

Abstract
Formaldehyde (FA), a major industrial chemical and ubiquitous environmental pollutant, has recently been classified by the International Agency for Research on Cancer as a human leukemogen. The major mode of action of FA is thought to be the formation of DNA-protein crosslinks (DPCs). Repair of DPCs may be mediated by the Fanconi anemia pathway; however, data supporting the involvement of this pathway is limited, particularly in human hematopoietic cells. Therefore, we assessed the role of FANCD2, a critical component of the Fanconi anemia pathway, in FA-induced toxicity in human lymphoblast cell models of FANCD2-deficiency (PD20 cells) and FANCD2-sufficiency (PD20-D2 cells). After treatment of the cells with 0-150 μM FA for 24 hours, DPCs were increased in a dose-dependent manner in both cell lines, with greater increases in FANCD2-deficient PD20 cells. FA also induced cytotoxicity, micronuclei, chromosome aberrations, and apoptosis in a dose-dependent manner in both cell lines, with greater increases in cytotoxicity and apoptosis in PD20 cells. Increased levels of γ-ATR and γ-H2AX in both cell lines suggested the recognition of FA-induced DNA damage; however, the induction of BRCA2 was compromised in FANCD2-deficient PD20 cells, potentially reducing the capacity to repair DPCs. Together, these findings suggest that FANCD2 protein and the Fanconi anemia pathway are essential to protect human lymphoblastoid cells against FA toxicity. Future studies are needed to delineate the role of this pathway in mitigating FA-induced toxicity, particularly in hematopoietic stem cells, the target cells in leukemia.
Keywords: DNA-protein crosslinks, micronuclei, chromosome aberrations, apoptosis, DNA damage and repair, Fanconi anemia
Introduction
Formaldehyde (FA, CAS Reg. No. 50-00-0) is a high production volume industrial chemical and also a ubiquitous environmental pollutant (NTP 2010). Though FA occurs naturally in living organisms, FA exposure has been associated with various toxic effects in humans, including eye, nose, throat, and skin irritation, allergic contact dermatitis, histopathological abnormalities of the nasal mucosa, occupational asthma, reduced lung function, altered immune response, hematotoxicity, and carcinogenicity (IARC 2006). The International Agency for Research on Cancer (IARC) has classified FA as a group 1 human carcinogen based on sufficient evidence that FA causes both nasopharyngeal cancer (IARC 2006) and leukemia (Baan et al. 2009) in humans.
Studies in vitro and in vivo have shown that FA is genotoxic and mutagenic to mammalian cells (Baan et al. 2009; IARC 1995; IARC 2006). The formation of DNA-protein crosslinks (DPCs) in target tissues has been proposed as the primary mechanism by which FA leads to DNA damage expressed as chromosomal changes, including chromosomal aberrations (CA), sister chromatid exchanges (SCEs), and micronuclei (MN) (Bauchinger and Schmid 1985; Jakab et al. 2010; NTP 2010; Thomson et al. 1984). Recently, we reported an increased frequency of monosomy of chromosome 7 and trisomy of chromosome 8 in myeloid blood progenitor cells cultured from workers exposed to high levels of FA, suggesting that exogenous FA might enter the systemic circulation of humans and damage stem cells in bone marrow or other targets (Zhang et al. 2010b). These chromosome changes, together with the observed suppression of peripheral blood cell counts (hematotoxicity), are consistent with an increased risk of leukemia.
Given that formation of DPCs is the major mode of action of FA toxicity, competent DPC repair mechanisms are critical in the mitigation of cancer risk in FA-exposed individuals. Although both nucleotide excision repair (NER) pathways (Grafstrom et al. 1984; Lorenti Garcia et al. 2009) and homologous recombination (HR) pathways (de Graaf et al. 2009; Ridpath et al. 2007) have been shown to be involved in the repair of FA-induced DPCs, current evidence suggests that HR-mediated repair may play a pivotal role, especially in chronic low-dose FA exposure (de Graaf et al. 2009). It has been suggested that most FA-induced DPCs can be removed without the involvement of DNA excision repair (Grafstrom et al. 1984). In addition, HR but not NER, plays a pivotal role in the tolerance of DPCs in mammalian cells (Nakano et al. 2009).
The Fanconi anemia repair pathway is an HR repair mechanism which is involved in the repair of DNA damage induced by alkylating agents and chromosome defects that occur during homologous recombination. Disruption of this pathway results in chromosome instability, increased sensitivity to DNA-DNA cross-linking agents, and a rare genetic cancer-susceptibility syndrome, Fanconi anemia (D'Andrea and Grompe 2003). The Fanconi anemia repair pathway consists of multiple interconnected “FANC” and related proteins that are subject to tight regulation (Noda et al. 2011; Thompson and Hinz 2009). The FANC proteins (A, B, C, E, F, G, L and M), together with FAAP24/100, comprise a nuclear core complex. In response to exogenous DNA damage, or during normal S phase progression, FANCD2 undergoes monoubiquitylation by the complex and is targeted into nuclear foci where it co-localizes with BRCA1, FANCD1/BRCA2, FANCN/PALB2, RAD51, FANCJ/BRIP1 and other proteins. FANCD2 and other FANC proteins, including FANCC, promote HR repair and together the Fanconi anemia factors are required for cellular resistance to DNA cross-linking agents.
Despite the critical role of the Fanconi anemia repair pathway in resistance to DNA cross-linking agents, evidence of its role in the repair of FA-induced DPCs is limited, particularly in human hematopoietic cells. One study showed that several Fanconi anemia pathway proteins, including FANCD2 and FANCD1/BRCA2, are essential to counteract DPCs caused by FA in a chicken B lymphocyte cell line (DT40) (Ridpath et al. 2007). Another study, however, reported finding no significant differences between normal human fibroblast cells and human fibroblast cells deficient in the Fanconi anemia repair pathway protein FANCA, with respect to the induction and removal of DPCs (Speit et al. 2000). A recent study in mouse and Chinese hamster cell lines suggests that FA-induced DNA damage may be repaired through the Fanconi anemia repair pathway independent of the Fanconi anemia nuclear core complex, and that FANCD2 plays a major role in this repair process (Noda et al. 2011).
We hypothesized that deficiency in the Fanconi anemia repair pathway, in particular FANCD2, sensitize human hematopoietic cells to FA-induced toxicicty. Therefore, we measured FA-induced DPCs, MN, CA, apoptosis and cytotoxicity, as well as protein levels of components of the Fanconi anemia repair pathway, in an in vitro human lymphoblastoid cell model of FANCD2 deficiency/sufficiency (Timmers et al. 2001).
Materials and Methods
Cell culture and chemical treatments
PD20 is a human lymphoblastoid cell line derived from a Fanconi anemia patient. It is deficient in the FANCD2 protein as a consequence of mutations in the FANCD2 gene. The PD20-D2 cell line was generated from the PD20 cell line to ectopically express the FANCD2 protein (Timmers et al. 2001). This was achieved by transducing PD20F cells with retroviral supernatants from the vector pMMP- puro into which the full-length FANCD2 cDNA had been subcloned, and selecting puromycin resistant cells.
Both cell lines were cultured at a starting cell density of 5 × 104 cells per ml in RPMI 1640 (Mediatech, Inc, Manassas, VA) with L-glutamine, 15% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin (Omega Scientific, San Diego, CA), under standard culturing conditions. Once established, cells were plated at a density of 5 × 105 cells/ml and treated with 0, 50, 75, 100, 125, 150 and 200 μM FA for 24 hours. FA was diluted in 1 PBS from a 37% FA solution (Sigma Aldrich, St. Louis, MO) immediately before the treatments.
Cytotoxicity assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was performed to compare the cell viability between PD20 and PD20-D2 cells after FA treatment. Cells were seeded in RPMI 1640 medium without phenol red (Invitrogen, Carlsbad, CA) at a density of 2 × 105 cells per ml, 100 μl per well, in 96-well plates. Twenty four hours after incubation with FA (6 replicates/FA dose), 10 μl of sterile MTT dye (Sigma Aldrich; 5 mg/ml) was added to each well and plates were incubated at 37 °C for 4 h. The culture medium was then removed and was thoroughly mixed for 10 min after the addition of 150 μl of DMSO. Spectrometric absorbance at 570 nm was measured using a microplate reader. Three independent experiments were conducted.
DPC assay
DPCs were measured using a previously described procedure (Costa et al. 1996; Zhitkovich and Costa 1992), with minor modifications. In this assay, protein-linked DNA is sequentially precipitated from cell lysates, and both protein-linked and non-linked DNA are measured. Briefly, 1×106 cells were washed twice with ice-cold 1×PBS and re-suspended in 100 μl 1×PBS. The cells were lysed by the addition of 0.5 ml of a 1% SDS solution and the DNA was sheared by passing the cell lysates through a 25-gauge needle 4 times. 0.5 ml of 100 mM KCl-20 mM Tris-HCl (pH 7.5) was added and the lysates were mixed by vigorous vortexing for 5s. The samples were heated in 65°C water bath for 10 min and then placed on ice for 10 min to allow the formation of Potassium Dodecyl Sulfate precipitates. After centrifugation at 6000g for 5 min at 4°C, the supernatants were collected. The resulting pellets were subjected to three rounds of precipitation: pellets were re-suspended in 0.5 ml of 100 mM of KCl-20 mM Tris-HCl (pH 7.5) by brief vortexing, heated at 65°C for 10 min, placed on ice for 10 min, and centrifuged at 6000g for 5 min at 4°C. Non-protein-linked DNA was measured in the collected supernatants using PicoGreen (Invitrogen Corporation, Carlsbad, CA). The protein-linked DNA, contained in the pellets following the last round of precipitation, was measured as follows. The pellets were resuspended in 0.5 ml proteinase K solution (0.2 mg/ml in 100 mM KCI / 20 mM Tris-HCl / 10 mM EDTA) by vortexing. The samples were then incubated in 50°C water bath for 3 hrs. After the incubation, 50 μl of 4 mg/ml BSA was added. The samples were vortexed and placed on ice for 10 min to form precipitates. The samples were then centrifuged at 12000g for 10 min at 4°C after which the supernatant was collected. The quantity of protein-linked DNA was measured using PicoGreen. The DPC rate was calculated as DPC (%) = protein-linked DNA / (protein-linked DNA + non-linked DNA) × 100%. Two independent experiments were conducted.
Cytokinesis-block micronuclei (CBMN) assay
The CBMN assay was performed as described previously (Fenech 2000), with minor modifications. Briefly, cytochalasin-B was added to the cultures at a final concentration of 6 μg/ml, three hours after the start of treatment. At the end of the 24 hr treatment period, cells were washed twice with 1×PBS and the slides were prepared by cyto-spin. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) and micronuclei were detected using a fluorescent Nikon microscope at 1000× magnification. The slides were coded to blind the scorer to the treatment status. For each sample, 2000 binucleated cells were scored for MN frequency and 1000 cells were scored for nuclear division index (NDI). NDI was calculated as NDI = (M1 + 2M2 + 3M3 + 4M4) / N, where M1, M2, M3, and M4 represent the numbers of cells with 1, 2, 3, and 4 nuclei, respectively, and N is the total number of viable cells scored (Fenech 2000). Two independent experiments were conducted.
CA analysis
For CA analysis, PD20 and PD20-D2 cells were exposed to FA for 24 hours. Colcemid (0.1 μg/ml, Invitrogen, Carlsbad, CA) was added to each culture 2 hours before harvesting to arrest cells at metaphase. After hypotonic treatment (0.075 M KCl) for 30 min at 37°C, the cells were fixed three times with freshly prepared Carnoy's fixative (methanol: glacial acetic acid = 3:1). The fixed cells were dropped onto glass slides, allowed to air dry and stored at -20°C. The cells on the slides were then stained with Giemsa and metaphase spreads were scanned and localized automatically using Metafer software (MetaSystems, Altlussheim, Germany). Metaphases were scored at 1000× magnification to detect structural chromosomal aberrations. Only metaphase spreads in which the cells appeared intact with the chromosomes condensed and well spread, and the centromeres and chromatids were readily visible, were scored. The structural chromosomal aberrations were defined according to An International System for Human Cytogenetic Nomenclature (2005). All slides were coded to prevent observer bias and at least 200 metaphases were scored for CA frequency and 2000 cells were scored for mitotic index (MI). MI was calculated as MI = M / (M + I) × 100%, where M and I represent the numbers of cells in mitosis and interphase, respectively. Two independent experiments were conducted.
Apoptosis analysis
Following FA treatment, cells were collected and stained with propidium iodide (PI) and Annexin V-FITC according to the manufacturer's protocol (BD Pharmingen, San Diego, CA). At least 1×104 cells were analyzed, from two independent experiments performed in triplicate, on a Beckman Coulter EPICS XL-MCL flow cytometer using System II software. Three independent experiments were conducted.
Protein immunoblot assay
Total cell lysates were collected from 5×106 cells at the end of the 24 hr treatment period using 300 μl of radioimmunoprecipitation assay (RIPA) lysis buffer. Protein concentrations were determined by the detergent compatible protein assay (Bio-Rad, Hercules, CA). Equal protein amounts were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto nitrocellulose membranes, and incubated with monoclonal antibodies specific to FANCD2 and FANCC (generously provided by Fanconi Anemia Research Fund, Eugene, OR), BRCA2 (Santa Cruz Biotch Inc, Santa Cruz, CA), Phospho-ATR (γATR), Phospho-Histone H2A.X (γH2AX), Phospho-P53 (γP53), cleaved PARP-1, cleaved caspase-9, -7 and -3, cytochrome c (Cell Signaling Technology, Danvers, MA), and β-actin (Sigma-Aldrich, St. Louis, MO). Proteins were visualized using the enhanced chemiluminescence (ECL) method as per the manufacturer's protocol (Amersham Biosciences, United Kingdom). Films were exposed and developed using the Konica SRX-101 developer (Konica Minolta Medical Imaging USA, Wayne, NJ).
Statistical analysis
Each measured protein was normalized to β-actin, the loading control, and quantified using ImageJ software (NIH, Bethesda, MD). Data is presented as the representative or the mean of at least two independent experiments. Error bars represent standard deviation. Linear regression was used to test the dose-response relationship between each endpoint and FA concentration. The levels of cytotoxicity, DPCs, micronuclei, CAs, and apoptosis induced by FA in the two cell lines were normalized by calculating the percentage in each treatment group relative to the controls. The differences in the normalized percentages of each endpoint between the two cell lines over FA doses were in normal distribution; thus, the paired t-test was applied to test the differences in sensitivity to FA toxicity between the two cell lines. The null hypothesis was that the mean of the differences in the percentages between the two cell lines over FA doses would be equal to zero (i.e. the induction of the toxic effects in the two cell lines would be at the same level). The significance level to reject the null hypothesis was 0.05.
Results
Cytotoxicity induced by FA in PD20 and PD20-D2 cells
The PD20 and PD20-D2 cell lines represent human hematopoietic models of FANCD2 deficiency and sufficiency, respectively. We confirmed the absence of FANCD2 protein in PD20 cells, and its presence at high levels in PD20-D2 cells by protein immunoblot assay (Fig. 1).
Fig. 1.
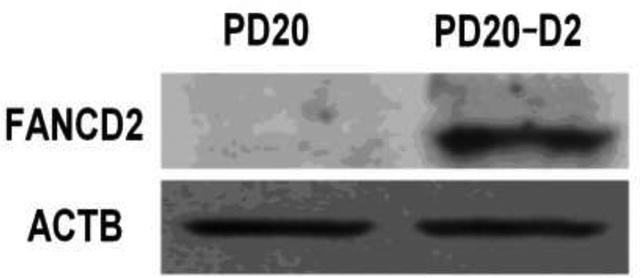
FANCD2 protein is expressed in PD20-D2 cells but not in PD20 cells.
Representative image of immunoblot analysis performed using antibodies against FANCD2 with β-actin (ACTB) used as loading control.
Untreated PD20 and PD20-D2 cell lines exhibited similar growth characteristics in culture. Treatment with FA for 24 hours, with a range of FA doses (50, 75, 100, 125, 150 and 200 μM) caused a dose-dependent decrease in cell viability in both cell lines (P trend (PD20) < 0.01 and P trend (PD20-D2) < 0.01), with greater decreases observed in FANCD2-deficient PD20 cells than the PD20-D2 cells (P (PD20 vs PD20-D2) < 0.01) (Fig. 2).
Fig. 2.

Cytotoxicity induced by FA in PD20 and PD20-D2 cells.
FA decreased cell viability in a dose-dependent manner in both cell lines (Ptrend (PD20) < 0.01 and P trend (PD20-D2) < 0.01) with greater effects in PD20 cells (P(PD20 vs PD20-D2) < 0.01).
Induction of DPCs and FANC/BRCA repair pathway components by FA in PD20 and PD20-D2 cells
DPC levels were increased in a dose-dependent manner in both the PD20 and PD20-D2 cell lines (P trend (PD20) < 0.01 and Ptrend (PD20-D2) < 0.01) after 24 hours of treatment with FA (50 -150 μM) and the induction was greater in PD20 cells (P(PD20 vs PD20-D2) < 0.01) at least at higher doses of FA treatment (Fig. 3A). For example, at 125 μM FA, the induced level of DPC was 88% higher in the PD20 than PD20-D2 cells, while at 150 μM FA, the increase was 176%.
Fig. 3.
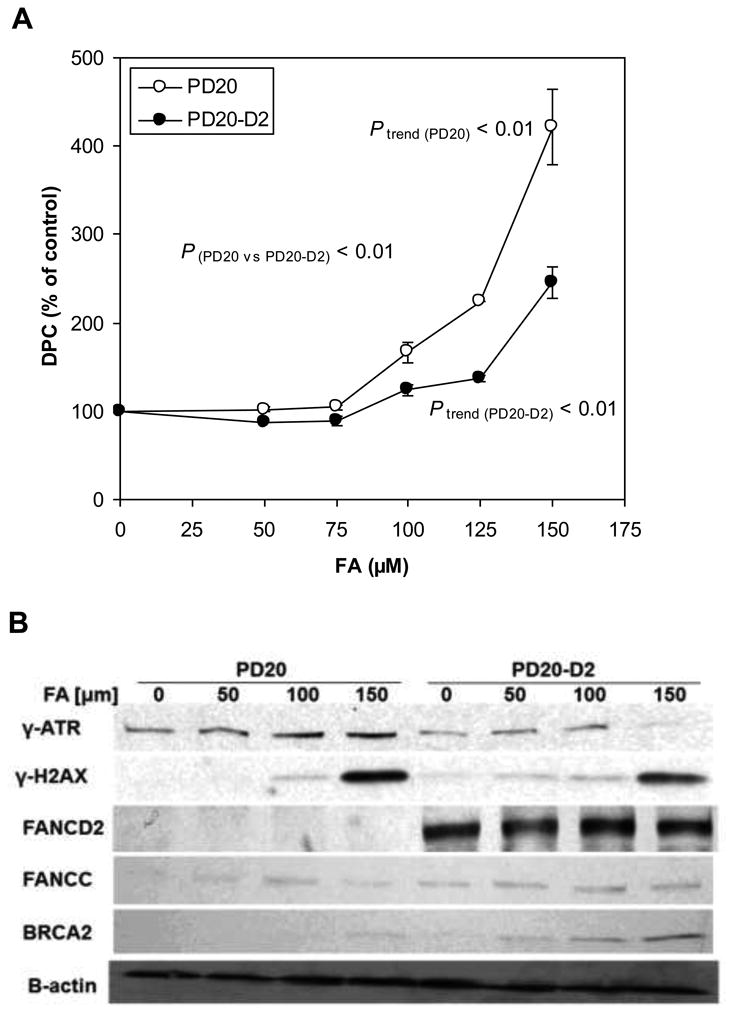
Induction of DPCs and FANC/BRCA repair pathway components by FA in PD20 and PD20-D2 cells.
(A) DPC levels in FA-treated PD20 and PD20-D2 cells as a percentage of untreated control cells. FA increased DPCs in a dose-dependent manner in both cell lines (Ptrend (PD20) < 0.01 and Ptrend (PD20-D2) < 0.01) with greater effects in PD20 cells (P(PD20 vs PD20-D2) < 0.01).
(B) Protein levels of γATR and γH2AX in FA-treated PD20 and PD20-D2 cells. FA did not alter the FANCD2 or FANCC protein levels in either cell line. BRCA2 protein level was increased only in PD20-D2 in a dose-dependent manner. β-actin was used as loading control.
The higher DPC induction in the PD20 cells suggested that repair of DPCs was impaired in these cells. Thus, we examined markers of DNA damage and repair. We found that levels of γATR and γH2AX, markers of DNA damage and DNA double strand breaks, respectively, were increased in a dose-dependent manner in both cell lines after 24 hours of FA treatment (Fig. 3B). FANCD2 and FANCC levels were unaltered by FA treatment in both cell lines (Fig. 3B). However, BRCA2, whose interaction with monoubiquitylated FANCD2 is an essential component of homologous recombination repair triggered by the Fanconi anemia pathway, was increased in a dose-dependent manner in FA-treated PD20-D2 cells, whereas in PD20 cells it was increased slightly only at 150 μM FA. Together, these data suggest that DPCs are recognized as FA-induced DNA damage within both cell lines but that in the absence of FANCD2, the induction of BRCA2 is compromised and may underlie the reduced capacity to repair DPCs in PD20 cells.
Chromosomal damage induced by FA in PD20 and PD20-D2 cells
MN is a widely used biomarker for chromosomal damage (Bonassi et al. 2011). After treatment with FA for 24 hours, the NDI in untreated PD20 cells was 1.43 and in cells treated with 50, 100, and 150 μM FA the NDIs were 1.38 (97%, percentage to the controls), 1.28 (90%), and 1.12 (78%), respectively. The NDI in untreated PD20-D2 cells was 1.40 and in cells treated with 50, 100, and 150 μM FA the NDIs were 1.41 (101%), 1.33 (95%), and 1.12 (80%), respectively. The baseline MN frequencies in PD20 and PD20-D2 cells were 3.43%, 2.40%, respectively, and dose-dependent increases in MN frequency were observed in both cell lines (Ptrend (PD20) < 0.05 and Ptrend (PD20-D2) < 0.05) (Fig. 4A). Although a paired t-test showed that the MN inductions in the two cell lines across FA doses were not significantly different (P(PD20 vs PD20-D2) = 0.53), a higher MN induction was observed in the PD20 cells (300% relative to the control) than in the PD20-D2 cells (183%) at 150 μM FA.
Fig. 4.

Chromosomal damage (MN and CA) induced by FA in PD20 and PD20-D2 cells.
(A) MN levels in FA-treated PD20 and PD20-D2 cells as a percentage of untreated control cells. FA increased MN in a dose-dependent manner in both cell lines (Ptrend (PD20) < 0.05 and Ptrend (PD20-D2) < 0.05), while MN inductions in the two cell lines across FA doses were not significantly different (P (PD20 vs PD20-D2) = 0.53). A higher MN induction was observed in the PD20 cells at 150 μM FA (300%) than in the PD20-D2 cells (183%).
(B) CA levels in FA-treated PD20 and PD20-D2 cells as a percentage of untreated control cells. FA increased CA in a dose-dependent manner in both cell lines (Ptrend (PD20) < 0.001 and Ptrend (PD20-D2) < 0.01), while induction of CA was not statistically different between the two cell lines.
We also examined CA induced by FA treatment. After treatment with FA for 24 hours, the MI in untreated PD20 cells was 6.00 and in cells treated with 50, 100, and 150 μM FA the MIs were 5.20 (87%, percentage to the controls), 5.60 (93%), and 2.88 (48%), respectively. The MI in untreated PD20-D2 cells was 4.43 and the MIs in cells treated with 50, 100, and 150 μM FA were 4.78 (108%), 4.75 (107%), and 2.30 (52%), respectively. The baseline CA frequencies in PD20 and PD20-D2 cells were 3.40%, 1.55%, respectively, and CA was increased in a dose-dependent manner in both cell lines (P trend (PD20) < 0.001 and Ptrend (PD20-D2) < 0.01) (Fig. 4B). The response between the two cell lines was not statistically different.
Apoptosis induced by FA in PD20 and PD20-D2 cells
The percentage of apoptotic cells in cell cultures treated with FA for 24h was measured by flow cytometry. A dose-dependent increase in apoptosis occurred in both cell lines (P trend (PD20) < 0.0001 and Ptrend (PD20-D2) < 0.001) (Fig. 5A). Apoptosis was induced to a greater degree by FA in PD20 cells than in PD20-D2 cells (P(PD20 vs PD20-D2) < 0.01).
Fig. 5.
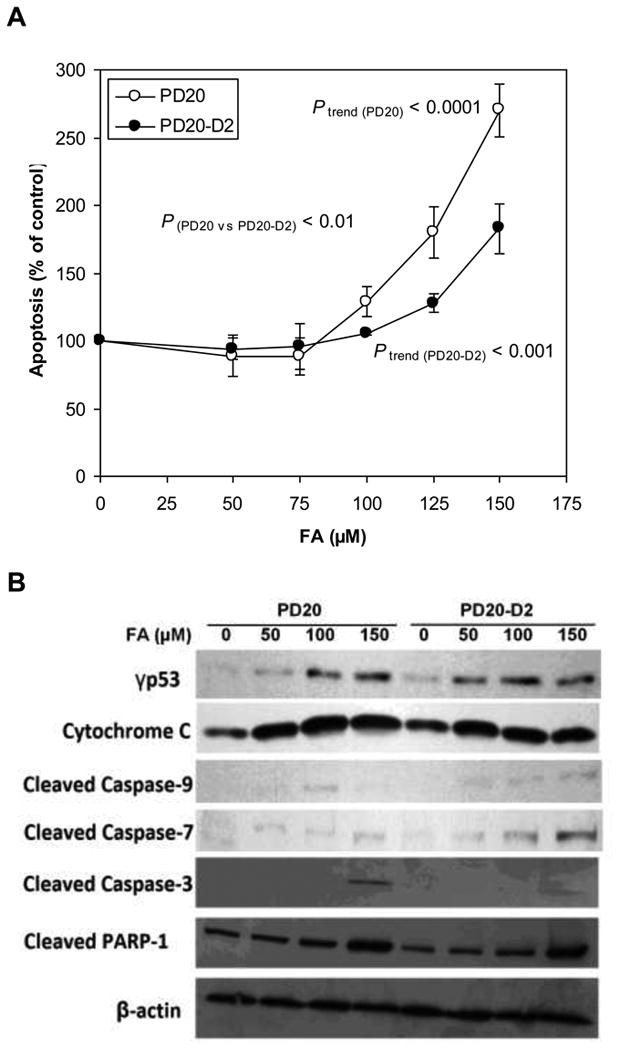
Apoptosis induced by FA in PD20 and PD20-D2 cells.
(A) Apoptosis in FA-treated PD20 and PD20-D2 cells as a percentage of untreated control cells. FA induced apoptosis in a dose-dependent manner in both cell lines (Ptrend (PD20) < 0.0001 and P trend (PD20-D2) < 0.001) with greater effects in PD20 cells (P(PD20 vs PD20-D2) < 0.01).
(B) Apoptosis-related protein expression in FA-treated PD20 and PD20-D2 cells as a percentage of untreated control cells. p53 was activated and caspase signaling pathway was induced by FA at similar levels in both PD20 and PD20-D2 cells. β-actin is used as loading control.
Apoptosis, a highly regulated process, is usually initiated by a p53-mediated response to DNA damage, followed by a series of well-characterized events including activation of the caspase cascade that ultimately leads to apoptotic cell death. Immunoblotting with antibodies to γP53, cytochrome-C, cleaved caspases-9, -7 and -3, and cleaved PARP-1, in PD20 and PD20-D2 cell lysates following 24 treatment with FA, revealed that these apoptotic effectors were induced to a similar degree in both cell lines (Fig. 5B).
Discussion
The formation of DPCs in target tissues is considered to be the primary action of FA and the inadequate repair of FA-induced DPCs could lead to further DNA damage, and consequently to chromosomal aberrations and carcinogenesis (Merk and Speit 1998; Speit et al. 2000). We hypothesized that, in hematopoietic cells, compromised functionality of FANCD2 and/or other components of the Fanconia anemia pathway involved in repair of DPCs could potentially modulate the cellular response to FA toxicity and influence leukemia risk. In the current study, we have shown that FA significantly induces DPCs, chromosomal damage (MN and CA), apoptosis and cytotoxicity in a dose-dependent manner in both PD20 and PD20-D2 cells, particularly at doses exceeding the human physiological level (∼80 μM) (Heck et al. 1985), and that FANCD2-deficient PD20 cells are more sensitive to most of these adverse effects. These data are consistent with previous findings showing that deficiency in FANCD2 leads to an increased sensitivity to FA exposure in vitro in mouse and Chinese hamster fibroblast cell lines and in chicken B lymphocyte cell lines (Noda et al. 2011; Ridpath et al. 2007).
Many FANC and FANC-related proteins are involved in the tightly regulated Fanconi anemia repair pathway and may be involved in repair of DPCs induced by FA, as recently reviewed by us (Zhang et al. 2010a). FANCC, together with other FANC proteins, is involved in HR repair. Co-localization of monoubiquitylated FANCD2 with several proteins including BRCA2 in nuclear foci is necessary for cellular resistance to DNA crosslinking agents. The importance of components of the Fanconi anemia pathway in the mitigation of FA-induced toxicity has been demonstrated in studies conducted in various cell types. Chicken B lymphocyte DT40 cells deficient or lacking in FANCD2, BRCA1, FANCD1/BRCA2 (Ridpath et al. 2007), FANCL, FANCB, FANCC, FANCJ, and FANCM (Rosado et al. 2011), were shown to be sensitive to exogenous FA toxicity. Inactivation of FA catabolism in DT40 cells by disruption of the main FA detoxifying gene, alcohol dehydrogenase 5 (ADH5) gene, resulted in synthetic lethality in conjunction with upstream (FANCL) or downstream (FANCD2) components of the Fanconi anemia pathway (Rosado et al. 2011). These data show that endogenous FA is genotoxic in Fanconia anemia-deficient cells. Human colorectal cancer cells deficient in FANCC or FANCG (Ridpath et al. 2007) and a FANCB-deficient human B cell line, NALM-6 (Rosado et al. 2011) were shown to be sensitive to FA toxicity. FANCA and FANCC were shown to play minor roles in the repair of FA-induced DNA damage in mouse embryonic fibroblast cell lines, while FANCD1/BRCA2 (Chinese hamster lung fibroblast), FANCG (Chinese hamster ovary), and FANCD2 (mouse embryonic fibroblast cell lines) appeared to be more important (Noda et al. 2011). Our current study extends these findings by showing that human lymphoblastoid cells deficient in FANCD2 are sensitive to FA-induced DPCs, possibly as a result of a failure to induce BRCA2. Further studies should examine the mechanism of DPC repair in these cells and delineate the factors involved.
As well as DPCs, cytotoxicity and apoptosis were also elevated by FA in PD20 cells in a dose-responsive manner compared with PD20-D2 cells, while chromosomal damage did not significantly differ between the two cell lines. Thus, cytotoxicity in FANCD2-deficient cells appears to be mediated by apoptosis occurring subsequent to DPC formation rather than chromosome damage. Additional DNA and/or chromosome repair factors, may prevent the formation of MN and CA in FANCD2-deficient cells. One possible candidate is FANCB which was shown to prevent FA-induced chromosome damage in human NALM6 cells (Rosado et al. 2011).
Deficiency in the Fanconi anemia repair pathway, in particular FANCD2, has been associated with carcinogenesis. For instance, Fancd2-knockout mice were found to have an increased incidence of epithelial cancers (Houghtaling et al. 2003), though to the best of our knowledge, incidence of leukemia has not been examined in such mice. Bone marrow from Fancd2-deficient mice has been shown to exhibit hematopoietic defects, including a decreased frequency and activity of hematopoietic stem cells (Parmar et al. 2010). Chromosomal breaks and DNA breaks have been demonstrated in the primary fibroblasts of Fancd2-/- mice (Reliene et al. 2010). Our current findings of elevated toxicity in FACND2 deficient PD20 cells compared to FACND2 sufficient PD20-D2 cells suggest that deficient Fanconi anemia repair pathway might contribute to FA-induced leukemogenesis. Confirmation of the observed effects in cell lines with different mutations in Fanconi anemia repair pathway and in primary cells, including the hematopoietic stem cells, is desirable.
Overall, our results indicate that the Fanconi anemia repair pathway plays a critical role in protecting human lymphoblastoid cells from FA-induced toxicity. Deficiency in components of the pathway, particularly FANCD2, may significantly increase the sensitivity of these cells to FA-induced DPCs and other toxicities. It has recently been hypothesized that individuals with heterozygous mutations in genes involved in DNA repair, including BRCA1/2 and Fanconi anemia pathway and ATM genes have an increased risk of myeloid leukemia through increased susceptibility to FA toxicity (Friedenson 2011). Future studies should seek to explore the role of such mutations, and polymorphisms in Fanconi anemia pathway-related genes, in susceptibility to FA-induced toxicity and leukemogenesis, in cell models and FA-exposed populations; investigate the mechanisms underlying DPC and DNA damage repair in hematopoietic stem cells exposed to FA; and, measure the incidence of leukemia in FANC protein deficient mice.
Acknowledgments
We are grateful to Prof. Toshiyasu Taniguchi from the Fred Hutchinson Cancer Research Center, Seattle, WA, for generously providing us with the human lymphoblast PD20 and PD20-D2 cells. This work was supported by National Institute of Environmental Health Sciences, National Institute of Health grant R01ES017452 to Dr. L Zhang.
References
- Baan R, Grosse Y, Straif K, Secretan B, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V. A review of human carcinogens--Part F: chemical agents and related occupations. Lancet Oncol. 2009;10:1143–4. doi: 10.1016/s1470-2045(09)70358-4. [DOI] [PubMed] [Google Scholar]
- Bauchinger M, Schmid E. Cytogenetic effects in lymphocytes of formaldehyde workers of a paper factory. Mutat Res. 1985;158:195–9. doi: 10.1016/0165-1218(85)90085-0. [DOI] [PubMed] [Google Scholar]
- Bonassi S, El-Zein R, Bolognesi C, Fenech M. Micronuclei frequency in peripheral blood lymphocytes and cancer risk: evidence from human studies. Mutagenesis. 2011;26:93–100. doi: 10.1093/mutage/geq075. [DOI] [PubMed] [Google Scholar]
- Costa M, Zhitkovich A, Gargas M, Paustenbach D, Finley B, Kuykendall J, Billings R, Carlson TJ, Wetterhahn K, Xu J, Patierno S, Bogdanffy M. Interlaboratory validation of a new assay for DNA-protein crosslinks. Mutat Res. 1996;369:13–21. doi: 10.1016/s0165-1218(96)90043-9. [DOI] [PubMed] [Google Scholar]
- D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- de Graaf B, Clore A, McCullough AK. Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair (Amst) 2009;8:1207–14. doi: 10.1016/j.dnarep.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech M. The in vitro micronucleus technique. Mutat Res. 2000;455:81–95. doi: 10.1016/s0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Friedenson B. A common environmental carcinogen unduly affects carriers of cancer mutations: carriers of genetic mutations in a specific protective response are more susceptible to an environmental carcinogen. Med Hypotheses. 2011;77:791–7. doi: 10.1016/j.mehy.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Grafstrom RC, Fornace A, Jr, Harris CC. Repair of DNA damage caused by formaldehyde in human cells. Cancer Res. 1984;44:4323–7. [PubMed] [Google Scholar]
- Heck HD, Casanova-Schmitz M, Dodd PB, Schachter EN, Witek TJ, Tosun T. Formaldehyde (CH2O) concentrations in the blood of humans and Fischer-344 rats exposed to CH2O under controlled conditions. Am Ind Hyg Assoc J. 1985;46:1–3. doi: 10.1080/15298668591394275. [DOI] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17:2021–35. doi: 10.1101/gad.1103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. Formaldehyde. IARC Monogr Eval Carcinog Risks Hum. 1995;62:217–375. [PMC free article] [PubMed] [Google Scholar]
- IARC. Formaldehyde, 2-butoxyethanol and 1-tert-butoxypropan-2-ol. IARC Monogr Eval Carcinog Risks Hum. 2006;88:1–478. [PMC free article] [PubMed] [Google Scholar]
- Jakab MG, Klupp T, Besenyei K, Biro A, Major J, Tompa A. Formaldehyde-induced chromosomal aberrations and apoptosis in peripheral blood lymphocytes of personnel working in pathology departments. Mutat Res. 2010;698:11–7. doi: 10.1016/j.mrgentox.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Lorenti Garcia C, Mechilli M, Proietti De Santis L, Schinoppi A, Kobos K, Palitti F. Relationship between DNA lesions, DNA repair and chromosomal damage induced by acetaldehyde. Mutat Res. 2009;662:3–9. doi: 10.1016/j.mrfmmm.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Merk O, Speit G. Significance of formaldehyde-induced DNA-protein crosslinks for mutagenesis. Environ Mol Mutagen. 1998;32:260–8. doi: 10.1002/(sici)1098-2280(1998)32:3<260::aid-em9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Nakano T, Katafuchi A, Matsubara M, Terato H, Tsuboi T, Masuda T, Tatsumoto T, Pack SP, Makino K, Croteau DL, Van Houten B, Iijima K, Tauchi H, Ide H. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J Biol Chem. 2009;284:27065–76. doi: 10.1074/jbc.M109.019174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Takahashi A, Kondo N, Mori E, Okamoto N, Nakagawa Y, Ohnishi K, Zdzienicka MZ, Thompson LH, Helleday T, Asada H, Ohnishi T. Repair pathways independent of the Fanconi anemia nuclear core complex play a predominant role in mitigating formaldehyde-induced DNA damage. Biochem Biophys Res Commun. 2011;404:206–10. doi: 10.1016/j.bbrc.2010.11.094. [DOI] [PubMed] [Google Scholar]
- NTP. Final Report on Carcinogens Background Document for Formaldehyde. Rep Carcinog Backgr Doc. 2010:i–512. [PubMed] [Google Scholar]
- Parmar K, Kim J, Sykes SM, Shimamura A, Stuckert P, Zhu K, Hamilton A, Deloach MK, Kutok JL, Akashi K, Gilliland DG, D'Andrea A. Hematopoietic stem cell defects in mice with deficiency of Fancd2 or Usp1. Stem Cells. 2010;28:1186–95. doi: 10.1002/stem.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reliene R, Yamamoto ML, Rao PN, Schiestl RH. Genomic instability in mice is greater in Fanconi anemia caused by deficiency of Fancd2 than Fancg. Cancer Res. 2010;70:9703–10. doi: 10.1158/0008-5472.CAN-09-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, Buerstedde JM, Gillespie DA, Sale JE, Yamazoe M, Bishop DK, Takata M, Takeda S, Watanabe M, Swenberg JA, Nakamura J. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007;67:11117–22. doi: 10.1158/0008-5472.CAN-07-3028. [DOI] [PubMed] [Google Scholar]
- Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat Struct Mol Biol. 2011;18:1432–4. doi: 10.1038/nsmb.2173. [DOI] [PubMed] [Google Scholar]
- Speit G, Schutz P, Merk O. Induction and repair of formaldehyde-induced DNA-protein crosslinks in repair-deficient human cell lines. Mutagenesis. 2000;15:85–90. doi: 10.1093/mutage/15.1.85. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutat Res. 2009;668:54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson EJ, Shackleton S, Harrington JM. Chromosome aberrations and sister-chromatid exchange frequencies in pathology staff occupationally exposed to formaldehyde. Mutat Res. 1984;141:89–93. doi: 10.1016/0165-7992(84)90016-2. [DOI] [PubMed] [Google Scholar]
- Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox B, Olson S, D'Andrea AD, Moses R, Grompe M. Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell. 2001;7:241–8. doi: 10.1016/s1097-2765(01)00172-1. [DOI] [PubMed] [Google Scholar]
- Zhang L, Freeman LE, Nakamura J, Hecht SS, Vandenberg JJ, Smith MT, Sonawane BR. Formaldehyde and leukemia: epidemiology, potential mechanisms, and implications for risk assessment. Environ Mol Mutagen. 2010a;51:181–91. doi: 10.1002/em.20534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Tang X, Rothman N, Vermeulen R, Ji Z, Shen M, Qiu C, Guo W, Liu S, Reiss B, Freeman LB, Ge Y, Hubbard AE, Hua M, Blair A, Galvan N, Ruan X, Alter BP, Xin KX, Li S, Moore LE, Kim S, Xie Y, Hayes RB, Azuma M, Hauptmann M, Xiong J, Stewart P, Li L, Rappaport SM, Huang H, Fraumeni JF, Jr, Smith MT, Lan Q. Occupational exposure to formaldehyde, hematotoxicity, and leukemia-specific chromosome changes in cultured myeloid progenitor cells. Cancer Epidemiol Biomarkers Prev. 2010b;19:80–8. doi: 10.1158/1055-9965.EPI-09-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A, Costa M. A simple, sensitive assay to detect DNA-protein crosslinks in intact cells and in vivo. Carcinogenesis. 1992;13:1485–9. doi: 10.1093/carcin/13.8.1485. [DOI] [PubMed] [Google Scholar]