

Abstract
In spite of considerable interest in the field, reprogramming induced pluripotent stem cells (iPSCs) directly from cancer cells has encountered considerable challenges, including the extremely low reprogramming efficiency and instability of cancer-derived iPSCs (C-iPSCs). In this study, we aimed to identify the main obstacles that limit cancer cell reprogramming. Through a detailed multidimensional kinetic optimization, a highly optimized protocol is established for reprogramming C-iPSCs using nonviral plasmid vectors. We demonstrated how the initial cancer cell density seeded could be the most critical factor ultimately affecting C-iPSCs reprogramming. We have consistently achieved an unprecedented high C-iPSC reprogramming efficiency, establishing stable colonies with typical iPSC morphology, up to 50% of which express the iPSC phenotypic (Oct3/4, Sox2, Nanog) and enzymatic (alkaline phosphatase) markers. Furthermore, established C-iPSC lines were shown to be capable of forming teratomas in vivo, containing cell types and tissues from each of the embryonic germ layers, fully consistent with their acquisition of pluripotency. This protocol was tested and confirmed in two completely unrelated human lung adenocarcinoma (A549) and mouse melanoma (B16f10) cancer cell lines and thus offers a potentially valuable method for generating effectively virus-free C-iPSCs for future applications.
Introduction
The successful demonstration of reprogramming somatic adult cells to acquire a stem cell–like pluripotency through ectopic expression of a few defined transcription factors has opened up a new era across many medical and biological fields (Chun et al., 2010; Ramos-Mejia et al., 2012; Shafa et al., 2012; Sharkis et al., 2012; Takahashi and Yamanaka, 2006; Yu et al., 2007). As a new-horizon discovery, these induced pluripotent stem cells (iPSCs) not only have changed conceptually our basic understanding in terms of developmental biology but also hold great promise in the areas of regenerative medicine, transplantation, oncology, and tumor immunology. For cancer research, attempts have also been made to induce iPSCs directly from tumor cells (C-iPSCs). This may help to reveal a possible link between pluripotency and oncogenic transformation, hence a better understanding of the oncogenic mechanisms or even the fundamental cause of malignancy (Ramos-Mejia et al., 2012). It may also contribute to the development of novel therapeutic options for cancer treatment, e.g., dendritic cells derived from specific C-iPSCs as antigen-presenting cells for tumor vaccine development in the future.
For reprogramming iPSCs from normal somatic adult cells, many different methods have been developed and tested on a variety of cell types in recent years. These experimental protocols, on the basis of their vector types, can be broadly classified as viral and nonviral approaches. In general, the use of viral vectors has achieved a relatively higher reprogramming efficiency (although widely variable, 0.05–11.8%) (Dewi et al., 2012). The main concern is that such vectors could potentially alter endogenous genomic construction and may even increase the risk of tumorigenesis by introducing additional mutations that might confound any insights gained into the original events that caused transformation.
Furthermore, the safety concern of using viruses is a significant hurdle for clinical applications. To overcome this issue, the use of nonintegrating viruses as the vector (Stadtfeld et al., 2008) and alternative virus-free methods (plasmid DNA, synthetic modified mRNA, or cell-penetrating peptides) (Kaji et al., 2009; Okita et al., 2010; Okita et al., 2008; Zhou et al., 2009; Zhu et al., 2010), have subsequently been developed. Among them, a protocol for reprogramming mouse embryonic fibroblasts (MEFs) into iPSCs by transfection using nonviral plasmid vectors encoding four essential transcription factors (Oct-3/4, Sox2, Klf4, and c-Myc) has recently been described (Okita et al., 2008). Although its relatively low reprogramming efficiency may still be a disadvantage, significant improvement can be achieved following multiple repeated transfections (Okita et al., 2008;Okita et al., 2010).
However, despite considerable interest in the field, only a few reports to date have demonstrated successful reprogramming of malignant cells to pluripotency, all of which adopt the viral approach (lentivirus or retrovirus) (Carette et al., 2010; Dewi et al., 2012; Lin and Chui, 2012; Lin et al., 2008; Miyoshi et al., 2010; Nagai et al., 2010). Thus, the generation of cancer-derived iPSCs remains a challenge, and there are many hurdles still to be addressed. The main issues, in addition to the captioned safety concern, include reprogramming inefficiency and relative instability of the resulting C-iPSCs (Ramos-Mejia et al., 2012).
The present study aimed to develop a highly optimized method for reprogramming iPSCs directly from cancer cells using the nonviral plasmid vector approach. To maximize reprogramming efficiency, the underlying basis of the variable susceptibility of different cancer cells to reprogramming needs to be better understood. Due to the transformed nature of cancer cells, conferring on them rapid and uncontrolled proliferation, cancer cells seeded at high density can quickly become overconfluent. The growth of those newly induced iPSC colonies at their initial low frequency may be hindered by the majority of those nontransformed parental tumor cells, particularly after the prolonged culture time required for iPSC reprogramming. On the other hand, if cells are seeded at too low a density, they may senesce, being less amenable to reprogramming. Therefore, it is important to understand the problems involved to remove the barriers preventing efficient C-iPSC induction.
Using a protocol previously developed by Kaji et al. (2009) for iPSCs induction from normal somatic adult cells using the nonviral approach, we aimed to achieve its optimization for efficient reprogramming of C-iPSCs in the present study. A detailed kinetic analysis was carried out, taking into account a combination of parameters considered to be most critical for C-iPSCs induction. The efficiency for cancer cell transfection and iPSC induction was found to be critically dependent on the initial cell density seeded, ratio of plasmid DNA (Oct-3/4, Sox2, Klf4, and c-Myc), as well as transfection reagent concentrations [plasmid to transfection reagent (P:TR) ratio], and detailed time kinetics.
We tested and compared these parameters by reprogramming two very different cancer cell types, the human lung adenocarcinoma (A549) and mouse melanoma (B16f10) cell lines, respectively. We show here that although correct timing and an optimized transfection reagent concentration/ratio are very important kinetic factors to be considered, the initial cancer cell density is the most critical determining factor, which is also very much cancer cell type–dependent. By developing a finely optimized protocol, we demonstrate here that high frequencies (around 50%) of A549-iPSCs and B16f10-iPSCs could be generated reliably, expressing a combination of the enzymatic (alkaline phosphatase [AP]) and phenotypic (Oct-3/4, Sox2, Nanog) markers of iPSCs, with typical iPSC morphology and long-term stability. The pluripotency of our established C-iPSC lines has also been further confirmed by their ability to form teratomas in vivo in immunocompromised mice. We believe that the same principles established in this study can be used to generate virus-free iPSCs from other cancer cells or cell lines, thereby providing a general protocol with widely applicable potential for future studies.
Materials and Methods
Cell lines and maintenance
The A549 human alveolar adenocarcinoma cell line was obtained from Invitrogen (cat. no. k1679), and maintained in RPMI-1640 (Nakalai Tesque, Kyoto) containing 10% fetal bovine serum (FBS). The B16f10 cell line was kindly provided by Dr. Jianguo Chai and Dr. Caroline Addey (Division of Immunology and Inflammation, Imperial College, London), and maintained in Dulbecco's Modified Eagle Medium (DMEM; Nakalai Tesque, Kyoto) containing 10% FBS. Mouse embryonic fibroblast (MEF) feeder cells were maintained in DMEM containing 10% FBS, penicillin/streptomycin, l-glutamine, nonessential amino acids (NEAA), sodium pyruvate, and 2-mercaptoethanol (2ME). The reprogrammed A549-iPSCs and B16f10-iPSCs were maintained in RPMI-1640 (Nakalai Tesque, Kyoto) and DMEM (Nakalai Tesque, Kyoto), respectively, both of which contained 15% KnockOut™ Serum Replacement (Invitrogen/Gibco), penicillin/streptomycin, l-glutamine, NEAA, sodium pyruvate, 2ME, and recombinant murine leukemia inhibitory factor (rmLIF), in the case of the B16f10 C-iPSC lines. All of the cell lines were maintained in cultures at 37°C and 5% CO2.
Plasmid vectors and transfection reagents
For the cell transfection experiments, three plasmid vectors were obtained commercially. These included pCX-OKS-2A (encoding Oct-3/4, Sox2, and Klf4) and pCX-cMyc (encoding c-Myc) from Addgene (cat. no. 19771, 19772) for iPSC induction and pIRES2-EGFP from Clontech (cat. no. 6029-1) for the assessment of cell transfection efficiency. The other main components of the transfection reagents used were Opti-MEM I Reduced Serum Medium (Invitrogen, cat. no. 31985-062) and X-tremeGENE Transfection Reagent (Roche, cat. no. 06366511001). For plasmid purification, a QIAGEN plasmid kit was used according to the standard protocol provided (QIAGEN, cat. no. 27106).
Initial assessment for cancer cell transfection efficiency using the plasmid vectors
A protocol for cell transfection using the virus-free plasmid vectors previously described by Okita et al. (2010) was adopted, but modified for cancer cell transfection in the present study. To optimize for transfection efficiency, an initial assessment was carried out using the pIRES2-EGFP plasmid encoding a fluorescent protein for tracking. Briefly, cancer cells were prepared in six-well plates containing 2 mL per well of fresh medium A549 or B16f10 (RPMI-1640 or DMEM containing 10% FCS) respectively. To prepare for the DNA/X-tremeGENE complexes, for each well, 50 μL of Opti-MEM were transferred into a 1.5-mL test tube. The pIRES2-EGFP plasmid was then added (0.5 μg/0.5 μL), together with the X-tremeGENE Transfection Reagent (TR), at different P:TR ratios (1:1 to 1:6 in volume). After gentle mixing and incubation for 20 min at room temperature, the DNA/X-tremeGENE complex preparation was added to the cancer cell cultures at a 1:1 ratio and incubated overnight at 37°C, 5% CO2.
Following the transfection procedures, at different time points, samples of the transfected cells were collected, washed with fluorescence-activated cell sorting (FACS) buffer and analyzed by flow cytometry to detect and to quantify for the frequency of green fluorescent protein–positive (GFP+) cells. An all-in-one-type fluorescence microscope (BZ-8000; Keyence, Osaka) with digital photographic capability was used to visualize cells at several magnifications, and the images were analyzed with Adobe Photoshop software. The growth rates of the cultured cancer cell lines were measured by counting the number of cells using Cell-Tac (Nihon Koden, Tokyo).
C-iPSC induction
The transfection protocol optimized above was then adopted to transfect A549 and B16f10 cancer cell lines for iPSC induction using the pCX-OKS-2A and pCX-cMyc plasmids. The cancer cells were seeded at a wide range of different cell densities in six-well culture plates and transfected with the pCX-OKS-2A and pCX-cMyc plasmids by the established protocol. Moreover, to enhance the transfection rate and to maintain the transgene expression, a total of four cycles of repeated transfections were carried out at 48-h intervals as previously described for reprogramming of somatic cells (Okita et al., 2008).
By day 10 after the first transfection, the transfected cells were resuspended in 10 mL of culture medium containing 10% KnockOut™ Serum Replacement, transferred to a culture dish covered with feeder cells, and incubated further at 37°C, 5% CO2. Medium changes were performed every other day, and cell passages were made when the cells became 80–90% confluent. By day 30, some iPSC-like colonies were observable that were morphologically different from their parental cancer cells under light microscopy. These cells were then resuspended and transferred to gelatin-coated plates and maintained in the culture medium containing 10% KnockOut™ Serum Replacement for a further period of 10–30 days (i.e., days 40–60). Human iPSCs/embryonic stem cells (ESCs), however, are known to survive poorly as single cells (Lerou et al., 2008). The initial passage of new colonies therefore had to be done mechanically, and several passages (approximately five to ten) were required before the cells could be adapted to enzymatic dissociation.
Following long-term culture, stable iPSC-like colonies were observed forming sizeable cell clusters in the cultures. These cell clusters in suspensions were then carefully collected, purified, and transferred back to new 90-mm culture dishes covered with feeder cells and cultured for a further 3 days at 37°C, 5% CO2. At this stage, the iPSC-like colonies with more defined morphology could be observed in the cultures. The cells were subsequently analyzed and their expression of iPSC enzymatic and phenotypic markers confirmed, as described below.
AP staining for initial iPSCs identification
The induction of iPSC-like cells was initially identified by their expression of AP using the StemTAG™ Alkaline Phosphatase Staining Kit (Cell Biolabs, Inc). According to the protocol, the cells were washed with phosphate-buffered saline containing 0.05% Tween 20 (PBS-T), and fixed at room temperature for 2 min in the fixative solution. After two washes in PBS-T, freshly prepared substrate StemTAG™ Alkaline Phosphate Staining Solution was added and incubated at room temperature for 30 min in the dark. The stained cells were then washed twice with PBS and analyzed by light microscopy. The numbers of AP+ and AP− colonies in 10 randomly selected views of each well were counted twice under an inverted microscope (X-400; Olympus, Japan) and shown as mean values and percentages of the AP+ colonies over the total numbers of colonies.
Flow cytometry analysis
Flow cytometry analysis was carried out throughout for quantification and phenotypic analysis of the GFP-transfected cells and the induced C-iPSCs. For phenotypic analysis, the cells were stained with the BD Stemflow™ human and mouse Pluripotent Stem Cell Transcription Factor Analysis Kits according to manufacturer's recommendations (BD Biosciences, cat. no. 560589, 560585). Briefly, single-cell suspensions were prepared and fixed with the BD Cytofix Fixation Buffer [4% paraformaldehyde (PFA)] and incubated for 20 min at room temperature. The cells were washed twice with 1×BD Perm/Wash Buffer and centrifuged at 500×g for 5 min. The fixed and permeabilized cells (1×106 cells) were then resuspended and stained at room temperature in the dark for 30 min with fluorescence-labeled antibodies specific for human or mouse Oct-3/4, Sox2, and Nanog. These included PE-mouse-anti-human Nanog, PerCP-Cy™5.5 mouse-anti-human Oct-3/4, and Alexa Fluor® 647-mouse-anti-human Sox2; and PE-mouse-anti-mouse Nanog, PerCP-Cy™5.5-mouse-anti-mouse Oct-3/4, and Alexa Fluor® 647-mouse-anti-mouse Sox2 (BD Biosciences). Control samples consisted of cells stained with respective isotype-matched control antibodies. After washes in 1×Perm/Wash buffer, the cells were resuspended in the staining buffer (FBS) for FACS analysis using a flow cytometer (FACSCaliber, BD Biosciences) equipped with the CellQuest software. FlowJo software was used for the data analysis.
Teratoma formation
Two distinct clones of the B16f10 C-iPSC, B16f10/1, and B16f10/2 (generated under the same conditions but from two separate experiments) were tested. The murine C-iPSCs were cultured on mitotically inactivated mouse fibroblast feeder cells in medium containing rmLIF and were dissociated to single-cell suspensions using TrypLE™ Express Dissociation Reagent (Life Technologies, Paisley, UK). Feeder cells, which attach to tissue culture plastic more readily than iPSCs, were removed from the cell suspension by two rounds of 30 min of culture in a minimal volume of medium. To form embryoid bodies (EBs), 0.5×106 cells were plated onto 90-mm bacteriological (low-attachment) petri dishes (Bibby Sterilin) in DMEM containing l-glutamine, NEAA, sodium pyruvate, penicillin/streptomycin, and 2ME plus 10% FBS. The medium and petri dishes were changed on days 3 and 8 following plating by collecting the cell clusters in suspension and allowing them to sediment for several minutes under unit gravity in a 50-mL Falcon tube. Most of the upper supernatant was discarded. The EBs were plated in fresh medium onto new low-attachment petri dishes.
On day 14, between four and six EBs were unilaterally implanted beneath the kidney capsule of immune-compromised CBA.RAG1−/− female mice. EBs from B16f10/1 and B16f10/2 cell lines were transplanted into three recipient mice per cell line. Teratoma tissue was harvested 18 days later and embedded in OCT compound (VWR Chemicals, UK). Frozen sections (7 μm thick) were cut for histological analysis, and representative sections stained with Hematoxylin (Harries) and Eosin (H&E) under standard conditions and imaged on a Nikon Coolscan microscope. All procedures were conducted in accordance with the Home Office Animals (Scientific Procedures) Act of 1986 and received local ethical committee approval.
Statistical Analysis
All data were expressed as the mean±standard deviation (SD). For continuous variables, data were analyzed by the nonparametric one-way analysis of variance (ANOVA) test followed by Tukey comparison (Graphpad Software, San Diego. CA). Student's t-test was also used for comparisons between two groups (Graphpad Software, San Diego. CA). p values ≤0.05 were considered statistically significant, and ≤0.01 highly significant.
Results
Optimization for transfection efficiency of cancer cells using nonviral plasmid vectors
To maximize the transfection efficiency, a detailed kinetic analysis was carried out first of all to determine the optimal condition for cancer cell transfection using the plasmid vector encoding enhanced GFP (eGFP) (pIRES2-EGFP). Data shown in Figure 1A–C are frequency (ratio) changes of the transfected (GFP+) A549 human lung cancer cells under different experimental conditions, including concentration of the transfection reagents [i.e., P:TR ratios] (Fig. 1A), time after the transfection (Fig. 1B), and the initial cancer cell densities seeded in the cultures (Fig. 1C). As indicated, the density of tumor cells initially seeded appears to be one of the most critical factors that largely determines the transfection efficiency. Under the highly optimized condition (initial cell density seeded, 6.25×103/mL; P:TR ratio, 1:3; time, 48 h), at its peak, around 25% of the A549 cells were GFP+ (transfected) (Fig. 1C arrow). The ratios of GFP+ cells to unlabeled cells, however, subsequently and quickly declined (Fig. 1D, left panel). This was found to be mainly due to those much faster proliferating nontransfected (GFP−) cells outcompeting their transfected (GFP+) counterparts, as clearly reflected by the respective total cell counts in cultures (Fig. 1D–E, left panels). Similar kinetic changes were also observed when the B16f10 mouse melanoma cell line was tested at its respective optimal conditions (Fig. 1D–E, right panels). Again, the initial cell density appears to be a critical factor for efficient B16f10 cell transfection although, due to their even faster proliferation rate, the optimal initial cell density was found to be five-fold lower (at 1.2×103/mL; data not shown) compared to their A549 counterparts (6.3×103/mL; Fig. 1C).
FIG. 1.


Detailed kinetic optimization for maximizing the efficacy of transfecting malignant cell lines. The two unrelated but well-established human lung adenocarcinoma (A549) and mouse melanoma (B16f10) cancer cell lines were transfected once with the pIRES2-EGFP expression vector as described in Materials and Methods, under different experimental conditions detailed below, and analyzed by flow cytometry. (A–C) Representative FACS profiles illustrating the effects of: (A) Different plasmid (pIRES2-EGFP) and transfection reagent concentrations (P:TR ratios, 1:1 to 1:6) at 48 h with a fixed initial cell density (105/mL). (B) Time kinetics with a fixed initial cell density (105/mL) and P:TR ratio (1:3). (C) A range of different cell densities seeded with a fixed P:TR ratio (1:3) and time (48 h), on the efficiency of transfecting A549 cells. The linear graphs plotted at the bottom of A, B, and C illustrate the respective kinetic changes from the results calculated and shown as the means (±SD) of triplicate samples. Arrows indicate the peaks of expression levels. (D) Time kinetic changes of GFP+ cell frequency shown as a percentage (GFP+%) detected in the cell cultures under the highly optimized conditions for iPSCs induction from A549 (initial cell density seeded at 6.3×103/mL; P:TR ratio, 1:3) and from B16f10 (initial cell density seeded at 1.2×103/mL; P:TR ratio, 1:3) cell lines (left and right panels), respectively. (E) Time kinetic changes of GFP+ and GFP− cell frequency shown as total cell counts (absolute number) detected in the same cultures mentioned in D. Data shown in D and E were of mean (±SD) calculated from triplicate samples. Statistical analysis (D): Nonparametric one-way ANOVA test, p ≤ 0.01 (highly significant).
Effects of initial cell density on iPSC induction from the A549 and B16f10 cancer cell lines
Following the initial optimization for transfection efficiency above, the efficiency for reprogramming iPSCs from two very different cancer cell lines was assessed and compared side by side, focusing on the effects of cancer cell density initially seeded in the cultures. Moreover, to enhance the reprogramming efficiency, the strategy of multiple repeated transfections, which have been shown to improve the reprogramming of somatic adult cells (Okita et al., 2010), was also applied in our experimental model. Data shown in Figure 2 were derived from representative experiments showing induction of iPSC-like cells from the A549 and B16f10 cancer cell lines, on the basis of their expression of the iPSCs enzymatic marker (AP). At day 10, i.e., after a total of four repeated transfections (48-h intervals), a number of AP+ cells were detected in the reprogrammed A549 cells, but only at very low frequency in the B16f10 cultures (Fig. 2A, B, left panels). At day 30, i.e., 24 days after the 4th (last) transfection procedure, although high frequencies of AP+ cells could be detected in the A549 cultures with a peak again at the optimized cell density (6.3×103/mL), significant numbers of AP+ cells also appeared in the B16f10 cultures (Fig. 2A, B, right panels). The differences were further analyzed quantitatively by counting the numbers of AP+ and AP− colonies in 10 randomly selected views of each culture condition/well under an inverted microscope (X-400; Olympus, Japan). The counting was repeated twice in a blinded manner, and the mean values shown as the percentages of AP+ clones over the total numbers of colonies detected in the cultures at day 10 and day 30, respectively (Fig. 2C, D, histograms).
FIG. 2.

Effects of initial cell density on iPSCs induction from the A549 human lung adenocarcinoma and B16f10 murine melanoma cell lines. A549 and B16f10 cells were reprogrammed for iPSCs induction using the pCX-OKS-2A and pCX-cMyc plasmids four times at 48-h intervals as described in Materials and Methods. The programming was carried out under the optimized conditions for cell transfection in terms of P:TR ratio (1:3), timing (48 h), and a range of different cell density initially seeded in the cultures (as indicated in graphs). By day 10 and day 30, the cell cultures were stained for the expression of the iPSC enzymatic marker, AP. (A and B) Representative photos (magnification, 400×) showing detection of AP+ cells (red) in the reprogrammed A549 (A) and B16f10 (B) cultures of day 10 and day 30, respectively, as indicated in the graphs. The percentages of AP+ cells were quantified and further analyzed under the microscope by counting cells in 10 randomly selected views and shown as the means (±SD) for the A549 (C) and B16f10 (D) cultures (n=3) of day 10 and day 30, respectively. Statistical analysis (C and D): Nonparametric one-way ANOVA test, p ≤ 0.05 (significant) and ≤ 0.01 (highly significant).
Under light microscopy (X-100, Olympus, Japan), these AP+ colonies also appeared to be different morphologically from their parental counterparts and started showing an iPSC-like morphology around days 20–30 with sharp and defined cellular margins (Fig. 3). The frequency of these cells in the cultures increased in a time-dependent manner. By day 60 and day 40, for the A547 (Fig. 3A) and B16f10 (Fig. 3B) cell cultures, respectively, high frequencies of these AP+ iPSC-like cells became readily detectable. At this late stage, they were also less amenable to being taken over by, and physically more separable from, their nontransformed counterparts.
FIG. 3.
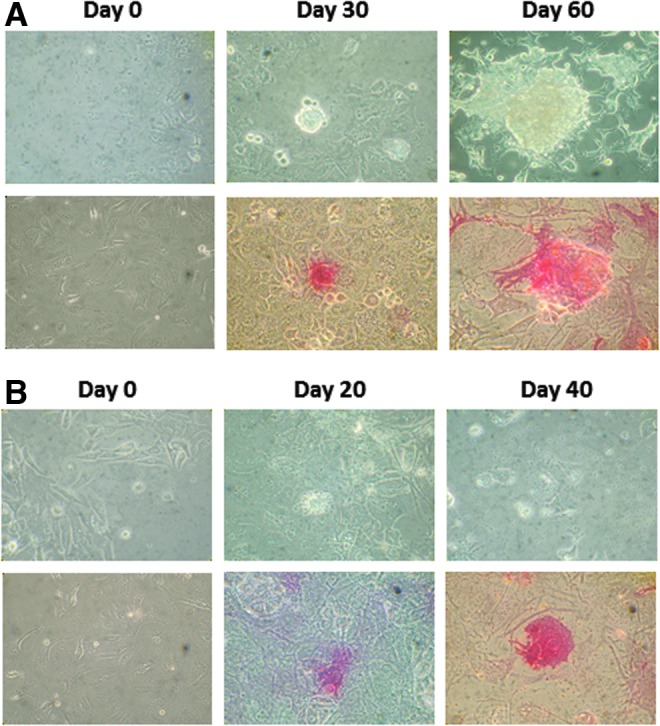
Establishment of stable iPSC-like colonies from the A549 and B16f10 cancer cell lines by using the highly optimized reprogramming protocol. iPSCs were induced from A549 (A) and B16f10 (B) cell lines as described in the Fig. 2 legend. Cell morphological changes and AP expression were monitored after the multiple reprogramming procedures (four times, at 48-h intervals). Representative graphic photos showing the morphological features (upper panels) and AP+ (in red, bottom panels) were taken under an inverted microscope (magnification, 400×) at days 0, 30, and 60 of the A549 (A) and days 0, 20, and 40 of the B16f10 (B) cultures.
Phenotypic characterization of the A549 and B16f10-derived iPSC-like cells
To confirm these results further phenotypically, the iPSC-like cells were harvested and stained for their expression of iPSC markers. Flow cytometry analysis revealed that these cancer-derived iPSC-like cells expressed several typical pluripotency markers, including Sox2, Oct-3/4, and Nanog (Fig. 4). The expression of these transcription factors, in terms of frequency and level, also increased in a time-dependent manner. Sox2, being an early marker of pluripotency, was clearly detectable at days 20–30, followed by the appearance of Oct-3/4 and Nanog at a relatively later stage (Fig. 4A, B). By day 60 and day 40 for the A549 and B16f10 lines, respectively, many of these cells, as high as 31.6% and 24.3% from those cultures with highly optimized reprogramming conditions (Fig. 4C, D, arrows), were found to be co-expressing Sox2 and Oct-3/4. As shown in Figure 4C and D, the initial cell density was again confirmed to be the most critical factor that determined the overall reprogramming efficacy of the C-iPSCs and cancer cell type-dependency. The experiments were repeated for more than three times, and in replicates for both cell lines under the described conditions, and with consistent results as defined by dose kinetics in terms of the optimal condition versus the initial cell density seeded. There is in general again a five-fold difference between the two tumor cell lines tested in terms of the optimal cell density initially seeded, which correlated with the proliferation rates of their parental tumor cells in the control cultures (three- to four-fold difference; data not shown).
FIG. 4.


Phenotypic characterization of the A549-iPSC and B16f10-iPSC cell lines. iPSCs were induced from A549 and B16f10 cancer cell lines as described in the Fig. 2 legend, under the optimized conditions (initial cell density seeded, P:TR ratio, and timing indicated in the graphs). The expression of the stem cell markers, including Oct-3/4, Sox2, and Nanog at day 60 and day 40 in A549-iPSCs (A, C) and B16f10-iPSCs (B, D), respectively, was determined by flow cytometry. The relative expression levels (FACS profiles as histograms in A and B; or dot plots in C and D) and frequencies of cells expressing each of these three markers individually (A, B), and cells co-expressing both Sox2 and Oct-3/4 (C, D), are shown. Arrows indicate the peak of expression level. Data shown are from one representative experiment in triplicate (mean±SD; n=3); the experiment was repeated for more than three times with consistent results under the respective conditions described.
Establishment of stable A549 and B16f10 C-iPSC lines
The results shown in Figures 2–4 indicate that there was a stable increase of these iPSC-like cells in the long-term cultures, in terms of the frequency and typical morphological features of the cells, and their expression of the typical enzymatic as well as intracellular pluripotency markers. Subsequently, these stable iPSC-like colonies, which formed sizeable cell clusters in suspension, were isolated and transferred back to the feeder cell–coated plates. After 3 more days in culture, typical iPSC colonies with more defined morphology were observed and further characterized by their expression of the iPSC markers. As shown in Figure 5, these replated cells expressed enhanced and stable levels of AP activity with high frequencies, around 50% of them co-expressing Oct-3/4 and Sox2 transcription factors (A549-iPSC, 51.1%, Fig. 5A; B16f10-iPSC, 43.4% Fig. 5B).
FIG. 5.
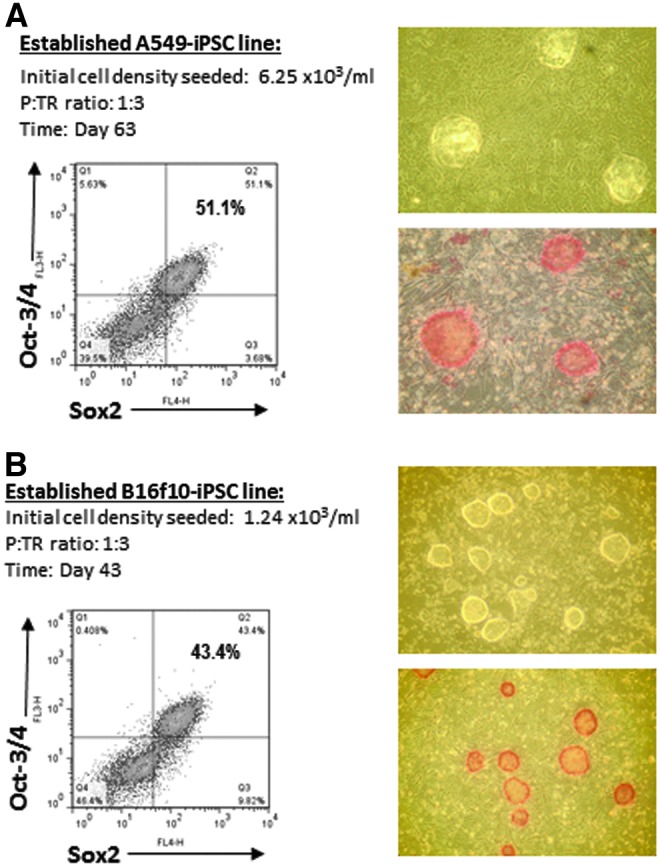
Morphological and phenotypic characteristics of the established (replated and purified) A549-iPSC and B16f10-iPSC cell lines. Following long-term cell culture, the reprogrammed A549 (day 60) and B16f10 (day 40) iPSC-like cells that formed large and floating colonies were isolated, replated, and further differentiated for 3 days in feeder-coated plates. These replated/differentiated cells were then collected and reanalyzed morphologically and phenotypically. Representative photos (magnification, 100×) and FACS profiles of the established A549-iPSC (A) and B16f10-iPSC (B) cell lines are shown, illustrating the typical iPSC morphology (upper photo) and the expression of iPSC enzymatic (AP, lower photo in red) as well as intracellular (Oct-3/4, Sox2) ESC markers.
To demonstrate the capacity of C-iPSC lines to support the differentiation of cell types from each of the three embryonic germ layers, we performed teratoma assays using two independent clones derived from the B16f10 murine cell line. EBs were formed in suspension cultures and were implanted under the kidney capsule of immune-compromised female CBA.RAG1−/− mice. All six recipients showed significant teratoma growth when sacrificed 18 days later. Figure 6 shows histological analysis of H&E-stained sections from representative teratomas from either of the two C-iPSC clones. Tissues derived from each of the three embryonic germ layers could be readily identified, including putative gut-like structures of endodermal origin (Fig. 6A, D), mesodermally derived cartilage and smooth muscle (Fig. 6B, E), and ectodermal tissues including epidermis of the skin (Fig. 6F). Interestingly, we were also able to identify in a teratoma derived from the B16f10/1 C-iPSC line a structure highly reminiscent of the developing eye with well-defined retinal pigmented epithelium (Fig. 6C and inset), retina, and putative lens, derived from the ectoderm. The ability of C-iPSC lines to give rise to structures from each of the embryonic germ layers is fully consistent with their acquisition of pluripotency,
FIG. 6.
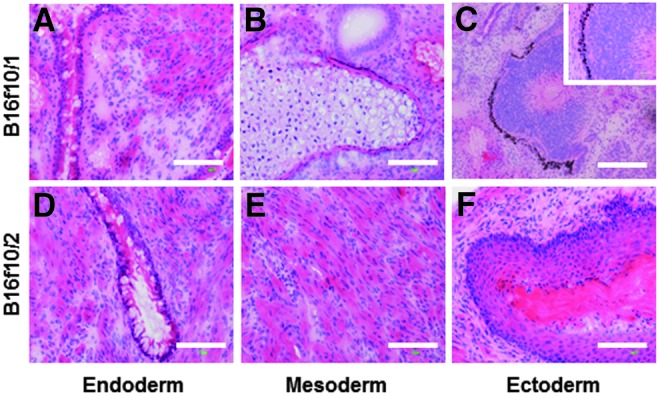
Pluripotency of C-iPSC lines demonstrated by teratoma formation. Representative sections from teratomas derived from the B16f10/1 (A–C) and the B16f10/2 (D–F) C-iPSC lines, stained with H&E. Pluripotency was verified by identification of tissues derived from each of the three embryonic germ layers. (A and D) Gut-like tissues of endodermal origin, including putative goblet cells within simple columnar epithelium. (B and E) Mesodermal differentiation represented by cartilage (B) and smooth muscle (E). (C and F) Ectodermal differentiation illustrated by the development of a structure reminiscent of the developing eye with well-defined retinal pigmented epithelium, retina, and lens (C and inset), and keratinized stratified epithelium of the epidermis (F). Scale bars, 100 μm.
Discussion
In this study, a highly optimized protocol has been developed for reprogramming cancer cells to pluripotency with high efficiency. Importantly, we show for the first time that this can be successfully and reliably achieved using a nonviral plasmid vector approach. To achieve this, we have carried out a detailed kinetic optimization focusing on key factors or aspects that could significantly impact on the efficiency of cancer cell transfection and reprogramming. The present findings have confirmed our hypothesis that the main obstacle preventing efficient iPSCs induction from cancer cells (C-iPSCs) is the rapid expansion of those parental tumor cells over their transfected and subsequently transformed counterparts. We demonstrate here that, due to the superiority of these parental tumor cells versus the length of time required for reprogramming to pluripotency (>10 days), the initial cell density seeded in culture is one of the most critical parameters to consider to achieve a high reprogramming efficacy for C-iPSCs induction and establishment.
The first issue being addressed here is about transfection efficiency. Cancer cells have been shown to share many biological features with stem cells in terms of their high and dynamic genomic activities required for self-renewal and potential differentiation into multiple cell types. This particular feature should endow them with a higher transfection rate over that of benign and normal adult cells. Indeed, we showed that their one-hit transfection rate at its peak (48 h) could be rather high (∼20% under the optimized conditions; Fig. 1D). However, due to the superiority and significantly faster expansion of those nontransfectants (Fig. 1E), the relative ratio of those transfected cells in the cultures soon declined. As a result, the overall frequency of transfected cells became very low in the cultures at later time points. In addition, for iPSC induction, the fast expanding parental tumor cells could also hinder the growth of new iPSC colonies by posing the risk of cell layer lifting, particularly after the prolonged culture times required for the reprogramming (Kaji et al., 2009). Together, these factors may contribute to and help explain the low success rate in C-iPSCs reprogramming, even by using the virus-based approaches as previously reported (Carette et al., 2010; Lin and Chui, 2012; Lin et al., 2008; Miyoshi et al., 2010; Nagai et al., 2010).
Certain cancer-specific genetic lesions have been shown to affect C-iPSCs programming, although whether and how these defects may explain the barriers responsible in the process is still unclear. Reduced expression of the tumor suppressor p53, for example, was found to result in a more efficient cellular reprogramming (Banito and Gil, 2010). However, because most tumors are linked with a defective p53 suppressor pathway, one would expect an enhanced rather reduced reprogramming efficiency of C-iPSCs. When different lines of evidence are drawn together, it was concluded that genetic defects on their own might be insufficient to impede the C-iPSCs reprogramming efficacy (Ramos-Mejia et al., 2012). Therefore, it is likely that other biological or technical barriers are involved in the process.
Our present findings demonstrate that the induction of C-iPSCs with high frequencies may be achieved reproducibly by using the nonviral approach, as long as a properly optimized protocol is employed. The most important parameter to be considered is the cell density initially seeded in the cultures. Different types of tumor cells may understandably vary in their rate of expansion. This suggests that the protocol also needs to be tailor-made in this regard, i.e., the inclusion of dose-kinetic analysis for each type of cancer cells being tested, to maximize the reprogramming efficiency for general practice. By taking this factor into account, and by employing the multiple repeated transfection strategy (Okita et al., 2010), we showed evidence that as high as 50% of the cells or more with typical iPSCs morphology and phenotypic markers could be consistently achieved. This was tested and confirmed in our study by reprogramming two totally unrelated but well-established cancer cell lines, i.e., the B16f10 mouse melanoma and A549 human adenocarcinoma cell lines, pointing to the general applicability of this method.
Furthermore, this virus-free technique not only provides an optimized method for reprogramming of cancer lines into iPSCs using plasmid vectors, but also reduces the safety concern for their potential future clinical applications (Lin and Chui, 2012). In the present study, we have also made an observation similar to that previously reported for iPSCs induction from mouse and human embryonic fibroblasts or somatic adult cells (Fluri et al., 2012; Kaji et al., 2009; Shafa et al., 2012). We observed that, while most of their nontransformed parental counterparts adhered to the gelatin-coated wells, the induced C-iPSC colonies could grow well in suspension. Such differences made it relatively easy to separate them physically. After subsequent transfer into a new well for further but brief expansion, we obtained a consistently high percentage and purity (∼50%) of cells expressing the typical iPSC enzymatic (AP) and intracellular phenotypic (Oct-3/4, Sox2, Nanog) markers. In conclusion, this highly optimized nonviral protocol for C-iPSCs reprogramming can be a very useful tool in generating effectively virus-free C-iPSCs for future studies and, potentially, clinical applications.
Acknowledgments
We would like to thank Dr. Jianguo Chai, Dr. Caroline Addey, Dr. Heidi Ling, Dr. Mitali Patel, and Dr. Sharmal Narayan at the Division of Immunology & Inflammation, Department of Medicine, Imperial College, London, for their kind help in providing the tumor cell lines and technical advice, as well as useful scientific discussions during the study. H.Z. was supported by the Pennine-Great Wall Ltd. Medical Research Fund, Y.C. by the Chinese Government Visiting Research Fellow Scheme, S.S. and F.P.H. by the ARUK Project Grant (18523). P.J.F. was supported by the Regenerative Medicine Initiative of the Britain–Israel Research and Academic Exchange Partnership (BIRAX).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
- Banito A., and Gil J. (2010). Induced pluripotent stem cells and senescence: Learning the biology to improve the technology. EMBO Rep. 11, 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J.E., Pruszak J., Varadarajan M., Blomen V.A., Gokhale S., Camargo F.D., Wernig M., Jaenisch R., and Brummelkamp T.R. (2010). Generation of iPSCs from cultured human malignant cells. Blood 115, 4039–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun Y.S., Chaudhari P., and Jang Y.Y. (2010). Applications of patient-specific induced pluripotent stem cells; focused on disease modeling, drug screening and therapeutic potentials for liver disease. Int. J. Biol. Sci. 6, 796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewi D., Ishii H., Haraguchi N., Nishikawa S., Kano Y., Fukusumi T., Ohta K., Miyazaki S., Ozaki M., Sakai D., Satoh T., Nagano H., Doki Y., and Mori M. (2012). Reprogramming of gastrointestinal cancer cells. Cancer Sci. 103, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluri D.A., Tonge P.D., Song H., Baptista R.P., Shakiba N., Shukla S., Clarke G., Nagy A., and Zandstra P.W. (2012). Derivation, expansion and differentiation of induced pluripotent stem cells in continuous suspension cultures. Nat. Methods 9, 509–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji K., Norrby K., Paca A., Mileikovsky M., Mohseni P., and Woltjen K. (2009). Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458, 771–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerou P.H., Yabuuchi A., Huo H., Miller J.D., Boyer L.F., Schlaeger T.M., and Daley G.Q. (2008). Derivation and maintenance of human embryonic stem cells from poor-quality in vitro fertilization embryos. Nat. Protoc. 3, 923–933 [DOI] [PubMed] [Google Scholar]
- Lin F.K., and Chui Y.L. (2012). Generation of induced pluripotent stem cells from mouse cancer cells. Cancer Biother. Radiopharm. 27, 694–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.L., Chang D.C., Chang-Lin S., Lin C.H., Wu D.T., Chen D.T., and Ying S.Y. (2008). Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA 14, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi N., Ishii H., Nagai K., Hoshino H., Mimori K., Tanaka F., Nagano H., Sekimoto M., Doki Y., and Mori M. (2010). Defined factors induce reprogramming of gastrointestinal cancer cells. Proc. Natl. Acad. Sci. USA 107, 40–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai K., Ishii H., Miyoshi N., Hoshino H., Saito T., Sato T., Tomimaru Y., Kobayashi S., Nagano H., Sekimoto M., Doki Y., and Mori M. (2010). Long-term culture following ES-like gene-induced reprogramming elicits an aggressive phenotype in mutated cholangiocellular carcinoma cells. Biochem. Biophys. Res. Commun. 395, 258–263 [DOI] [PubMed] [Google Scholar]
- Okita K., Nakagawa M., Hyenjong H., Ichisaka T., and Yamanaka S. (2008). Generation of mouse induced pluripotent stem cells without viral vectors. Science 322, 949–953 [DOI] [PubMed] [Google Scholar]
- Okita K., Hong H., Takahashi K., and Yamanaka S. (2010). Generation of mouse-induced pluripotent stem cells with plasmid vectors. Nat. Protoc. 5, 418–428 [DOI] [PubMed] [Google Scholar]
- Ramos-Mejia V., Fraga M.F., and Menendez P. (2012). iPSCs from cancer cells: Challenges and opportunities. Trends Mol. Med. 18, 245–247 [DOI] [PubMed] [Google Scholar]
- Shafa M., Day B., Yamashita A., Meng G., Liu S., Krawetz R., and Rancourt D.E. (2012). Derivation of iPSCs in stirred suspension bioreactors. Nat. Methods 9, 465–466 [DOI] [PubMed] [Google Scholar]
- Sharkis S.J., Jones R.J., Civin C., and Jang Y.Y. (2012). Pluripotent stem cell-based cancer therapy: Promise and challenges. Sci. Transl. Med. 4, 127ps9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M., Nagaya M., Utikal J., Weir G., and Hochedlinger K. (2008). Induced pluripotent stem cells generated without viral integration. Science 322, 945–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., and Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R., Slukvin I.I., and Thomson J.A. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 [DOI] [PubMed] [Google Scholar]
- Zhou H., Wu S., Joo J.Y., Zhu S., Han D.W., Lin T., Trauger S., Bien G., Yao S., Zhu Y., Siuzdak G., Schöler H.R., Duan L., and Ding S. (2009). Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4, 381–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Li W., Zhou H., Wei W., Ambasudhan R., Lin T., Kim J., Zhang K., and Ding S. (2010). Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 7, 651–655 [DOI] [PMC free article] [PubMed] [Google Scholar]