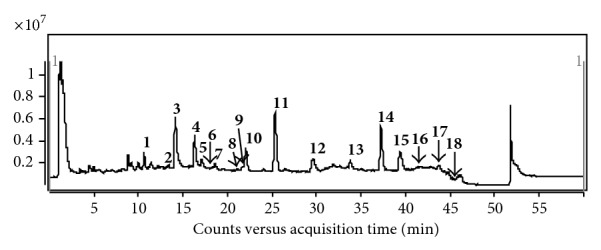
UHPLC-Q-TOF fingerprint of extract of Eclipta prostrata L.
3.3.1. Identification of Flavonoids
Seven flavonoids including two isoflavonoids were unambiguously or tentatively identified according to the UV spectra and MS fragmentation pathway. Compounds 1 and 7 showed [M–H]− at m/z 447.0949 and 461.1094, respectively (Table 2). Similar diagnostic fragment Y0 ions were observed in their MS/MS spectra (Figure 3), suggesting these compounds are O-glycosyl flavonoids. Based on the UV, MS fragmentation pathway, and retention times, compounds 1 and 7 were identified as luteolin-7-O-β-D-glucoside and 7-O-methylorobol-4′-O-β-D-glucoside, respectively.
Table 2.
Characterization of compounds from E. prostrata by UHPLC-DAD-Q-TOF.
| Number | RT (min) | UV λ max (nm) | Formula | m/z (experimental) | Error (ppm) | Product ions (m/z, relative abundance, diagnostic ions) | Structural elucidation |
|---|---|---|---|---|---|---|---|
| 1 | 10.93 | 228, 254, 350 | C21H20O11 | 447.0949 [M–H]− | −3.66 | 285.0381 (100) [Y0] | Luteolin-7-O-β-D-glucoside |
|
| |||||||
| 2 | 14.13 | 230, 289, 330 | C9H10O4 | 181.0514 [M–H]− | −4.30 | 108.0256 (100) [M-CO2-C2H5] | 3,4-Dihydroxy-benzoic acid ethyl ester |
|
| |||||||
| 3 | 14.53 | 226, 254, 350 | C15H10O9S | 364.9990 [M–H]− | −4.80 | 285.0385 (100) [Y0] | Luteolin sulfate |
|
| |||||||
| 4 | 16.70 | 228, 268, 340 | C15H10O8S | 349.0034 [M–H]− | −3.08 | 269.0434 (100) [Y0] | Apigenin sulfate |
|
| |||||||
| 5 | 17.45 | 246, 350 | C15H10O6 | 285.0409 [M–H]− | −1.48 | 175.0378 (10) [0,4B-2H] | Luteolin |
| 133.0277 (100) [1,3B] | |||||||
| 107.0128 (20) [0,4A] | |||||||
|
| |||||||
| 6 | 18.31 | 228, 260 | C16H10O7 | 313.0363 [M–H]− | −3.04 | 298.0112 (100) [M-CH3] | Wedelolactone |
| 270.0158 (20) [M-CH3-CO] | |||||||
| 242.0212 (10) [M-CH3-2CO] | |||||||
|
| |||||||
| 7 | 18.81 | 245 | C22H22O11 | 461.1094 [M–H]− | −0.92 | 299.0573 (100) [Y0] | 7-O-Methylorobol-4′-O-β-D-glucoside |
| 284.0336 (80) [Y0-CH3] | |||||||
|
| |||||||
| 8 | 21.61 | 228, 267, 340 | C15H10O5 | 269.0451 [M–H]− | 1.69 | 159.0431 (30) [0,4B-2H] | Apigenin |
| 117.0332 (100) [1,3B] 107.0131 (24) [0,4A] | |||||||
|
| |||||||
| 9 | 22.32 | 270, 328, 350 | C16H12O6 | 299.0556 [M–H]− | 1.77 | 284.0330 (100) [M-CH3] | 3′-Hydroxybiochanin A |
| 256.0372 (40) [M-CH3-CO] | |||||||
| 255.0303 (50) [M-CH2-CO-H2] | |||||||
| 239.0350 (18) [M-CH4O-CO] | |||||||
| 227.0353 (67) [M-CH2-2CO-H2] | |||||||
| 211.0402 (29) [M-CH4O-2CO] | |||||||
|
| |||||||
| 10 | 22.32 | 210 | C48H78O19 | 957.5055
[M–H]−
(CE 35) |
0.91 | 837.4584 (4) [ 0,2X0β] | Eclalbasaponin III |
| 795.4470 (100) [Y0β] | |||||||
| 733.4425 (1) [Y0β-H2O-CO2] | |||||||
| 633.3962 (1) [Y0β-glc] | |||||||
| 957.5055
[M–H]−
(CE 70) |
0.91 | 795.4489 (50) [Y0β] | |||||
| 733.4421 (4) [Y0β-H2O-CO2] | |||||||
| 633.3951 (22) [Y0β-glc] | |||||||
| 471.3486 (10) [Y0β-2glc] | |||||||
| 161.0449 (31) [B1α] | |||||||
| 113.0237 (62) [B1α-CH2O-H2O] | |||||||
| 101.0243 (100) [M-0,2X1α-H2O] | |||||||
| 1003.5110 [M + HCOO]−
(CE 35) |
1.10 | 837.4583 (6) [ 0,2X0β] | |||||
| 795.4481 (100) [Y0β] | |||||||
| 733.4424 (1) [Y0β-H2O-CO2] | |||||||
| 633.3934 (1) [Y0β-glc] | |||||||
| 1003.5110 [M + HCOO]−
(CE 70) |
1.10 | 795.4496 (100) [Y0β] | |||||
| 733.4477 (8) [Y0β-H2O-CO2] | |||||||
| 633.3968 (32) [Y0β-glc] | |||||||
| 615.3851 (6) [Y0β-glc-H2O] | |||||||
| 571.3985 (5) [Y0β-glc-H2O-CO2] | |||||||
| 471.3447 (10) [Y0β-2glc] | |||||||
| 161.0444 (36) [B1α] | |||||||
| 113.0239 (56) [B1α-CH2O-H2O] | |||||||
| 101.0240 (89) [M- 0,2X1α-H2O] | |||||||
|
| |||||||
| 11 | 25.75 | 210 | C42H68O14 | 841.4590 [M + HCOO]−
(CE 35) |
0.19 | 675.4077 (4) [ 0,2X0β] | Ecliptasaponin C |
| 633.3986 (100) [Y0β] | |||||||
| 841.4590 [M + HCOO]−
(CE 70) |
0.19 | 633.3990 (54) [Y0β] | |||||
| 587.3935 (13) [Y0β-HCOOH] | |||||||
| 407.3315 (1) [Y0α-HCOOH-H2O] | |||||||
| 161.0448 (10) [B0α] | |||||||
| 113.0243 (43) [B0α-CH2O-H2O] | |||||||
| 101.0246 (100) [M- 0,2X0α-H2O] | |||||||
|
| |||||||
| 12 | 30.27 | 210 | C42H68O17S | 875.4116 [M–H]−
(CE 35) |
−1.28 | 875.4062 (100) [M–H] | Eclalbasaponin VI |
| 713.3533 (3) [Y0β] | |||||||
| 875.4116 [M–H]−
(CE 70) |
−1.28 | 875.4063 (16) [M–H] | |||||
| 713.3551 (100) [Y0β] | |||||||
| 651.3543 (2) [Y0β-H2O-CO2] | |||||||
| 241.0013 (21) [B0α] | |||||||
|
| |||||||
| 13 | 34.04 | 210 | C42H68O14 | 795.4550 [M–H]−
(CE 35) |
−1.77 | 795.4491 (100) [M–H] | Eclalbasaponin IV |
| 733.4492 (4) [M–H-H2O-CO2] | |||||||
| 633.3971 (16) [Y1α] | |||||||
| 471.3453 (3) [Y0α] | |||||||
| 795.4550 [M–H]−
(CE 70) |
−1.77 | 795.4509 (5) [M–H] | |||||
| 633.4065 (4) [Y1α] | |||||||
| 615.3855 (3) [Y1α-H2O] | |||||||
| 471.3446 (16) [Y0α] | |||||||
| 407.3301 (31) [Y0α-HCOOH-H2O] | |||||||
| 161.0431 (10) [B1α] | |||||||
| 113.0234 (57) [B1α-CH2O-H2O] | |||||||
| 101.0237 (100) [M- 0,2X1α-H2O] | |||||||
| 841.4605 [M + HCOO]−
(CE 35) |
−1.79 | 795.4495 (100) [M–H] | |||||
| 733.4480 (2) [M–H-H2O-CO2] | |||||||
| 633.3971 (5) [Y1α] | |||||||
| 471.3432 (1) [Y0α] | |||||||
| 841.4605 [M + HCOO]−
(CE 70) |
−1.79 | 795.4476 (7) [M–H] | |||||
| 633.3989 (12) [Y1α] | |||||||
| 615.3834 (2) [Y1α-H2O] | |||||||
| 471.3439 (20) [Y0α] | |||||||
| 407.3295 (18) [Y0α-HCOOH-H2O] | |||||||
| 161.0450 (11) [B1α] | |||||||
| 113.0236 (54) [B1α-CH2O-H2O] | |||||||
| 101.0241 (100) [M- 0,2X1α-H2O] | |||||||
|
| |||||||
| 14 | 37.60 | 210 | C36H58O9 | 633.4023
[M–H]−
(CE 35) |
−0.19 | 633.3985 (100) [M–H] | Eclalbasaponin A |
| 587.3922 (4) [M–H-HCOOH] | |||||||
| 571.3966 (1) [M–H-H2O-CO2] | |||||||
| 633.4023
[M–H]−
(CE 70) |
−0.19 | 587.3916 (3) [M–H-HCOOH] | |||||
| 585.3706 (5) [M–H-HCOOH-H2] | |||||||
| 407.3281 (12) [Y0α-HCOOH-H2O] | |||||||
| 161.0434 (1) [B0α] | |||||||
| 113.0235 (32) [B0α-CH2O-H2O] | |||||||
| 101.0241 (100) [M- 0,2X0α-H2O] | |||||||
|
| |||||||
| 15 | 36.79 | 210 | C36H58O12S | 713.3582 [M–H]−
(CE 35) |
−0.77 | 713.3545 (100) [M–H] | Eclalbasaponin V |
| 713.3582
[M–H]−
(CE 70) |
−0.77 | 713.3544 (62) [M–H] | |||||
| 683.3073 (17) [M–H-CO-2H] | |||||||
| 651.3541 (10) [M–H-H2O-CO2] | |||||||
| 471.3464 (1) [Y0α] | |||||||
| 407.3306 (3) [Y0α-HCOOH-H2O] | |||||||
| 241.0007 (100) [B0α] | |||||||
| 113.0234 (7) [B0α-CH2O-H2O] | |||||||
| 101.0241 (10) [M- 0,2X0α-H2O] | |||||||
|
| |||||||
| 16 | 41.86 | 210 | C36H58O9 | 679.4053 [M + HCOO]−
(CE 35) |
1.59 | 471.3430 (100) [Y0α] | Echinocystic acid 28-O-β-D-glucopyranoside |
| 407.3280 (3) [Y0α-HCOOH-H2O] | |||||||
| 679.4053 [M + HCOO]−
(CE 70) |
1.59 | 471.3393 (18) [Y0α] | |||||
| 425.3376 (11) [Y0α-HCOOH] | |||||||
| 407.3301 (100) [Y0α-HCOOH-H2O] | |||||||
| 391.2945 (12) [Y0α-CO2-2H2O] | |||||||
|
| |||||||
| 17 | 44.61 | 210 | C30H48O4 | 471.3470
[M–H]−
(CE 35) |
2.06 | 471.3439 (84) [M–H] | Echinocystic acid |
| 407.3281 (100) [M–H-HCOOH-H2O] | |||||||
| 425.3407 (10) [M–H-HCOOH] | |||||||
|
| |||||||
| 18 | 44.75 | 210 | C30H46O4 | 469.3311
[M–H]−
(CE 35) |
2.51 | 469.3283 (9) [M–H] | 3-Oxo-16α-hydroxy-olean-12-en-28-oic acid |
| 423.3222 (1) [M–H-HCOOH] | |||||||
| 407.3296 (100) [M–H-HCOOH-H2O] | |||||||
Figure 3.

MS/MS spectra of flavonoids. ((a) compound 1; (b) compound 3; (c) compound 4; (d) compound 5; (e) compound 8; and (f) compound 7).
Compounds 3 and 4 produced [M–H]− at m/z 364.9990 and 349.0034 which correspond to C15H10O9S and C15H10O8S, respectively. In MS/MS spectra, Y0 ions at m/z 285.0385 and 269.0434 were obtained. According to UV, MS, and literature [15], 3 and 4 were tentatively identified as luteolin sulfate and apigenin sulfate.
Three flavone aglycones were identified corresponding to the diagnostic fragment ions 1,3B, 0,4A, and 0,4B-2H. Finally, Compounds 5, 8, and 9 were unambiguously identified as luteolin, apigenin, and 3′-hydroxybiochanin A by comparing their retention times, MS/MS fragment data, and UV spectra with reference compounds.
3.3.2. Identification of Triterpenoids
The triterpenoids were the major components isolated from E. prostrata and compounds 10–16 were identified as triterpenoid saponins. In order to assist in structural elucidation, compound 10 was chosen as an example to elucidate the nomenclature (Figure 4). The ions retaining the charges on the main core structures were termed Y representing glycosidic cleavages and X for cross-ring cleavages [16]. Cross ring cleavage ions were designated by superscript numbers indicating cleavage of the two bonds. The oligosaccharide chain at C-3 was defined as theα-chain whereas the one at C-28 was the β-chain.
Figure 4.
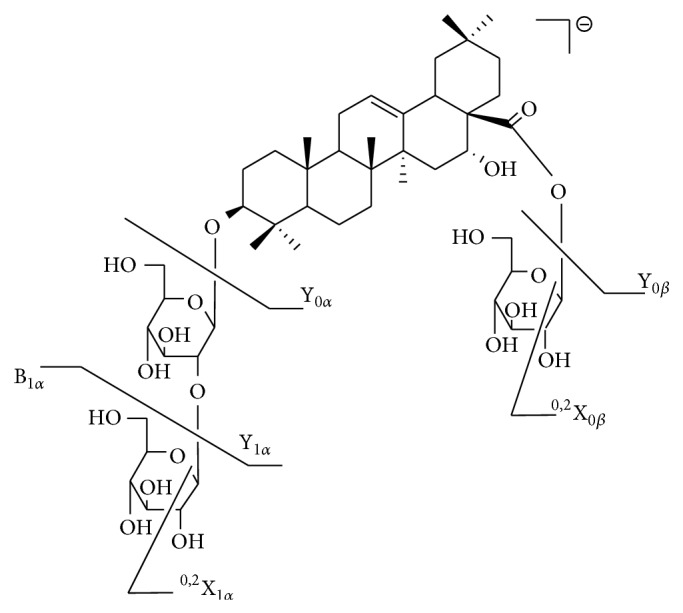
The fragmentation nomenclature of compound 10.
Since the molecular masses of saponins were usually large, we used two collision energies (CE) to elucidate the structures. Compound 13 showed m/z 795.4550 [M–H]− and 841.4605 [M+HCOO]− which correspond to C42H68O14 in MS spectrum (Figure 5(b)). As shown in Figures 5 (c, c′, d, d′), the MS/MS spectra of [M–H]− and [M+HCOO]− of 13 were significantly different due to different CE. A collision energy of 35 V produced a base peak of [M–H]− (m/z 795.4491) whereas that of 70 V produced M-0,2X1α-H2O (m/z 101.0237). As shown in Figure 5(c′), diagnostic fragment ions such as Y0α, Y1α, Y1α-H2O, Y0α-HCOOH, Y0α-HCOOH-H2O, B1α, B1α-CH2O-H2O, and M-0,2X1α-H2O were clearly observed. Figure 6 shows the elucidation of the detailed fragment cleavage pathway and similar fragmentation pathways were observed in the MS/MS spectrum of [M+HCOO]− (Figure 5(d′)). Through UV, MS fragmentation pathway, and retention times, compound 13 was unambiguously identified as eclalbasaponin IV.
Figure 5.

MS and MS/MS spectra of compounds 10, 11, and 13. ((a) MS spectrum of 11; (b) MS spectrum of 13; (c) MS/MS spectrum of [M–H]− of 13, CE 35 V; (c′) MS/MS spectrum of [M–H]− of 13, CE 70 V; (d) MS/MS spectrum of [M+HCOO]− of 13, CE 35 V; (d′) MS/MS spectrum of [M+HCOO]− of 13, CE 70 V; (e) MS/MS spectrum of [M–H]− of 10, CE 35 V; (e′) MS/MS spectrum of [M–H]− of 10, CE 70 V; (f) MS/MS spectrum of [M+HCOO]− of 11, CE 35 V; (f′) MS/MS spectrum of [M+HCOO]− of 11, CE 70 V; (g) MS/MS spectrum of [M–H]− of 12, CE 35 V; (g′) MS/MS spectrum of [M–H]− of 12, CE 70 V).
Figure 6.

Fragmentation pattern of compound 13.
Compound 11 was the isomer of 13. However only [M+HCOO]− ion (m/z 841.4590) was observed in its MS spectrum (Figure 5(a)). In the MS/MS spectra (CE 70 V), similar diagnostic fragment ions (Y0β, Y0β-HCOOH, Y0α-HCOOH-H2O, B0α, B0α-CH2O-H2O, and M-0,2X0α-H2O) were observed. Compound 11 was subsequently identified as ecliptasaponin C.
Compound 10 produced [M–H]− and [M+HCOO]− ions at m/z 957.5055 and 1003.5110 (corresponding to C48H78O19), which were 162 Da higher than that of compound 13. From the MS/MS fragment ions, UV spectrum, and literatures [9, 15], compound 10 was tentatively identified as eclalbasaponin III. Based on the UV, MS fragmentation pathway, and retention times, compounds 14 and 16 were unambiguously identified as isomers, namely, eclalbasaponin A and echinocystic acid 28-O-β-D-glucopyranoside, respectively.
Compound 15 showed [M–H]− ion at m/z 713.3582 (corresponding to C36H58O12S) which is 80 Da higher than that of 14 (m/z 633.4023). From the MS spectrum, we could deduce that compound 15 was the sulphate of compound 14. An m/z of 241.0007 was a significant diagnostic ion which indicated the location of SO3 on the glucoseα-chain. In the MS/MS spectra of [M–H]− (CE 70 V), diagnostic fragment ions, such as M-H-CO-2H, M-H-H2O-CO2, Y0α, Y0α-HCOOH-H2O, B0α, B0α-CH2O-H2O, and M-0,2X0α-H2O, were observed. According to the UV, MS fragmentation pathway, and literature [7], compound 15 wasidentified as eclalbasaponin V tentatively and compound 12 was 162 Da higher than 15. From the MS/MS fragmentation pathway (Table 2 and Figure 5(g′)), the 162 Da could be deduced as a β-chain linked glucose. Finally, compound 12 was tentatively identified as eclalbasaponin VI [15].
Two aglycones were also identified according to their UV, MS fragmentation pathway, and retention times. Compounds 17 and 18 were identified as echinocystic acid and 3-oxo-16α-hydroxy-olean-12-en-28-oic acid, respectively.
3.3.3. Identification of Other Compounds
Two compounds (2 and 6) were unambiguously identified as 3,4-dihydroxy-benzoic acid ethyl ester and wedelolactone, respectively, by comparing their UV, exact molecular masses, MS/MS spectra (Table 2), and retention times.
3.4. Validation of Quantitative Method
3.4.1. Linearity
A series of standard solutions with seven different concentrations were analyzed by an established method in triplicate. Every calibration curve was plotted based on linear regression analysis of the integrated peak areas (Y) versus concentrations (X, μg/mL) as listed in Table 3. Calibration curves were linear with correlation coefficients (R 2) above 0.9990 for all analytes.
Table 3.
Calibration curves, LOD, and LOQ of nine compounds.
| Compounds | Calibration curves | R 2 | Linear range (μg/mL) | LOD (ng/mL) | LOQ (ng/mL) |
|---|---|---|---|---|---|
| 1 | Y = 8272X + 203 | 0.9996 | 0.028–20.17 | 3.37 | 11.22 |
| 11 | Y = 6347.7X + 4182.9 | 0.9992 | 0.070–52.28 | 1.10 | 3.68 |
| 5 | Y = 3350X + 4.63 | 0.9998 | 0.012–8.59 | 1.77 | 5.90 |
| 13 | Y = 1190X + 462.4 | 0.9993 | 0.060–43.89 | 4.41 | 14.71 |
| 8 | Y = 5271X + 245.1 | 0.9996 | 0.008–5.78 | 1.94 | 6.47 |
| 14 | Y = 822.7X + 261.7 | 0.9990 | 0.033–23.89 | 2.24 | 7.47 |
| 16 | Y = 27335.1X − 318.5 | 0.9996 | 0.013–9.63 | 1.36 | 4.54 |
| 17 | Y = 6640.4X + 865.8 | 0.9994 | 0.013–9.39 | 2.00 | 6.69 |
| 18 | Y = 2577.4X + 393.6 | 0.9991 | 0.010–7.13 | 2.47 | 8.23 |
3.4.2. LOD and LOQ
The stock solutions containing nine reference compounds were diluted to a series of appropriate concentrations, using 50% methanol (v/v), and injected into LC/MS for analysis. The LOD and LOQ under the chromatographic conditions were determined at approximate signal-noise (S/N) ratios of 3 and 10, respectively. The results were given in Table 3.
3.4.3. Precision and Repeatability
Intra- and interday precisions were performed by repetitive injections on the same day (intraday) for a total of six injections and on three consecutive days (interday). RSD values for both intra- and interday precision were below 2.5% (Table 4).
Table 4.
Precision, recovery, and repeatability of nine compounds.
| Compounds | Precision (RSD, %, n = 6) | Recovery (%, n = 6) | Repeatability (%) (RSD, n = 6) | ||
|---|---|---|---|---|---|
| Intraday | Interday | Mean | RSD | ||
| 1 | 1.00 | 1.16 | 97.21 | 2.39 | 1.75 |
| 11 | 0.66 | 1.98 | 95.41 | 2.63 | 1.12 |
| 5 | 1.56 | 1.60 | 97.82 | 3.99 | 2.99 |
| 13 | 0.97 | 2.49 | 101.28 | 3.84 | 2.35 |
| 8 | 1.83 | 2.50 | 96.00 | 3.88 | 2.45 |
| 14 | 2.37 | 2.46 | 103.25 | 2.85 | 1.99 |
| 16 | 1.64 | 2.13 | 104.76 | 3.49 | 2.44 |
| 17 | 0.57 | 2.45 | 96.44 | 2.37 | 2.95 |
| 18 | 1.64 | 2.42 | 103.56 | 4.20 | 2.64 |
The analytic repeatability was examined by the injection of six different samples (S9), which were prepared with the same sample preparation procedure. The repeatability of the solution was less than 2.99% (Table 4).
3.4.4. Accuracy
The accuracy of the method was determined by spiking an appropriate amount of each crude E. prostrata extract sample (S9) with accurate amounts of the nine reference standards and each sample was analyzed in triplicate. The results showed good accuracy with average recovery from 95.41% to 104.76% for the compounds concerned with RSD < 4.20% (Table 4).
These results show that the LC/MS method was precise, accurate, and sensitive enough for simultaneously quantitative evaluation of the nine compounds from the aerial part of E. prostrata.
3.5. Application of Quantitative Method
The method can be used for simultaneous analysis of thirteen E. prostrata samples from different habitats of China (S1: Hunan; S2: Hebei; S3: Henan; S4: Henan; S5: Hebei; S6: unknown; S7: Jiangsu; S8: unknown; S9: Hebei; S10: unknown; S11: unknown; S12: Hebei; and S13: Anhui) (Figure 7). The saponins (11, 13, 14, and 16) were the major constituents among the nine compounds analysed and they showed relatively high variations (Table 5). Compound 11 was the most abundant among the nine compounds and was also recording high variations (426.16–13056.45 μg/g). This high degree of contents variability among the thirteen samples from different geographical locations could be due to various factors such as geographical source, climate, harvest time, and storage condition.
Figure 7.

Typical MRM chromatograms of mix standard (a) and sample (b).
Table 5.
Average contents of nine compounds in E. prostrata (mean ± SD, μg/g, n = 3).
| 1 | 11 | 5 | 13 | 8 | 14 | 16 | 17 | 18 | |
|---|---|---|---|---|---|---|---|---|---|
| S1 | 12.15 ± 0.06 | 8827.39 ± 29.38 | 7.29 ± 0.09 | 473.81 ± 4.18 | ND | 1438.43 ± 6.66 | 342.60 ± 3.45 | 342.60 ± 3.33 | 354.75 ± 3.84 |
| S2 | 724.00 ± 10.42 | 13056.45 ± 20.30 | 154.09 ± 1.91 | 1369.73 ± 5.54 | ND | 3032.97 ± 8.80 | 286.18 ± 3.34 | 454.95 ± 4.56 | 369.34 ± 4.96 |
| S3 | 2237.32 ± 14.13 | 3128.35 ± 15.19 | 350.57 ± 0.98 | 326.22 ± 3.23 | 8.82 ± 0.06 | 1188.04 ± 2.15 | 12.17 ± 0.09 | 17.04 ± 0.09 | ND |
| S4 | 1198.61 ± 10.25 | 2674.59 ± 12.14 | 210.50 ± 1.23 | 304.61 ± 2.02 | ND | 973.25 ± 2.23 | 64.39 ± 1.11 | 173.35 ± 1.10 | 118.87 ± 2.83 |
| S5 | 104.063 ± 1.21 | 426.16 ± 8.23 | 74.33 ± 0.46 | 411.30 ± 2.01 | ND | 1194.25 ± 4.56 | 14.87 ± 0.10 | 374.13 ± 3.23 | 421.21 ± 3.75 |
| S6 | 921.77 ± 6.48 | 2092.68 ± 13.15 | 201.79 ± 1.56 | 465.87 ± 4.09 | ND | 1542.10 ± 5.14 | 24.91 ± 0.07 | 146.99 ± 1.18 | 89.69 ± 1.56 |
| S7 | 27.241 ± 0.25 | 5757.80 ± 15.20 | 44.58 ± 0.20 | 928.68 ± 6.28 | ND | 2439.33 ± 5.85 | 136.21 ± 1.89 | 307.08 ± 3.33 | 222.88 ± 2.34 |
| S8 | 392.41 ± 0.86 | 10565.27 ± 40.59 | 193.72 ± 2.02 | 1204.55 ± 7.93 | ND | 2799.03 ± 7.19 | 139.08 ± 1.93 | 317.90 ± 3.44 | 206.14 ± 3.32 |
| S9 | 3748.13 ± 18.18 | 4099.054 ± 10.12 | 428.07 ± 2.88 | 380.79 ± 2.89 | 14.37 ± 0.10 | 1057.74 ± 6.34 | 17.42 ± 0.09 | 69.69 ± 0.69 | 7.47 ± 0.06 |
| S10 | 665.41 ± 2.23 | 3160.69 ± 10.89 | 151.46 ± 1.45 | 322.77 ± 2.26 | ND | 1104.88 ± 5.25 | 52.14 ± 0.21 | 216.01 ± 3.74 | 136.56 ± 1.54 |
| S11 | 556.11 ± 4.56 | 8980.05 ± 13.15 | 226.93 ± 2.77 | 1017.46 ± 4.87 | ND | 2496.26 ± 8.29 | 127.18 ± 1.10 | 192.02 ± 3.21 | 137.18 ± 1.34 |
| S12 | 698.32 ± 7.19 | 8260.18 ± 20.86 | 177.08 ± 1.23 | 905.33 ± 4.98 | ND | 2239.63 ± 9.10 | 67.34 ± 0.42 | 207.00 ± 2.34 | 114.73 ± 1.09 |
| S13 | 531.12 ± 4.42 | 7837.12 ± 19.38 | 269.30 ± 2.56 | 1269.20 ± 6.30 | 33.19 ± 0.22 | 2822.66 ± 14.14 | 119.69 ± 1.85 | 321.66 ± 5.02 | 182.03 ± 1.21 |
ND: not detected; S1: Hunan; S2: Hebei; S3: Henan; S4: Henan; S5: Hebei; S6: unknown; S7: Jiangsu; S8: unknown; S9: Hebei; S10: unknown; S11: unknown; S12: Hebei; S13: Anhui.
4. Conclusion
The two different LC/MS methods of analyses could be used for the rapid profiling and determination of major constituents from E. prostrata. The qualitative and quantitative methods could also be reliable tools for quality control analyses.
Acknowledgments
This work was supported by Program for New Century Excellent Talents in University (NCET-12-1069), Important Drug Development Fund, Ministry of Science and Technology of China (nos. 2012ZX09304007 and 2013ZX09401-004), National Natural Science Foundation of China (NSFC81125024), Research Fund for the Doctoral Program of Higher Education of China (20121210110007), and Tianjin Municipal Higher School Science and technology development fund program (20120203).
Conflict of Interests
The authors declare that there is no conflict of interests in the paper.
References
- 1.Pharmacopoeia Commission of People’s Republic of China. Pharmacopoeia of the People’s Republic of China. Vol. 1. Beijing, China: People Health Press; 2010. [Google Scholar]
- 2.Lee M. K., Ha N. R., Yang H., Sung S. H., Kim Y. C. Stimulatory constituents of Eclipta prostrata on mouse osteoblast differentiation. Phytotherapy Research. 2009;23(1):129–131. doi: 10.1002/ptr.2560. [DOI] [PubMed] [Google Scholar]
- 3.Tewtrakul S., Subhadhirasakul S., Tansakul P., Cheenpracha S., Karalai C. Antiinflammatory constituents from eclipta prostrata using RAW264.7 macrophage cells. Phytotherapy Research. 2011;25(9):1313–1316. doi: 10.1002/ptr.3383. [DOI] [PubMed] [Google Scholar]
- 4.Kim D.-I., Lee S.-H., Choi J.-H., Lillehoj H. S., Yu M.-H., Lee G.-S. The butanol fraction of Eclipta prostrata (Linn) effectively reduces serum lipid levels and improves antioxidant activities in CD rats. Nutrition Research. 2008;28(8):550–554. doi: 10.1016/j.nutres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Kumari C. S., Govindasamy S., Sukumar E. Lipid lowering activity of Eclipta prostrata in experimental hyperlipidemia. Journal of Ethnopharmacology. 2006;105(3):332–335. doi: 10.1016/j.jep.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 6.Liu Q.-M., Zhao H.-Y., Zhong X.-K., Jiang J.-G. Eclipta prostrata L. phytochemicals: isolation, structure elucidation, and their antitumor activity. Food and Chemical Toxicology. 2012;50(11):4016–4022. doi: 10.1016/j.fct.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Yahara S., Ding N., Nohara T. Oleanane glycosides from Eclipta alba. Chemical & Pharmaceutical Bulletin. 1994;42(6):1336–1338. doi: 10.1248/cpb.42.1336. [DOI] [Google Scholar]
- 8.Sun Z.-H., Zhang C.-F., Zhang M. A new benzoic acid derivative from Eclipta prostrata . Chinese Journal of Natural Medicines. 2010;8(4):244–246. doi: 10.3724/SP.J.1009.2010.00244. [DOI] [Google Scholar]
- 9.Zhao Y.-P., Tang H.-F., Jiang Y.-P., Wang Z.-Z., Yi Y.-H., Lei Q.-Y. Triterpenoid saponins from Eclipta prostrata L. Acta Pharmaceutica Sinica. 2001;36(9):660–663. [PubMed] [Google Scholar]
- 10.Han L. F., Zhao J., Zhang Y., Kojo A., Liu E. W., Wang T. Chemical constituents from Eeclipta prostrata . Chinese Herbal Medicines. 2013;5(4):313–316. [Google Scholar]
- 11.Ogunbinu A. O., Flamini G., Cioni P. L., Ogunwande I. A., Okeniyi S. O. Essential oil constituents of Eclipta prostrata (L.) L. and Vernonia amygdalina Delile. Natural Product Communications. 2009;4(3):421–424. [PubMed] [Google Scholar]
- 12.Xia A. J., Li L., Dong X., Lou Z. Y., Liang Y., Zhang Q. Studies on chemical constituents of Eclipta prostrata by UHPLC-Q-TOF/MS. Pharmaceutical Journal of Chinese People's Liberation Army. 2012;28(5):404–407. [Google Scholar]
- 13.Yang H. Y., Wang X. M., Du G., Wang J. P., Li G. J., Gao Y. T. HPLC determination of luteolin and apigenin in herba ecliptae. Lishizhen Medicine and Materia Medica Research. 2008;19(12):2994–2995. [Google Scholar]
- 14.Yuan H. X., Zhao Y. L., Wang X. Y., Tang Q., Gao X. X., Yu Z. G. Simultaneous determination of wedelolactone and isodemethylwedelolactone in Herba ecliptae by reversed-phase high performance liquid chromatography. Chinese journal of chromatography. 2007;25(3):371–373. [PubMed] [Google Scholar]
- 15.Lee K. Y., Ha N. R., Kim T. B., Kim Y. C., Sung S. H. Characterization of triterpenoids, flavonoids and phenolic acids in Eclipta prostrata by high-performance liquid chromatography/diode-array detector/electrospray ionization with multi-stage tandem mass spectroscopy. Natural Product Sciences. 2010;16(3):164–168. [Google Scholar]
- 16.Han L., Pan G., Wang Y., et al. Rapid profiling and identification of triterpenoid saponins in crude extracts from Albizia julibrissin Durazz. by ultra high-performance liquid chromatography coupled with electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Journal of Pharmaceutical and Biomedical Analysis. 2011;55(5):996–1009. doi: 10.1016/j.jpba.2011.04.002. [DOI] [PubMed] [Google Scholar]