

Abstract
Deer bone has been used as a health-enhancing food as well as an antiaging agent in traditional Oriental medicine. Recently, the water extract of deer bone (DBE) showed a neuroprotective action against glutamate or Aβ1–42-induced cell death of mouse hippocampal cells by exerting antioxidant activity through the suppression of MAP kinases. The present study is to examine whether DBE improves memory impairment induced by scopolamine. DBE (50, 100 or 200 mg/kg) was administered orally to mice for 14 days, and then scopolamine (2 mg/kg, i.p.) was administered together with DBE for another 7 days. Memory performance was evaluated in the Morris water maze (MWM) test and passive avoidance test. Also, brain acetylcholinesterase (AChE) and choline acetyltransferase (ChAT) activity, biomarkers of oxidative stress and the loss of neuronal cells in the hippocampus, was evaluated by histological examinations. Administration of DBE significantly restored memory impairments induced by scopolamine in the MWM test (escape latency and number of crossing platform area), and in the passive avoidance test. Treatment with DBE inhibited the AChE activity and increased the ChAT activity in the brain of memory-impaired mice induced by scopolamine. Additionally, the administration of DBE significantly prevented the increase of lipid peroxidation and the decrease of glutathione level in the brain of mice treated with scopolamine. Also, the DBE treatment restored the activities of antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, and glutathione reductase to control the level. Furthermore, scopolamine-induced oxidative damage of neurons in hippocampal CA1 and CA3 regions were prevented by DBE treatment. It is suggested that DBE may be useful for memory improvement through the regulation of cholinergic marker enzyme activities and the suppression of oxidative damage of neurons in the brain of mice treated with scopolamine.
Key Words: : antioxidant defense system, cholinergic enzymes, deer bone extract, memory, scopolamine
Introduction
As life expectancy increases in humans, elderly people tend to be more susceptible to neurodegenerative diseases.1,2 In aged people over 65 years old, Alzheimer's disease (AD) has been estimated to 50–60% of dementia cases.3 Therefore, AD is a major and increasing public health concern.4 AD is characterized by a progressive decline in cognitive function due to the deficits of cholinergic nervous system.5,6
Acetylcholine (ACh), cholinergic neurotransmitter, regulates the learning and memory process.5,6 The level of ACh is mainly regulated by cholinergic enzymes in the cholinergic neurons.7 Elevated acetylcholinesterase (AChE) activity in the brain of AD was involved in the increased oxidative stress derived from mitochondria dysfunction,8,9 which accelerates Aβ aggregation9,10 as well as memory decline.11,12 In this respect, the restoration of cholinergic function, accompanied by the suppression of oxidative damage, remains a rational strategy for the effective treatment of AD.12–15
Scopolamine, an anticholinergic agent, is well known to inhibit central cholinergic neuronal activity and induces the deficit in the learning, acquisition, and memory tasks in various animals, and is widely used as an inducer in experimental model of AD.5,16–18 Moreover, scopolamine significantly increased the AChE activity, and triggers reactive oxygen species formation and induces oxidative stress.19–21
Deer bone as well as deer antler has been used as a health-enhancing food as well as an antiageing agent in traditional Oriental medicine. Moreover, deer antler extract was reported to possess an antiamnesic activity.22 Very recently, we reported that the water extract of deer bone (DBE) showed a neuroprotective action against glutamate or Aβ1–42-induced cell death of mouse hippocampal cells by exerting antioxidant activity through the suppression of MAP kinases.23 However, the protective effect of DBE on memory impairment in mice has not yet been examined.
Therefore, the present study is to examine whether DBE improves memory impairment caused by scopolamine in mice. For this purpose, the behavioral parameters were evaluated using the Morris water maze (MWM) test and passive avoidance test. In addition, the change of biomarkers of oxidative stress and AChE activity in brain tissue was determined, and the loss of neuronal cells in the hippocampus was evaluated by histological examinations.
Materials and Methods
Materials
Deer bone extract
DBE was kindly provided by the Nongshim Food Company (Seoul, Korea). DBE was prepared according to the previously described method with slight modification of extraction.23 DBE contained 80.94% of crude protein, 12.47% of crude fat, 3.52% of crude ash, and 3.07% of moisture. Total ganglioside content of DBE as sialic acid was 0.11%. Total amino acid and free amino acid amounts of DBE were 831.09 and 5.48 mg/g, respectively.
Reagents
Scopolamine hydrobromide, tacrine (9-Amino-1,2,3,4-tetrahydroacridine hydrochloride hydrate), acetylthiocholine iodide, 5, 5′-dithiobis[2-nitrobezoic acid] (DTNB), tetramethoxypropane, thiobarbituric acid (TBA), sodium dodecyl sulfate (SDS), glutathione reductase (GR, Type III from baker's yeast), NADPH, glutathione disulfide (GSSG), and bovine serum albumin were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other materials were of the highest grade available.
Animals
Male ICR mice (28–30 g) were obtained from Deahan Biolink Co., Ltd. (Eumseong, Korea). All experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and (NIH Publication No. 85-23, 1985, revised in 1996) were approved by the Institutional Animal Care and Use Committee at Chungnam National University (Code number: CNU-00293). The mice housed in three per cage were allowed access to water and food ad libitum, maintained at an ambient temperature of 24±2°C, and 55±10% humidity under a 12 h diurnal light cycle (light at 06:00–18:00 o'clock) before testing. The mice were habituated for 5 days before the drug administration. All behavioral experiments were carried out in a room adjacent to the housing room under the same ambient conditions.
To assess the effect of DBE on scopolamine-induced changes, mice were divided into six groups with nine mice in each group; normal control group and scopolamine group were administrated with vehicle solution (Con, 0.9% saline, p.o.) and scopolamine (Sco, 2 mg/kg, i.p.), respectively. Tacrine group, a positive control, was administrated with tacrine (Tac, 10 mg/kg, p.o.), a centrally acting cholinesterase inhibitor and scopolamine, and DBE group was injected with DBE (50, 100, 200 mg/kg, p.o.) and scopolamine. DBE and scopolamine were dissolved in physiological saline solution for use. Memory impairment was induced by scopolamine.
After a 5-day habituation period, mice were given DBE for a total of 21 days. First, mice were treated with DBE alone for 14 days, and then with scopolamine 30 min after the administration of each drug (tacrine or DBE) or vehicle solution (0.9% saline) for the following 7 days. Mice underwent the MWM test for 5 days, starting from the 15th day of DBE treatment, and the probe test was conducted after the last training trial of the MWM test. Starting from the day when the probe test was completed, the passive avoidance test was conducted for additional 2 days. On the final day of passive avoidance test, the mice were sacrificed and brains were removed for biochemical analyses.
Behavior tests
MWM test
MWM test was performed as reported previously.21,24 Briefly, the MWM for mice consisted of a circular pool (90 cm diameter and 50 cm height) filled to a depth of 30 cm with water (25±1°C), which was made opaque with nontoxic white-colored dye. A submerged platform (6 cm diameter and 6 cm height), painted in white, was placed inside the target quadrants of the pool 1 cm below water surface, so that the position of platform was kept unaltered. The first day of MWM test experiment was dedicated to swimming training for 120 s in the absence of the platform. During the four subsequent days, the mice were given four trial sessions per day with the platform in place. The time interval between each trial session was 20 min. For four trial sessions, mice were placed in the water facing the pool wall in one of the pool quadrants. The entry point was changed in a different order each day. Drug or vehicle was administered 1 h before the first training trial on each day. When a mouse located the platform, it was permitted to remain on it for 10 s. If the mouse did not locate the platform within 120 s, it was placed on the platform for 10 s. The animal was returned to its home cage and allowed to be dried up with a hair dryer after each trial. One day after the last training trial sessions, mice were subjected to a probe trial session, in which the platform was removed from the pool, and mice were allowed to swim for 120 s to search for it. The escape latency (EL, the time taken to cross the platform for the first time) and the number of crossing platform area in 120 s were recorded using a video camera (TGCAM-2000STA, Sambo Electronic Co., Ltd., Seoul, Korea) connected to the EyeLine Video system.
Passive avoidance test
Passive avoidance task was performed using a passive avoidance apparatus (Jungdo Bio and Plant Co. Ltd, Seoul, Korea) to find the appropriate dose among several doses for in vivo experiment.4,21 This apparatus is comprised of two equal compartments (20×20×20 cm) separated by a guillotine door (5×5 cm). For the acquisition trial, mice were initially placed in the illuminated compartment and the door between the two compartments was opened 20 s later. The time (step-through latency) taken for a mouse to enter the dark compartment was measured. Upon entering the dark compartment, the door was closed and an electrical foot shock (0.5 mA for 3 s) was delivered through the stainless steel rods. Twenty-four hours after this training trial, the mouse was again placed in the illuminated compartment and the latency period to firstly enter the dark compartment was defined as retention and described as step-through latency21. Latency for entering the dark compartment was recorded up to 300 s. If a mouse did not enter the dark compartment within 300 s, the mouse was removed and assigned a latency score of 300 s.
Biochemical analyses
Following the passive avoidance test, mice were sacrificed. Whole brain was carefully removed from the skull, and stored at −80°C until used.
Determination of AChE activity
AChE activity was determined using the method of Ellman et al.25 The brains were homogenized in a glass–teflon homogenizer containing 10 volumes of homogenization buffer (12.5 mM sodium phosphate buffer pH 7.0, 400 mM NaCl), and then centrifuged at 1000 g for 10 min at 4°C. The supernatant so obtained was used as a source of enzyme for the assay. Briefly, 200 μL of supernatant was included in a test tube containing 0.1 M phosphate buffer (pH 7.4) containing 15 mM acetylthiocholine iodide and 3 mM DTNB. Activity was determined as measuring the change of absorbance at 412 nm.
Determination of choline acetyltransferase activity
Choline acetyltransferase (ChAT) activity was determined using the commercial kit (Nanjing JianCheng Bioengineering Institute, Nanging, China) according to the manufacturer's direction.
Measurement of lipid peroxidation
The brain was placed on ice, and homogenized with 50 mM sodium phosphate buffer using a tissue homogenizer with a Teflon® (Dupont, Wilmington, DE, USA) pestle. To the brain homogenate (1.0 mL) was added 1.0 mL of 8.1% SDS, 2 mL of 20% acetic acid, and 1 mL of 0.75% TBA. The mixture was boiled, and placed on ice after 30 min boiling. The mixture was centrifuged (17,000 g for 10 min). The absorbance of the malondialdehyde (MDA)-TBA adduct formed in supernatant was measured colorimetrically at 533 nm as previously described.26 The values of MDA were calculated from a standard curve that was prepared with tetramethoxypropane and expressed as TBA reactive substances values.
Total glutathione content
Brain tissue was pulverized in a cooled ceramic percussion motor with 6% metaphosphoric acid, and the mixture was centrifuged (55,000 g for 30 min) at 4°C. Total glutathione (GSH) level was determined enzymatically according to a previous procedure.27 The supernatant (0.05 mL) was mixed with 100 mM phosphate buffer containing 5 mM ethylenediaminetetraacetic acid (EDTA), 10 mM 5,5′-dithiobis-(2-nitrobenzoic acid), and 5 mM NADPH. After 3 min of equilibration at 25°C, the reaction was started by adding 2 U of GR. The formation of DTNB was continuously recorded at 412 nm with a UV/VIS spectrophotometer. The total amount of GSH in the samples was determined from a standard curve obtained by plotting the known amount of GSH versus the rate of change of absorbance at 412 nm.
Antioxidant enzyme activities
Brain tissue was homogenized in nine volumes of 20 mM phosphate buffer containing 0.1 M KCl, 1 mM EDTA, and 0.5% Triton X-100 (pH 7.4). The homogenate was centrifuged (55,000 g at 4°C) for 30 min, and the supernatant was used for the following enzyme assays. GR was determined using the method described by Pinto et al.28 The supernatant was mixed with 1 M GSSG and 5 mM NADPH in 0.1 M phosphate/0.5 mM EDTA buffer (pH 7.0), and the formation of NADP+ was monitored with a spectrophotometer at 340 nm. To determine glutathione peroxidase (GPx) activity, the supernatant was mixed with 1 mM EDTA, 100 mM GSH, 5 mM NADPH, and 1 U of GR in 0.1 M phosphate buffer (pH 7.0), and incubated for 3 min. After cumene hydroperoxide (10 mM) was added into the reaction mixture, the oxidation of NADPH into NADP+ was monitored spectrophotometrically by a decrease in absorbance at 340 nm. One unit of GPx causes the formation of 1 μmole NADP+ per min.29 For superoxide dismutase (SOD) assay, the homogenate was mixed with 1 mM xanthine, 0.2 mM cytochrome, and 0.05 M potassium cyanide in 0.05 M potassium phosphate/0.1 mM EDTA buffer, and then xanthine oxidase was added into the reaction mixture. SOD activity was measured as the inhibition of the reduction rate of cytochrome by superoxide radical, observed spectrophotometrically at 550 nm.30 The activities of GR, GPx, and SOD were expressed as an international unit/mg of liver tissue.
Morphological examination
For the microscopic evaluation, a part of the brain was fixed in 10% neutral formalin solution, and the formalin-fixed brain tissues were processed and embedded in paraffin. Serial coronal sections (4 μm in thickness), including dorsal hippocampus, were obtained, and stained with Hematoxylin and Eosin. The histopathological changes were assessed under a light microscope. For statistical analysis of the degree of neuronal injury in the hippocampus, a part of mice was subjected to histopathological examination, and the surviving pyramidal neurons in CA1 (per 50 μm) and CA3 (under a 400-fold-magnification filed) regions were counted bilaterally and averaged.31
Statistical analysis
All the results were expressed as means±standard error of the mean. The statistical analyses were performed using the SPSS software (SPSS 12.0 KO for Windows; SPSS, Inc., Chicago, IL, USA). Data were analyzed by one-way analysis of variation (ANOVA) followed by LSD test for multiple comparisons. Especially, the EL in the MWM test was analyzed using the one-way repeated ANOVA. A difference of P<.05 was regarded as being statistically significant.
Results
Effects of DBE on MWM test
To determine the effect of DBE on spatial learning memory impairment, the MWM test was performed using mice treated with scopolamine. For this purpose, the effect of DBE (50, 100 or 200 mg/kg, p.o.) on the EL and the cross times (the total times of mice cross the platform area) in mice treated with scopolamine, an inducer of memory impairment, was determined. As shown in Figure 1A, the EL of mice treated with scopolamine only was significantly longer than that of the normal control mice during all trial sessions (P<.01), proving the successfully constructed model of scopolamine-induced memory impairment in mice. However, oral administration of DBE (100 or 200 mg/kg) significantly recovered the EL prolonged by scopolamine (P<.01), similar to the observation with tacrine (P<.01). On the day following the final day of training trial sessions, the probe trial session was performed by removing the platform to estimate the spatial working memory. Figure 1B showed that the EL of the DBE-treated mice was significantly shorter than that of the scopolamine group (P<0.01). Normal control mice took about 14.7 s to find the platform quadrant, whereas scopolamine group spent about 84.2 s in that quadrant. Administration of DBE at a dose of 50, 100, and 200 mg/kg dose dependently reversed the extension of EL induced by scopolamine. Especially, DBE of 100 and 200 mg/kg restored the delay of EL induced by scopolamine to that of tacrine group. Figure 1C demonstrated that the cross times of mice treated with 100 and 200 mg/kg DBE was significantly increased, compared with that of scopolamine-treated mice (P<.01). Figure 1D depicts the representative swim paths of these mice on the fifth day of the MWM test. Normal control mice (a, Con) swam a shorter distance to find the platform, compared with scopolamine-treated mice (b, Sco). Tacrine (c, Sco+Tac) and DBE 50 (d, Sco+DBE 50 mg/kg), DBE 100 (e, Sco+DBE 100 mg/kg) or DBE 200 (e, Sco+DBE 200 mg/kg) shortened the distance required to find the platform. Thus, DBE (100 or 200 mg/kg, p.o.) showed a significant improvement of spatial learning ability in mice treated with scopolamine (P<.01), providing an evidence that DBE may exert an ameliorating effect on scopolamine-induced impairment of learning and memory in mice.
FIG. 1.

Effect of deer bone extract (DBE) on mean latency time (A), probe trial (B), and number of crossing platform area (C) in trial sessions of the Morris water maze test. DBE (50, 100, or 200 mg/kg, p.o.), tacrine (10 mg/kg, p.o.) or saline was administered to the mice. Thirty minutes later, the mice of DBE, tacrine, or control groups were treated with scopolamine (Sco, 2 mg/kg, i.p.) and subjected to the Morris water maze test. Probe trial sessions were performed for 120 s. Representative swimming paths of mice from each group subjected to Morris water maze test on the fifth training trial day (D). Data represent mean±standard error of the mean (SEM) (n=9/group); *P<.05, and **P<.01, compared with the control group. ##P<.01, compared with the scopolamine-treated group.
Effects of DBE on step-through passive avoidance test
Next, retention of the passive avoidance response was measured to confirm the effects of DBE on memory impairment (Fig. 2). Scopolamine was administered 30 min before the acquisition trial and the retention trial. The step-through latency of scopolamine-treated mice was significantly shorter, compared with that of the normal control mice in retention trial (P<.01), indicative of scopolamine-induced memory impairment. Mice treated with scopolamine showed a step-through latency of ∼102.3 s in the retention trial, whereas DBE treatment showed a remarkable mitigating effect in a dose-dependent manner (P<.01). Especially, the improving effect of DBE (100 or 200 mg/kg) on the memory impairment was similar to that of tacrine or vehicle. Thus, it is demonstrated that oral administration of DBE restores the reduction of passive avoidance response induced by scopolamine. This result suggests that an ameliorating effect of DBE on scopolamine-induced memory impairment may be mediated through cholinergic nervous system.
FIG. 2.
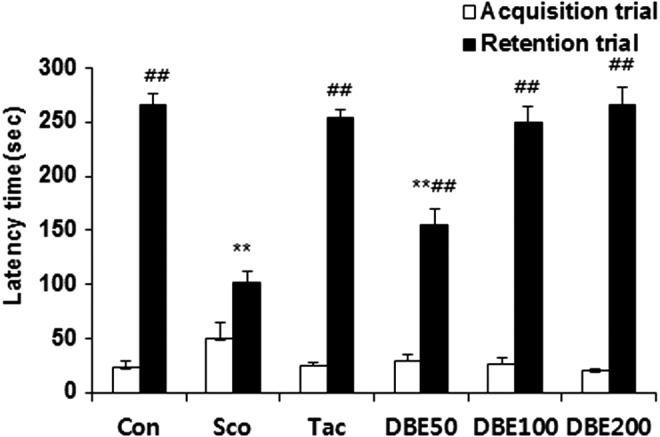
Effect of DBE on the step-through passive avoidance test. At 1 h before the test, DBE (50, 100 or 200 mg/kg, p.o.) or tacrine (10 mg/kg, p.o.) was administered to the mice. Thirty minutes later, the mice were treated with scopolamine (Sco, 2 mg/kg, i.p.) and tested for passive avoidance. To assess the effect of DBE on passive avoidance, DBE (50, 100 or 200 mg/kg, p.o.) was administered to mice 60 min before the tests. Data represent mean±SEM (n=9/group). **P<.01 compared with the control group. ##P<.01 compared with the scopolamine-treated group.
Effects of the DBE on the activities of AChE and ChAT
ACh is an important neurotransmitter responsible for learning and memory. To elucidate the underlying mechanism of DBE in ameliorating memory impairment induced by scopolamine, the activities of cholinergic marker enzymes were determined following the passive avoidance test. As shown in Figure 3, the activity of AChE in brain of the mice treated with scopolamine was higher, compared with the normal control group (P<.05), suggesting that the dysfunction of cholinergic nervous system may promote the process of memory impairment. Meanwhile, the treatment with DBE significantly restored the AChE activity in three DBE-treated groups to the normal level (P<.05). The ChAT activity was significantly increased in the brain of mice treated with DBE (100 mg/kg), compared with scopolamine-treated mice (P<.05). Thus, DBE may have the capability to improve the cholinergic dysfunction, and improve the behavioral dysfunction.
FIG. 3.
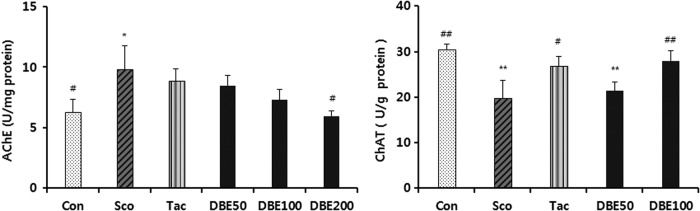
Effects of DBE on acetylcholinesterase (AChE) and choline acetyltransferase (ChAT) activities in brains of mice with memory impairment induced by scopolamine. Animals were decapitated 60 min after the passive avoidance test, and the brain was homogenized to assay AChE and ChAT activities. Data represent mean±SEM (n=6/group). *P<.05 and **P<.01 compared with the control group. #P<.05 and ##P<.01 compared with the scopolamine-treated group.
Effects of DBE on antioxidant defense system
To further elucidate the mechanism of the memory-improving effect of DBE, MDA level, total GSH level, and activities of antioxidant enzymes in the brain were measured following the passive avoidance test. First, the effect of DBE on the level of MDA, a marker of lipid peroxidation, in the brain of mice treated with scopolamine was examined. As shown in Figure 4, the level of MDA in the brain of the mice treated with scopolamine significantly increased, compared with normal control group (P<.01). In the meantime, DBE significantly decreased the MDA level in a dose-dependent manner. Moreover, the lipid peroxidation-suppressing potency of DBE at a dose of 100 or 200 mg/kg was greater than that of tacrine. Next, when the effect of DBE on the GSH level was investigated, it was found that scopolamine treatment significantly decreased the level of total GSH level (Fig. 5). An oral administration of DBE (50 or 200 mg/kg) dose dependently reversed the decrease of total GSH induced by scopolamine; the level of total GSH of mice treated with 200 mg/kg DBE was significantly increased, compared with scopolamine-treated mice (P<.05). Furthermore, DBE at 200 mg/kg demonstrated a similar potency with tacrine in restoring the GSH level. In an additional experiment, activities of antioxidant enzymes such as SOD, GPx, and GR significantly decreased in mice treated with scopolamine, compared with normal control group, whereas DBE restored the scopolamine-induced decrease of these enzyme activities to normal control group (Fig. 6). Of note, the activities of DBE-treated group at 200 mg/kg were similar to those of tacrine-treated group. From these results, it is suggested that DBE may be able to scavenge ROS or increase the antioxidant capacity in brain, thereby preventing the neurons from oxidative stress.
FIG. 4.
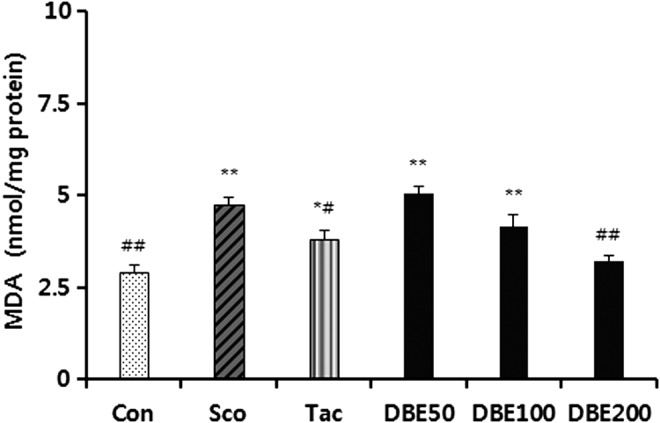
Effects of DBE on malondialdehyde (MDA) level in brains of mice with memory impairment induced by scopolamine. Animals were decapitated 60 min after the passive avoidance test, and the brain was homogenized to assay TBA reactive substances activity. Data represent mean±SEM (n=6/group). *P<.05 and **P<.01 compared with the control group. #P<.05 and ##P<.01 compared with the scopolamine-treated group.
FIG. 5.
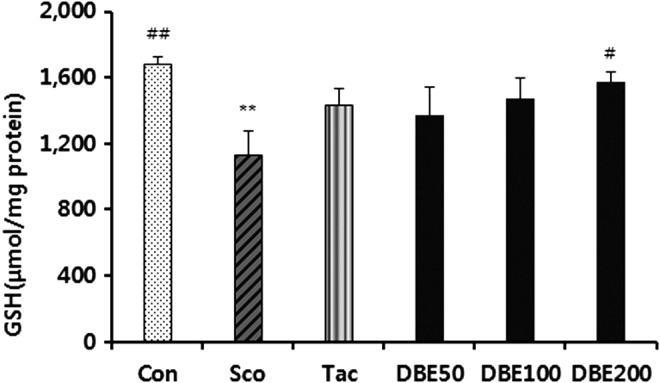
Effects of DBE on glutathione (GSH) level in brains of mice with memory impairment induced by scopolamine. Animals were decapitated 60 min after the passive avoidance test, and the brain was homogenized to assay GSH level. Data represent mean±SEM (n=6/group). **P<.01 compared with the control group. #P<.05 and ##P<.01 compared with the scopolamine-treated group.
FIG. 6.

Effect of DBE on the activities of antioxidant enzymes (A) SOD, (B) GPx, and (C) GR in the brains of mice with memory impairment induced by scopolamine. Data represent mean±SEM (n=6/group). *P<.05 and **P<.01 compared with the control group. #P<.05 and ##P<.01 compared with the scopolamine-treated group.
Effect of DBE on hippocampal CA1 and CA2 regions
Memory impairment induced by scopolamine is known to cause extensive injuries of pyramidal cells in hippocampal CA1 and CA3 regions. In this regard, the effect of DBE on the morphological change in hippocampus of mice treated with scopolamine was examined. As representative morphological change in hippocampus of mice treated with scopolamine was demonstrated in Figure 7, the injured cells exhibited a dark degeneration and shrinkage, leading to a pericellular halo and spongiform change of neurophils. Such degenerate neurons were markedly increased in CA1 and CA3 regions in mice treated with scopolamine alone, in contrast to a few dead cells in normal mice. In comparison, a significant reduction in the number of degenerate neurons was observed in CA1 and CA3 regions in mice administered with DBE (100 or 200 mg/kg). A significant recovery in the number of surviving cells was observed in mice administered with DBE (100 or 200 mg/kg), in comparison with the cell number in mice treated with scopolamine alone (Fig. 8). Thus, DBE appears to attenuate the scopolamine-induced neuronal injuries. On the final day following scopolamine treatment, only 47% and 58% of the neurons survived in CA1 and CA3 regions, respectively, indicative of excessive oxidative stress in brain tissues. Meanwhile, such a loss of hippocampal neurons was remarkably reduced in mice treated with DBE in a dose-dependent manner.
FIG. 7.

Effect of DBE on hippocampal CA1 (A) and CA3 (B) regions of brains from mice treated with scopolamine. Histological sections of the brain tissue showing neurological lesions (A–F). A part of the brain tissue was fixed in 10% neutral formalin solution, and the formalin-fixed brain tissues were processed and embedded in paraffin. Serial coronal sections (4 μm in thickness) were obtained, and stained with Hematoxylin and Eosin. The histopathological examination was assessed under a light microscope (a 400-fold-magnification).
FIG. 8.
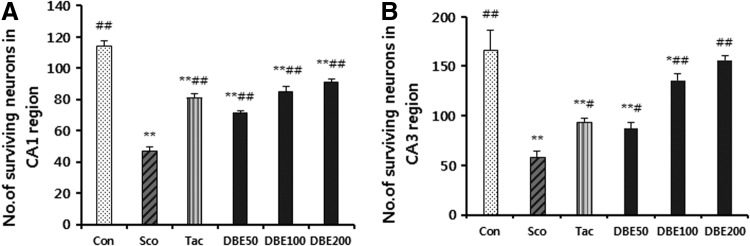
The number of surviving neurons in hippocampal CA1 (A) and CA3 (B) regions. Data represent mean±SEM (n=6/group). *P<.05 and **P<.01 compared with the control group. #P<.05 and ##P<.01 compared with the scopolamine-treated group.
Discussion
In an early pathological feature of AD, synaptic deficit is closely correlated to impaired cognitive function and memory loss.9,10 Recently, it was suggested that the mitochondrial dysfunction is associated with elevated oxidative stress, which triggered amyloidogenic amyloid precursor protein processing and resulted in the elevation of Aβ level in AD. In turn, this might lead to a vicious cycle further impairing synaptic mitochondrial dysfunction, leading to apoptosis, synaptic dysfunction, and memory decline.9,10 It is well known that scopolamine, a cholinergic receptor antagonist, impairs learning and memory processing.17 In particular, scopolamine-induced memory impairment in animals is similar to those observed in the dysfunction of AD or age-related senile central nervous system.16–18 Therefore, the scopolamine-induced memory impairment model has served as a useful tool to investigate the learning and memory processes that involve the cholinergic system.5,18,19,32
In the present study, DBE was found to improve memory impairment induced by scopolamine in vivo test. First, the memory-enhancing effect of DBE was confirmed by MWM test, which was used to evaluate spatial learning ability and long-term spatial memory.33 DBE treatment significantly decreased the EL prolonged by scopolamine during the trial sessions, and increased the number of crossing over the platform site, indicating that mice with a prior experience in the MWM test showed an improvement of spatial memory.34 Thus, it is suggested that DBE can ameliorate scopolamine-induced memory impairment. Additionally, such a beneficial effect of DBE on the long- term memory was further confirmed by a passive avoidance test, which was used to evaluate the effect of treatments on three stages of memory, such as learning acquisition, memory retention, and retrieval process.35 That is, DBE elongated the step-through latency, which was decreased by scopolamine. Thus, it suggested that the antiamnesic effect of DBE on scopolamine-induced memory impairment may be related to mediation of the cholinergic nervous system. In a neurochemical analysis to elucidate the mechanisms of memory-enhancing effects of DBE, the activities of AChE and ChAT as cholinergic markers in brain homogenates were assessed. In a model of memory impairment induced by scopolamine, cholinergic neurotransmission is obstructed, leading to an increase of AChE activity, responsible for the hydrolysis of ACh and the subsequent reduction of ACh level responsible for impaired cognition.14 Cholinergic transmission is terminated mainly by the hydrolysis of ACh by AChE in the synaptic cleft.7,11 It is well known that the antiamnestic effect of tacrine, a representative antiamnestic compound, is due to the inhibition of AChE in the brain. ChAT, which is a synthetic enzyme of ACh in the cholinergic neurons, is the most specific cholinergic marker.13 Therefore, AChE inhibitors and ChAT activators may compensate for reduced ACh level in brains with AD disease. In this study, DBE treatment demonstrated not only significant inhibition of AChE activity, but also promotion of ChAT activity, consistent with the previous reports.16,21 Thus, it could be explained that the antiamnesic effect of DBE on scopolamine-induced impairment of memory may be related to the modification of cholinergic neuronal systems.
Separately, scopolamine-induced memory impairment has been reported to be associated with increased oxidative stress in the brain of mice.18 This was confirmed by the present finding that scopolamine significantly increased lipid peroxidation in the brain, while reducing GSH level.4,18–20 Recently, the increased oxidative damage was reported to directly affect the cholinergic dysfunction in synaptic mitochondria; ACh level showed a significant positive correlation with the activity of ChAT, SOD, and GPx, whereas it demonstrated a significant inverse correlation with MDA and protein carbonyls.36 Meanwhile, the administration of DBE significantly prevented against the increase of lipid peroxidation and the decrease of GSH level in the brain of the mice treated with scopolamine. Furthermore, the DBE treatment restored the activities of antioxidant enzymes such as SOD, GPx, and GR to control level, consistent with the previous studies.20,21
These results indicate that DBE may restore the cholinergic function by preventing scopolamine-induced oxidative damage in brain tissue. This might be further supported by another finding that scopolamine-induced oxidative injuries of the neurons in hippocampal CA1 and CA3 regions were prevented by DBE treatment. Probably consistent with this, our previous study showed that DBE exerted a neuroprotective action against glutamate-induced oxidative damage of hippocampal cell lines by suppressing glutamate-induced phosphorylation of MAPKs.23
Our previous study23 demonstrated that the DBE preparation contained gangliosides and antioxidant peptides, probably responsible for the neuroprotective action of DBE. Earlier, some gangliosides had been reported to show a neuroprotective action.37 Therefore, some part of a neuroprotective action of DBE might be due to the existence of such lipid components. Alternatively, it is feasible that a neuroprotective action of DBE may be partly ascribed to the presence of some proteins or peptides in DBE preparation. In support of this, DBE was observed to contain antioxidant peptides showing a protective action against glutamate-induced oxidative cytotoxicity in HT-22 cells (Unpublished data). Nonetheless, gangliosides and antioxidant peptides may be required to convert to small molecules, readily transported into brain tissue, through a metabolic pathway. That may be why the pretreatment of DBE is required for the efficient neuroprotection in animal experiments. To our knowledge, this is the first report providing evidence that DBE may exert a neuroprotective action against scopolamine-induced amnesia in mice.
In conclusion, memory-enhancing activities of DBE might result from the regulation of cholinergic marker enzyme activities such as AChE and ChAT as well as the suppression of oxidative damage of neurons in the brain of mice treated with scopolamine. These results suggest that DBE may be useful for preventing memory impairment in AD.
Acknowledgment
This work was supported by Nongshim Co., Ltd., Seoul, South Korea.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Coyle JT, Puttfarcken P: Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993;262:689–695 [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B: Oxidative stress and neurodegeneration: Where are we now. J Neurochem 2006;97:1634–1658 [DOI] [PubMed] [Google Scholar]
- 3.Francis PT, Palmer AM, Snape M, et al. : The cholinergic hypothesis of Alzheimer's disease: A review of progress. J Neurol Neurosurg Psychiatry 1999;66:137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwon SH, Ma SX, Joo HJ, et al. : Inhibitory effects of Eucommia ulmoides Oliv. bark on scopolamine-induced learning and memory deficits in mice. Biomol Ther 2013;21:462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collerton D: Cholinergic function and intellectual decline in Alzheimer's disease. Neuroscience 1986;19:1–28 [DOI] [PubMed] [Google Scholar]
- 6.Hasselmo ME: The role of acetylcholine in learning and memory. Curr Opin Neurobiol 2006;16:710–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballard CG, Greig NH, Guillozet-Bongaarts AL, et al. : Cholinesterases: Roles in the brain during health and disease. Curr Alzheimer Res 2005;2:307–318 [DOI] [PubMed] [Google Scholar]
- 8.Melo JB, Agostinho P, Oliveira CR: Involvement of oxidative stress in the enhancement of acetylcholinesterase activity induced by amyloid beta-peptide. Neurosci Res 2003;45:117–127 [DOI] [PubMed] [Google Scholar]
- 9.Leuner K, Schütt T, Kurz C, et al. : Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 2012;16:1421–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leuner K, Müller WE, Reichert AS: From mitochondrial dysfunction to amyloid beta formation: Novel insights into the pathogenesis of Alzheimer's disease. Mol Neurobiol 2012;46:186–193 [DOI] [PubMed] [Google Scholar]
- 11.Chang Q, Gold PE: Switching memory systems during learning: Changes in patterns of brain acetylcholine release in the hippocampus and striatum in rats. J Neurosci 2003;22:3001–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fadda F, Cocco S, Stancampiano R: Hippocampal acetylcholine release correlates with spatial learning performance in freely moving rats. Neuroreport 2000;11:2265–2269 [DOI] [PubMed] [Google Scholar]
- 13.Oda Y: Choline acetyltransferase: The structure, distribution and pathologic changes in the central nervous system. Pathol Int 1999;49:921–937 [DOI] [PubMed] [Google Scholar]
- 14.Levey AI: Muscarinic acetylcholine receptor expression in memory circuits: Implications for treatment of Alzheimer disease. Proc Natl Acad Sci USA 1996;93:13541–13546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bores GM, Huger FP, Petko W, et al. : Pharmacological evaluation of novel Alzheimer's disease therapeutics: Acetylcholinesterase inhibitors related to galanthamine. J Pharmacol Exp Ther 1996;277:728–738 [PubMed] [Google Scholar]
- 16.Ahmed T, Gilani AH: Inhibitory effect of curcuminoids on acetylcholinesterase activity and attenuation of scopolamine-induced amnesia may explain medicinal use of turmeric in Alzheimer's disease. Pharmacol Biochem Behav 2009;91:554–559 [DOI] [PubMed] [Google Scholar]
- 17.Beatty WW, Butters N, Janowsky DS: Patterns of memory failure after scopolamine treatment: Implications for cholinergic hypotheses of dementia. Behav Neural Biol 1986;45:196–211 [DOI] [PubMed] [Google Scholar]
- 18.Kopelman MD, Corn TH: Cholinergic ‘blockade’ as a model for cholinergic depletion. A comparison of the memory deficits with those of Alzheimer-type dementia and the alcoholic Korsakoff syndrome. Brain 1988;111:1079–1110 [DOI] [PubMed] [Google Scholar]
- 19.El-Sherbiny DA, Khalifa AE, Attia AS, et al. : Hypericum perforatum extract demonstrates antioxidant properties against elevated rat brain oxidative status induced by amnestic dose of scopolamine. Pharmacol Biochem Behav 2003;76:523–533 [DOI] [PubMed] [Google Scholar]
- 20.Kumar H, Kim BW, Song SY, et al. : Cognitive enhancing effects of alpha asarone in amnesic mice by influencing cholinergic and antioxidant defense mechanisms. Biosci Biotechnol Biochem 2012;76:1518–1522 [DOI] [PubMed] [Google Scholar]
- 21.Lee MR, Yun BS, Park SY, et al. : Anti-amnesic effect of Chong-Myung-Tang on scopolamine-induced memory impairments in mice. J Ethnopharmacol 2010;132:70–74 [DOI] [PubMed] [Google Scholar]
- 22.Lee MR, Sun BS, Gu LJ, et al. : Effects of the deer antler extract on scopolamine-induced memory impairment and its related enzyme activities. J Korean Soc Food Sci Nutr 2009;38:409–414 [Google Scholar]
- 23.Kim CR, Jeon HL, Shin SK, et al. : Neuroprotective action of deer bone extract against glutamate or Aβ1-42-Induced oxidative stress in mouse hippocampal cells. J Med Food 2014;17:226–235 [DOI] [PubMed] [Google Scholar]
- 24.Morris R: Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 1984;11:47–60 [DOI] [PubMed] [Google Scholar]
- 25.Ellman GL, Courtney KD, Andres V Jr., et al. : A new and rapid colormetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95 [DOI] [PubMed] [Google Scholar]
- 26.Bidlack WT, Tappel AL: Damage to microsomal membrane by lipid peroxidation. Lipids 1973;8:177–182 [DOI] [PubMed] [Google Scholar]
- 27.Lee JH, Felipe P, Yang YH, et al. : Effects of dietary supplementation with red-pigmented leafy lettuce (Lactuca sativa) on lipid profiles and antioxidant status in C57BL/6J mice fed a high-fat high-cholesterol diet. Br J Nutr 2009;101:1246–1254 [DOI] [PubMed] [Google Scholar]
- 28.Pinto MC, Mata AM, Lopes-Barea J: Reversible inactivation of Sacchromyces cerevasiae glutathione reductase under reducing conditions. Arch Biochem Biophys 1984;228:1–12 [DOI] [PubMed] [Google Scholar]
- 29.Tappel AL, Fleischer S, Packer L: Glutathione peroxidase. Methods Enzymol 1978;52:506–523 [DOI] [PubMed] [Google Scholar]
- 30.McCord JC, Fridovich I: Superoxide dismutase. An enzymatic function for erythrocuprein. J Biol Chem 1969;244:6049–6055 [PubMed] [Google Scholar]
- 31.Sok DE, Oh SH, Kim YB, et al. : Neuroprotection by extract of Petasites japonicus leaves, a traditional vegetable, against oxidative stress in brain of mice challenged with kainic acid. Eur J Nutr 2006;45:61–69 [DOI] [PubMed] [Google Scholar]
- 32.Ebert U, Kirch W: Scopolamine model of dementia: Electroencephalogram findings and cognitive performance. Eur J Clin Invest 1998;28:944–949 [DOI] [PubMed] [Google Scholar]
- 33.D'Hooge R, De Deyn PP: Applications of the Morris water maze in the study of learning and memory. Brain Res Rev 2001;36:60–90 [DOI] [PubMed] [Google Scholar]
- 34.Blokand A, Geraerts E, Been A: A detailed analysis of rat's spatial memory in a probe trial of a Morris task. Behav Brain Res 2004;154:71–75 [DOI] [PubMed] [Google Scholar]
- 35.Lorenzini CA, Baldi E, Bucherelli C, et al. : Role of dorsal hippocampus in acquisition, consolidation and retrieval of rat's passive avoidance response: A tetrodotoxin functional inactivation study. Brain Res 1996;730:32–39 [DOI] [PubMed] [Google Scholar]
- 36.Zhang W, Bai M, Xi Y, et al. : Early memory deficits precede plaque deposition in APPswe/PS1dE9 mice: Involvement of oxidative stress and cholinergic dysfunction. Free Radic Biol Med 2012;52:1443–1452 [DOI] [PubMed] [Google Scholar]
- 37.Silva RH, Felicio LF, Frussa-Filho R: Ganglioside GM1 attenuates scopolamine-induced amnesia in rats and mice. Psychopharmacol 1999;141:111–117 [DOI] [PubMed] [Google Scholar]