

Abstract
Objective
To determine whether a combination of B cell depletion and BAFF blockade is more effective than monotherapy in treating models of spontaneous or accelerated systemic lupus erythematosus (SLE) in (NZB × NZW)F1 mice.
Methods
Clinical parameters such as disease progression–free survival, proteinuria, and renal injury were assessed in models of spontaneous, interferon-α (IFNα)–accelerated, or pristane-accelerated lupus in (NZB × NZW)F1 mice. Treatment arms included anti-CD20 (B cell depletion), B lymphocyte stimulator receptor 3 fusion protein (BR-3-Fc) (BAFF blockade), the combination of anti-CD20 and BR-3-Fc, isotype control, or cyclophosphamide. In models of spontaneous, IFNα-accelerated, or pristane-accelerated lupus, mice were treated for 24 weeks, 8 weeks, or 12 weeks, respectively. Peripheral and resident B cell subsets and various autoantibodies were examined.
Results
Compared to B cell depletion or BAFF blockade alone, combined therapy significantly improved disease manifestations in all 3 lupus models. In addition, marginal zone B cells, plasmablasts, and circulating and tissue plasma cells were decreased more effectively. Dual B cell immunotherapy also reduced multiple classes of pathogenic autoantibodies, consistent with its observed effectiveness in reducing immune complex–mediated renal injury.
Conclusion
Dual immunotherapy via B cell depletion and BAFF blockade is more efficacious than single agent immunotherapy in murine SLE models, and this combination treatment is predicted to be an effective strategy for immunotherapy in human SLE.
Systemic lupus erythematosus (SLE) is a relapsing autoimmune disease affecting multiple organs including the kidney, skin, and central nervous system; it manifests in a diverse pathology depending on the target tissue involved (1). In SLE, B cell homeostasis is severely disturbed and accompanied by an overactive germinal center (GC) reaction likely due to a failure to maintain B cell tolerance and to cull autoreactive B cells (2–4). Substantial preclinical and clinical data suggest that pathogenic B cells contribute to SLE pathogenesis by complex mechanisms including autoantibody production, antigen presentation, and cytokine generation (5). With the advancement of clinical proof-of-concept studies in human SLE, B cells are emerging as a validated pathogenic cell target. Moreover, we are beginning to understand which B cell subsets might be involved in disease, allowing generation of improved, rational therapeutic hypotheses.
Recently, belimumab, an antagonist of B lymphocyte stimulator (BLyS), has shown clinical efficacy in the treatment of active autoantibody-positive SLE, providing a rationale for further investigating B cell–targeted therapeutics (6). In human SLE, belimumab treatment led to decreased naive and transitional B cells with modest reduction of plasma cells (PCs) (7). Rituximab is a chimeric anti-CD20 monoclonal antibody (mAb) that depletes transitional and naive B cells but not PCs, which lack CD20 (5,8). Rituximab more effectively reduces circulating B cells compared to tissue-resident B cells (5). With rituximab treatment, several positive results have been generated in open-label SLE trials (9–11), although rituximab did not demonstrate efficacy in a randomized SLE trial (12).
Studies of anti-CD20–mediated B cell depletion in mice have shown that there is a tissue- and subset-specific hierarchy of sensitivity of B cells to depletion (13–15). It has been shown that circulating B cells are most sensitive to rapid depletion as compared to the slow and incomplete depletion of B cells in spleen, lymph nodes, or bone marrow (BM). In addition, survival cues provided by BAFF such as activation of the B cell survival pathway have been implicated in resistance to B cell depletion therapy (14). The reduced depletion of various B cell subsets might be explained in part by lower expression of CD20, B cell intrinsic factors, bioavailability of antibodies, and survival cues (13–15). In (NZB × NZW)F1 mice, anti-CD20 efficiently depletes naive B cells; however, marginal zone (MZ) B cells and PCs are somewhat more resistant (15). In contrast, BAFF blockade can have a profound effect on the survival and maturation of transitional, follicular, and MZ B cells (14,15), and BAFF itself can selectively prolong the survival of plasmablasts (16,17). Autoreactive B cells may be more dependent on BAFF than naive mature B cells (18,19). Short-course treatment (4 weeks) with the combination of anti-CD20 and BAFF blockade provides improved efficacy in (NZB × NZW)F1 mice with spontaneous lupus (15). Therefore, distinct and/or overlapping mechanisms contribute to the beneficial effects seen with either anti-CD20– or BAFF antagonist–based treatment modalities. Correspondingly, maximum preclinical efficacy and B cell depletion may require a broader impact on various B cell subsets.
The (NZB × NZW)F1 preclinical mouse model has been successfully used to bridge animal studies to the clinic, including validation of anti-CD20 as well as BAFF antagonists (20,21). In the (NZB × NZW)F1 mouse model of spontaneous lupus, various aspects of SLE develop, including lymphadenopathy, splenomegaly, and elevated autoantibodies. Immune complex–mediated glomerulonephritis appears at the approximate age of 5 months and leads to kidney failure and death at the approximate age of 10 months (22). Antinuclear antibodies (ANAs) are abundant, including anti–double-stranded DNA (anti-dsDNA) IgG antibodies, a majority of which are IgG2a and IgG3; however, certain autoantibodies such as anti-RNP are not present (22). In the model of pristane-accelerated lupus in (NZB × NZW)F1 mice, the production of anti-RNP autoantibodies is accompanied by rapid precipitation of lupus-like disease in which pathogenic cytokines such as interferon-α (IFNα) and downstream target genes are remarkably elevated, capturing other aspects of human SLE (23,24). Ectopic expression of IFNα also results in the rapid manifestation of SLE-like symptoms in an (NZB × NZW)F1 mouse model (25).
We report how the combination of a B cell depletion strategy with BAFF blockade impacts various aspects of B cell–driven pathogenesis in (NZB × NZW)F1 mouse models of spontaneous, IFNα-accelerated, or pristane-accelerated lupus after a long-term treatment course. Our data support the hypothesis that this combination therapy has the potential to provide a wider umbrella to cover overlapping and/or nonoverlapping depletion mechanisms to mediate better efficacy than that achievable using single therapeutics.
MATERIALS AND METHODS
Animal models
Female (NZB × NZW)F1 mice were purchased from The Jackson Laboratory and housed under specific pathogen–free conditions. All procedures were approved by the Institutional Animal Care and Use Committee. Models used rely on the syndrome that occurs in (NZB × NZW)F1 mice and thus, depending on the model variant, recapitulate at least 4 features of human SLE (genetic predisposition, glomerulonephritis, ANAs, and anti-dsDNA antibodies) (22). In spontaneous lupus studies, female, 7–8-month-old (NZB × NZW)F1 mice with moderate-to-severe proteinuria (100–300 mg/dl) were randomly enrolled in therapeutic groups (n = 20) and treated for 6 months (Figure 1A).
Figure 1.
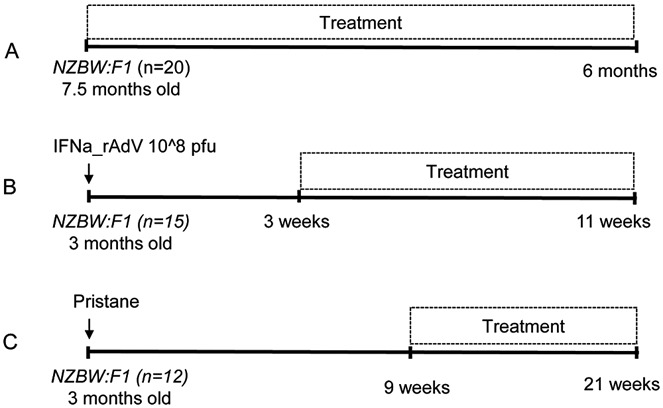
Schematic of preclinical disease models in (NZB × NZW)F1 (NZBW:F1) mice. A, Model of spontaneous lupus. B, Model of interferon-α (IFNα)–accelerated lupus. C, Model of pristane-accelerated lupus. IFNa_rAdV = adenovirus expressing recombinant murine IFNα; PFU = plaque-forming units.
Accelerated lupus in (NZB × NZW)F1 mice was precipitated by IFNα overexpression (25). Adenovirus expressing murine IFNα (IFNα-rAdv) or LacZ control (LacZ-rAdv) was used to induce lupus-like disease (Figure 1B). IFNα-rAdv or LacZ-rAdv was generated in-house and stored at 4–5 × 1012 particles/ml in 20 mM HEPES, 150 mM NaCl, and 10% glycerol. Twelve-week-old (NZB × NZW)F1 mice were administered 108 plaque-forming units (PFU) of IFNα-rAdv (n = 15) or LacZ-rAdv (n = 15) by tail vein injection. Elevated serum IFNα peaked 3 weeks after induction at an average of 10 ng/ml, then gradually declined to 4 ng/ml at week 6. Enrollment for treatment started 3 weeks after IFNα induction for a treatment duration of 8 weeks.
Pristane, a hydrocarbon derived from the metabolism of chlorophyll, is an environmental trigger and can precipitate and synchronize the lupus syndrome in (NZB × NZW)F1 mice (26). Pristane-accelerated lupus (26) was induced in 12-week-old (NZB × NZW)F1 mice (n = 12) by intraperitoneal (IP) injection of 150 μl pristane (Sigma-Aldrich) on day 0. At week 9, animals were randomized and enrolled for 12 weeks of treatment (Figure 1C).
Therapeutic reagents
Mouse anti-mouse CD20 mAb (clone 5D2, mouse IgG2a), a murine surrogate for rituximab, was dosed at 10 mg/kg intravenously (IV) once per week. Mouse BLyS receptor 3 fusion protein (BR-3-Fc) (mouse IgG2a) to mimic belimumab was given at 5 mg/kg subcutaneously (SC) 3 times per week. Animals receiving a combination of anti-CD20 and BR-3-Fc were given anti-CD20 at 10 mg/kg IV once per week and BR-3-Fc at 5 mg/kg SC 3 times per week. Controls included groups receiving anti-ragweed (mouse IgG2a) at 10 mg/kg IV once per week or at 5 mg/kg SC 3 times per week. Cyclophosphamide (CYC; Baxter) was used as a reference treatment and dosed at 30 mg/kg IP every 10 days. Treatment doses were chosen to achieve maximal B cell reduction as previously described (14). Antibodies and the BR-3-Fc fusion protein reagent were made in-house. Endotoxin concentrations of reagents used in vivo were <0.2 endotoxin units/mg.
Assessment of proteinuria
Proteinuria was assessed biweekly by colorimetric measurement using dipstick Multistix 10 SG (Bayer Diagnostics). Measured protein concentrations were binned as trace, ≤30 mg/dl, ≤100 mg/dl, and ≥300 mg/dl. Mice were considered to have severe proteinuria if 2 consecutive urine samples had a protein concentration of ≥300 mg/dl. Disease progression–free survival of treated animals was defined as the time from initial remission (proteinuria ≤300 mg/dl) to exacerbation (proteinuria >300 mg/dl) or as survival time if there was no exacerbation of proteinuria.
Pathology and immunohistochemistry
Treatment effect on terminal lymphocyte populations in the spleen was evaluated by immunohistochemistry for CD45R (B220) and CD3. Positive cell counts per section area were quantified for each of the markers using whole-slide scans. Renal lesions were evaluated on 2-μm paraffin sections. Sections were stained with hematoxylin and eosin or periodic acid–Schiff. Glomerulopathy, arteritis, and interstitial nephritis were assessed using an arbitrary scoring system (0–3) and evaluated as an average combined lesion score.
Serum serology and autoantibody assay
Serum IgM, IgG (IgG1 and IgG2b), and IgA were quantified using a Luminex kit (Millipore). Results were expressed in μg/ml by referring to a standard curve with known Ig concentration. Serum IgG or IgM against dsDNA was detected by enzyme-linked immunosorbent assay (ELISA) by measuring binding to coated calf thymus DNA (Sigma). Pooled sera from aged or young (NZB × NZW)F1 mice served as controls. The absorbance of the negative control was multiplied by a factor of 3 to yield a cutoff. The log titer for each sample was calculated as the log of the dilution needed to reach the absorbance of the cutoff point. A value of 3 means that a 1:1,000 dilution was required to reach this point. Given the minimum sample dilution of 1:25, the minimum quantifiable log titer value was 1.4. Quantitation of total Ig (IgG, IgA, IgM) anti-Sm antibodies and ANAs was determined by ELISA kits (Alpha Diagnostic).
Antibodies and fluorescence-activated cell sorting (FACS)
Lymphocyte populations in spleen, kidney, BM, and peripheral blood were analyzed by flow cytometry. Fluorescein isothiocyanate–, phycoerythrin (PE)–, PerCP-, allophycocyanin (APC)–, APC–Cy7–, PE–Cy7–, and Pacific Blue–conjugated anti-mouse B220, CD21, CD23, CD38, CD138, CD5, CD4, CD8, and CD44 were from BD Biosciences. Fluorochrome-conjugated anti-mouse IgM, IgG1, IgG2a, IgG2b, and IgD antibodies were from SouthernBiotech. FACS was conducted on an LSRII flow cytometer (BD Biosciences). B cell subsets were identified as described in the figure legends.
Statistical analysis
GraphPad Prism software, version 5.01 and Aabel software, version 3.0.6 (www.gigawiz.com) were used for statistical analysis. A Mann-Whitney nonparametric one- or two-way analysis of variance was used and compared with Dunnett's test. The log rank test was used for proteinuria and survival curves. An unpaired t-test was used for dual versus single treatment comparison. P values less than 0.05 were considered significant.
RESULTS
Greater improvement of clinical parameters with dual B cell–targeted therapy than with individual B cell monotherapy
To investigate the efficacy and mechanism of action of anti-CD20 and/or BAFF blockade, we used 3 (NZB × NZW)F1 mouse models of spontaneous and accelerated lupus. The overall design of the 3 animal models is shown in Figure 1. In the model of spontaneous lupus (22), (NZB × NZW)F1 mice with moderate-to-severe SLE (proteinuria 100–300 mg/dl) were treated for 6 months (Figure 1A). In IFNα-accelerated lupus in (NZB × NZW)F1 mice, disease was precipitated by IFNα overexpression (25). In this model, IFNα was ectopically expressed by infecting 3-month-old (NZB × NZW)F1 mice with adenovirus expressing IFNα, resulting in elevated serum IFNα and rapid lupus manifestation. Enrollment for treatment started 3 weeks after IFNα induction for a treatment duration of 8 weeks (Figure 1B). Alternatively, pristane-accelerated lupus (26) was induced in 3-month-old (NZB × NZW)F1 mice, and after 9 weeks animals were randomized and enrolled for a 12-week treatment (Figure 1C). For all 3 lupus models, disease progression–free survival and overall survival were used as parameters to assess efficacy of treatment (27). In addition, proteinuria was measured as a reflection of renal damage, which is a major contributor to disease progression in SLE (1).
In all 3 models tested, combination therapy or single therapies provided significantly greater therapeutic benefit compared to isotype control in disease progression–free survival rate, overall survival rate, or proteinuria (Figure 2). B cell–targeted dual therapy resulted in markedly greater improvement in clinical parameters as compared to B cell–targeted monotherapy (Figure 2). In the model of spontaneous lupus, B cell–targeted dual therapy significantly attenuated progression of proteinuria compared to treatment with anti-CD20 alone (Figure 2A). In IFNα-accelerated lupus, superior efficacy was obtained for disease progression–free survival with dual B cell therapy as compared to anti-CD20 (P < 0.008) and BR-3-Fc (P < 0.001). Dual B cell therapy also significantly increased overall survival rate compared to BR-3-Fc (P < 0.03) (Figure 2B). Similarly, in pristane-accelerated lupus, dual B cell therapy was superior to CYC treatment in all measures except for overall survival, where dual B cell therapy was slightly less efficacious (Figure 2C).
Figure 2.

Improved overall survival and disease progression–free survival (PFS) and decreased proteinuria resulting from combination B cell therapy (anti-CD20 plus B lymphocyte stimulator receptor 3 fusion protein [BR-3-Fc]). Three parameters were used to measure clinical efficacy in 3 lupus models. Disease progression–free survival of treated animals was defined as the time from initial remission (proteinuria ≤300 mg/dl) to exacerbation (proteinuria >300 mg/dl) or as survival time if there was no exacerbation of proteinuria. Overall survival was defined as the animals' survival during the disease course, which correlated with treatment and proteinuria. Proteinuria was assessed biweekly. Mice were considered to have severe proteinuria if 2 consecutive urine samples had a protein concentration of ≥300 mg/dl. A, Spontaneous lupus. B, Interferon-α (IFNα)–accelerated lupus. C, Pristane-accelerated lupus. Cyclophosphamide (CYC) was used as a reference treatment. Indicated P values are for treatment versus anti-ragweed (anti-RW) control. In spontaneous lupus, dual B cell therapy increased disease progression–free survival compared to anti-CD20 alone (∗ = P < 0.05). In IFNα-accelerated lupus, dual B cell therapy increased disease progression–free survival compared to treatment with BR-3-Fc (∗∗ = P < 0.001) and compared to treatment with anti-CD20 (∗∗ = P < 0.008). Dual B cell therapy also increased overall survival compared to treatment with BR-3-Fc (∗ = P < 0.03) in IFNα-accelerated lupus. Values for proteinuria are the mean ± SD.
Reduction of both circulating and tissue B cell subsets after dual B cell therapy
We then determined how various B cell subsets were modulated in these models. Depending on timing, regimen, and B cell status, rituximab-mediated depletion is not effective for all B cell subsets, with MZ B cells, GC B cells, and long-lived PCs being spared (16). Likewise, belimumab can decrease activated and naive B cells as well as PCs, but not memory B cells (7). In the model of spontaneous lupus, circulating B cells were significantly depleted in all treatment groups, and, unsurprisingly, monotherapy with either anti-CD20 or BR-3-Fc reduced some B cell subsets more than others. However, anti-CD20 combined with BR-3-Fc resulted in B cell depletion superior to that of either therapy alone (Figure 3A). In the model of IFNα-accelerated lupus, this effect of combination therapy was again observed, and only dual B cell therapy significantly reduced peripheral B cells compared to BR-3-Fc alone (Figure 3B). In addition, in the model of pristane-accelerated lupus, dual treatment was superior to both BR-3-Fc alone and anti-CD20–mediated depletion alone (Figure 3C).
Figure 3.

Improved B cell depletion in periphery and tissues with combination therapy compared to anti-CD20 or BR-3-Fc monotherapy. CYC was used as a reference treatment. B cell numbers are shown in the 3 lupus models. A, Spontaneous lupus 6 months after treatment. B, IFNα-accelerated lupus. No fluorescence-activated cell sorting data were collected for the anti-CD20–treated group 2 months after treatment. C, Pristane-accelerated lupus 2 months after treatment. Symbols represent individual mice; bars show the mean. ∗ = P < 0.05; ∗∗ = P < 0.001; ∗∗∗ = P < 0.0001 versus anti-ragweed control (red asterisks). ∗ = P < 0.05; ∗∗ = P < 0.001 (black asterisks). FOB = follicular B cells; MZ = marginal zone B cells; GC = germinal center B cells; PC = plasma cells (see Figure 2 for other definitions).
In the model of spontaneous lupus, splenic follicular B cells were significantly reduced with single or combination immunotherapies compared to isotype control. Interestingly, in models of accelerated lupus, combination treatment significantly reduced follicular B cells compared to single therapeutic modalities (Figures 3B and C). CYC only modestly reduced follicular B cells in all 3 models.
MZ B cells (identified as CD21highCD23lowIgMhigh) are enriched for autoreactive capacity and differentiate rapidly into PCs (10). In spontaneous lupus, dual B cell therapy showed a robust depletion of MZ B cells compared to B cell monotherapy (Figure 3A). In both IFNα-accelerated and pristane-accelerated lupus, dual B cell therapy was more efficacious at eliminating MZ B cells than B cell monotherapy or even reference treatment with CYC (Figures 3B and C).
GC activation and autoantibody-secreting PC expansion are hallmarks of SLE (10). In all models, GC B cells (B220+CD38dim) were significantly reduced in all treatment groups (Figure 3). However, dual B cell therapy consistently decreased GC B cells to a greater degree than other modalities. In spontaneous and IFNα-accelerated lupus, combination treatment resulted in significant GC B cell reduction compared to BR-3-Fc alone (Figures 3A and B).
PCs (B220lowCD138+) are classified as long- or short-lived and are localized primarily in the BM or spleen, respectively. IgG1 and IgG3 PCs are 2 major sources of autoantibodies in SLE (21). Long-lived PCs in the BM are known to be less sensitive to immunotherapy (16). However, splenic B220lowCD138+ PCs were depleted in all treatment groups in all models (Figure 3). IgM- or IgG-producing splenic PCs were significantly depleted in all treatment groups, although dual B cell therapy resulted in the highest level of depletion (Figure 3) (further information is available at http://research-pub.gene.com/lin_et_al_2014). FACS analysis of kidneys showed that in spontaneous lupus, dual B cell therapy could significantly reduce renal infiltration of activated T cells and PCs compared to the control antibody–treated group, but not compared to single agent therapy (further information is available at http://research-pub.gene.com/lin_et_al_2014). In pristane-accelerated lupus, while all therapies resulted in a reduction of BM PCs, only dual B cell therapy caused a significant reduction of IgG1 PCs (further information is available at http://research-pub.gene.com/lin_et_al_2014). In summary, B cell depletion via anti-CD20 plus BAFF blockade provided superior efficacy compared to single agent therapy and resulted in more effective depletion of circulating and tissue-resident B cells.
Significant reduction of various autoantibodies and renal injury by dual B cell–targeted treatment
Renal injury in the models evaluated is mainly caused by immune complex deposition of autoantibodies (22). In the model of spontaneous lupus, dual B cell therapy significantly reduced serum anti-Sm IgG and anti-dsDNA IgG autoantibody levels (P < 0.001 compared to anti-CD20), while neither B cell monotherapy nor CYC was effective in reducing anti-Sm autoantibodies (Figure 4A). Although dual B cell therapy did not result in improvement of ANA Ig levels in spontaneous lupus, these autoantibodies were significantly reduced in IFNα-accelerated lupus (Figure 4B) and showed a trend toward greater reduction in pristane-induced lupus (Figure 4C). For anti-dsDNA IgG, dual B cell therapy caused a significant reduction compared to monotherapies across the 3 models (Figure 4). Interestingly, dual B cell therapy significantly reduced anti-dsDNA IgM levels in IFNα-accelerated and pristane-accelerated lupus, but not in spontaneous lupus, indicating that the IgG subclass is the major source of autoantibodies in spontaneous lupus in (NZB × NZW)F1 mice (22). Combination treatment also effectively reduced anti-RNP antibodies in pristane-accelerated lupus (Figure 4C). Autoantibodies are associated with severity and activity of SLE. The greater reduction of various autoantibodies with dual immunotherapy is consistent with greater depletion of various B cell subsets.
Figure 4.

Impact of anti-CD20 and/or BAFF blockade on autoantibodies. Serum autoantibodies including anti-Sm, antinuclear antibodies (ANAs), and anti–double-stranded DNA (anti-dsDNA) were assessed in 3 lupus models in (NZB × NZW)F1 mice. A, Spontaneous lupus. B, IFNα-accelerated lupus. C, Pristane-accelerated lupus. Symbols represent individual mice; bars show the mean. ∗ = P < 0.05; ∗∗ = P < 0.001; ∗∗∗ = P < 0.0001 versus anti-ragweed control (red asterisks). ∗ = P < 0.05; ∗∗ = P < 0.001; ∗∗∗ = P < 0.0001 (black asterisks). Anti-nRNP = anti–nuclear RNP (see Figure 2 for other definitions).
Hypergammaglobulinemia is a hallmark of SLE, and differential elevations of IgG subclasses may be relevant to aspects of disease pathogenesis such as deposition of glomerular immune complexes (1). We examined the impact of B cell therapy on various Ig isotypes in the 3 animal models of lupus (Figure 5). In spontaneous lupus, dual therapy significantly reduced all Ig isotypes compared to monotherapy and also significantly reduced IgG1 and IgA levels compared to control antibody (Figure 5A). In IFNα-accelerated lupus, all serum immunoglobulins were significantly reduced with dual B cell therapy (Figure 5B). However, with BR-3-Fc monotherapy, IgA and IgG2b were significantly reduced (Figure 5B). In pristane-accelerated lupus, dual B cell therapy significantly reduced IgG3 and IgA, while IgM, IgG1, and IgG2b showed a trend toward reduction. However, dual therapy significantly reduced IgG and IgA isotypes compared to anti-CD20 monotherapy (Figure 5C). BR-3-Fc alone caused a significant reduction only in IgA (Figure 5C). In summary, dual B cell immunotherapy was efficacious in reducing various Ig isotypes in all 3 models.
Figure 5.

Impact of anti-CD20 and/or BAFF blockade on serum immunoglobulin levels in (NZB × NZW)F1 mice. Serum IgM, IgG1, IgG2b, IgA, and IgG3 levels were measured in mice with spontaneous lupus after 6 months of treatment (A), mice with IFNα-accelerated lupus after 8 weeks of treatment (B), and mice with pristane-accelerated lupus after 12 weeks of treatment (C). IgG2a levels were not measured since the treatment reagents were of the mouse IgG2a isotype. Symbols represent individual mice; bars show the mean. ∗ = P < 0.05; ∗∗ = P < 0.001; ∗∗∗ = P < 0.0001 versus anti-ragweed control (red asterisks). ∗ = P < 0.05; ∗∗ = P < 0.001; ∗∗∗ = P < 0.0001 (black asterisks). See Figure 2 for definitions.
Attenuation of renal pathology was most pronounced with dual B cell–targeted therapy, even though in pristane-accelerated lupus all treatment modalities except for the anti-ragweed control had similar efficacy (further information is available at http://research-pub.gene.com/lin_et_al_2014). Compared to the reference treatment (CYC), the combination of anti-CD20 and BR-3-Fc yielded a further reduction of severity, indicating an additive effect of the 2 agents. Dual B cell therapy was comparable to BR-3-Fc but superior to anti-CD20 in reducing glomerulopathy, arteritis, and interstitial nephritis in spontaneous lupus (further information is available at http://research-pub.gene.com/lin_et_al_2014). However, in IFNα-accelerated lupus, combination therapy provided a greater attenuation of overall renal injury as compared to monotherapies (further information is available at http://research-pub.gene.com/lin_et_al_2014). In summary, dual B cell immunotherapy significantly reduced different classes of pathogenic autoantibodies, consistent with its effectiveness in reducing immune complex–mediated renal injury.
DISCUSSION
We demonstrate that the combination of anti-CD20 B cell depletion with BAFF receptor blockade provides a significant improvement in mouse models of spontaneous and accelerated SLE as assessed by overall survival, disease progression–free survival, proteinuria, and renal injury. The models of spontaneous, IFNα-accelerated, and pristane-accelerated lupus in (NZB × NZW)F1 mice each capture various aspects of SLE including clinical manifestations, autoantibodies, and IFN gene signature. Of the various treatment modalities evaluated, dual B cell immunotherapy achieved greatest depletion of circulating and tissue-resident B cell subsets including MZ B cells, plasmablasts, and PCs. In addition, this therapeutic strategy effectively reduced a broad range of autoantibodies including several IgG isotypes. Comparison of various parameters between combination therapy and single agent treatments showed that maximal therapeutic benefit was only achieved when these clinically relevant end points were profoundly reduced. The improved efficacy and survival across all 3 preclinical lupus models substantiates the therapeutic potential of the dual immunotherapy approach. While our study is limited to preclinical mouse models of SLE, it furthers understanding of the utility of these known pathways in human disease, thus providing insights into the design of B cell immunotherapies in SLE.
Previous studies in normal mice or mouse models of lupus have suggested that treatment with anti-CD20 depletes B cells in a tissue- and subset-restricted manner (14,15,28). While anti-CD20 can effectively deplete circulating B cells, it affects MZ B cells, GC B cells, or long-lived PCs to a lesser extent (14,15,28). In contrast, BAFF antagonism is effective in blocking the survival and maturation of transitional, follicular, and MZ B cells (14,15). Our cellular analyses suggest that maximizing efficacy in (NZB × NZW)F1 mouse models requires not only targeting the components of MZ B cells but also depleting other B cell subsets such as naive/activated B cells. The clinical response to rituximab is not uniform in SLE (5) or even B cell lymphomas (29). The heterogeneity of CD20 expression or its down-regulation on various B cell subsets might be a contributing factor to the observed variability in the clinical response (29). In addition, the kinetics of B cell repopulation and autoantibody relapse after anti-CD20 therapy suggest that different B cell–mediated pathologies exist in SLE (3).
In active SLE, plasmablasts are increased, reflecting severe immune activation (3). In addition, the VH regions of anti-dsDNA–producing B cells are heavily mutated, and the frequency of these B cells correlates with active disease (30,31). After anti-CD20 treatment, relapse with high anti-dsDNA antibody levels is associated with an increased percentage of plasmablasts (32). Therefore, it is possible that certain pathogenic B cells with negative/low CD20 expression could escape treatment. It is known that rituximab efficiently depletes naive and memory B cells, where expression of CD20 is highest (5). In addition, using BAFF blockade to inhibit survival of B cells expressing low levels of CD20 might allow for more effective depletion of these B cells. While expression of BR-3 on plasmablasts is lower than on naive B cells, it has been shown that BAFF can prolong the survival of plasmablasts (16,17,33). In the present study, the observed benefit of complementary BAFF blockade in preclinical models clearly suggests that optimal clinical improvement requires modulation of additional pathways, such as inhibition of B cell survival, to ensure an effective response to treatment with anti-CD20. Thus, combination therapy may allow for a broader impact on pathogenic B cells than treatment with a single therapeutic agent.
BAFF receptor signaling is the main axis for B cell survival, and its blockade reduces populations of naive, transitional, and activated GC B cells as well as PCs to different degrees (6,34). Increased serum BAFF levels in SLE patients correlate with serologic and clinical parameters of disease activity (35,36). Following anti-CD20 B cell depletion therapy and B cell repopulation, BAFF levels were found to be significantly higher during relapse and correlated positively with anti-dsDNA antibodies (37). These results suggest that BAFF-dependent pathways may support the survival of certain pathogenic B cell subsets that lack or have reduced CD20 expression (37). Collectively, escape from CD20 depletion combined with increased BAFF levels could provide additional survival signals to residual pathogenic B cells during rituximab therapy. It is also possible that BAFF blockade impacts immune cells other than B cells such as macrophages and dendritic cells (38).
Our data suggest that combined B cell targeted therapy via anti-CD20 and BAFF blockade is an attractive treatment strategy in SLE and perhaps also in other B cell–mediated autoimmune diseases. Broader reduction and blockade of various B cell subsets including PCs provided improved efficacy in models of spontaneous or accelerated lupus in (NZB × NZW)F1 mice. Our findings confirm and expand initial observations in the model of spontaneous lupus in (NZB × NZW)F1 mice, in which impacts on B cell lineage and proteinuria were observed with short-term (4-week) treatment (15). Our analyses of kidney histology, various autoantibodies, and B cell subsets clearly demonstrate the benefit of long-term combination immunotherapy. Our data suggest that combination therapy could provide improved efficacy and should be attempted in the clinic in severely affected patients in whom therapeutic options are limited. As rituximab is used in clinical practice every 6–12 months while belimumab is administered more often, one can envision clinical regimens of rituximab induction and belimumab maintenance therapy. Alternation of treatments might provide a means of reducing safety concerns associated with long-term combination therapy. Our data contribute to the foundation for these hypotheses and provide support for testing and expanding them in human disease. Given the broader immunosuppressive effect of combined anti-CD20 and BAFF blockade therapy in preclinical models, it will be critical to investigate potential adverse effects of this dual B cell–targeted immunotherapy in future studies.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Martin and Zarrin had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. W. Lin, Lee, Balazs, Martin, Zarrin.
Acquisition of data. W. Lin, Seshasayee, Lee, Caplazi, Suto, Nguyen, Z. Lin, DeForge.
Analysis and interpretation of data. W. Lin, Seshasayee, Lee, Caplazi, McVay, Suto, Nguyen, Z. Lin, Sun, DeForge, Balazs, Martin, Zarrin.
ADDITIONAL DISCLOSURES
All authors were employees of Genentech, Inc. at the time of this study. Drs. Balazs and Martin are currently employees of Amgen.
REFERENCES
- 1.Putterman C, Caricchio R, Davidson A, Perlman H. Systemic lupus erythematosus. Clin Dev Immunol. 2012;2012:437282. doi: 10.1155/2012/437282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Odendahl M, Keitzer R, Wahn U, Hiepe F, Radbruch A, Dorner T, et al. Perturbations of peripheral B lymphocyte homoeostasis in children with systemic lupus erythematosus. Ann Rheum Dis. 2003;62:851–8. doi: 10.1136/ard.62.9.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005;105:1614–21. doi: 10.1182/blood-2004-07-2507. [DOI] [PubMed] [Google Scholar]
- 4.Dorner T, Jacobi AM, Lee J, Lipsky PE. Abnormalities of B cell subsets in patients with systemic lupus erythematosus. J Immunol Methods. 2011;363:187–97. doi: 10.1016/j.jim.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 5.Townsend MJ, Monroe JG, Chan AC. B-cell targeted therapies in human autoimmune diseases: an updated perspective. Immunol Rev. 2010;237:264–83. doi: 10.1111/j.1600-065X.2010.00945.x. [DOI] [PubMed] [Google Scholar]
- 6.Furie R, Nicholls K, Cheng TT, Houssiau F, Burgos-Vargus R, Chen SL, et al. Efficacy and safety of abatacept over 12 months in patients with lupus nephritis: results from a multicenter, randomized, double-blind, placebo-controlled phase II/III study. Arthritis Rheum. 2011;63(Suppl):S962–3. [abstract] [Google Scholar]
- 7.Jacobi AM, Huang W, Wang T, Freimuth W, Sanz I, Furie R, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2010;62:201–10. doi: 10.1002/art.27189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tedder TF, Streuli M, Schlossman SF, Saito H. Isolation and structure of a cDNA encoding the B1 (CD20) cell-surface antigen of human B lymphocytes. Proc Natl Acad Sci U S A. 1988;85:208–12. doi: 10.1073/pnas.85.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004;50:2580–9. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 10.Leandro MJ, Cambridge G, Edwards JC, Ehrenstein MR, Isenberg DA. B-cell depletion in the treatment of patients with systemic lupus erythematosus: a longitudinal analysis of 24 patients. Rheumatology (Oxford) 2005;44:1542–5. doi: 10.1093/rheumatology/kei080. [DOI] [PubMed] [Google Scholar]
- 11.Smith KG, Jones RB, Burns SM, Jayne DR. Long-term comparison of rituximab treatment for refractory systemic lupus erythematosus and vasculitis: remission, relapse, and re-treatment. Arthritis Rheum. 2006;54:2970–82. doi: 10.1002/art.22046. [DOI] [PubMed] [Google Scholar]
- 12.Reddy V, Jayne D, Close D, Isenberg D. B-cell depletion in SLE: clinical and trial experience with rituximab and ocrelizumab and implications for study design. Arthritis Res Ther. 2013;15(Suppl 1):S2. doi: 10.1186/ar3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol. 2005;174:817–26. doi: 10.4049/jimmunol.174.2.817. [DOI] [PubMed] [Google Scholar]
- 14.Lin WY, Gong Q, Seshasayee D, Lin Z, Ou Q, Ye S, et al. Anti-BR3 antibodies: a new class of B-cell immunotherapy combining cellular depletion and survival blockade. Blood. 2007;110:3959–67. doi: 10.1182/blood-2007-04-088088. [DOI] [PubMed] [Google Scholar]
- 15.Bekar KW, Owen T, Dunn R, Ichikawa T, Wang W, Wang R, et al. Prolonged effects of short-term anti-CD20 B cell depletion therapy in murine systemic lupus erythematosus. Arthritis Rheum. 2010;62:2443–57. doi: 10.1002/art.27515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells [published erratum appears in J Clin Invest 2004;113:1069] J Clin Invest. 2003;112:286–97. doi: 10.1172/JCI18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moreaux J, Legouffe E, Jourdan E, Quittet P, Reme T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103:3148–57. doi: 10.1182/blood-2003-06-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–98. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–53. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 20.Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–9. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 21.Haas KM, Watanabe R, Matsushita T, Nakashima H, Ishiura N, Okochi H, et al. Protective and pathogenic roles for B cells during systemic autoimmunity in NZB/W F1 mice. J Immunol. 2010;184:4789–00. doi: 10.4049/jimmunol.0902391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry D, Sang A, Yin Y, Zheng YY, Morel L. Murine models of systemic lupus erythematosus. J Biomed Biotechnol. 2011;2011:271694. doi: 10.1155/2011/271694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–23. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niewold TB. Interferon α-induced lupus: proof of principle. J Clin Rheumatol. 2008;14:131–2. doi: 10.1097/RHU.0b013e318177627d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leiss H, Niederreiter B, Bandur T, Schwarzecker B, Bluml S, Steiner G, et al. Pristane-induced lupus as a model of human lupus arthritis: evolvement of autoantibodies, internal organ and joint inflammation. Lupus. 2013;22:778–92. doi: 10.1177/0961203313492869. [DOI] [PubMed] [Google Scholar]
- 27.Sprangers B, Monahan M, Appel GB. Diagnosis and treatment of lupus nephritis flares: an update. Nat Rev Nephrol. 2012;8:709–17. doi: 10.1038/nrneph.2012.220. [DOI] [PubMed] [Google Scholar]
- 28.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. 2007;179:3351–61. doi: 10.4049/jimmunol.179.5.3351. [DOI] [PubMed] [Google Scholar]
- 29.Davis TA, Czerwinski DK, Levy R. Therapy of B-cell lymphoma with anti-CD20 antibodies can result in the loss of CD20 antigen expression. Clin Cancer Res. 1999;5:611–5. [PubMed] [Google Scholar]
- 30.Jacobi AM, Hansen A, Burmester GR, Dorner T, Lipsky PE. Enhanced mutational activity and disturbed selection of mutations in VH gene rearrangements in a patient with systemic lupus erythematosus. Autoimmunity. 2000;33:61–76. doi: 10.3109/08916930108994110. [DOI] [PubMed] [Google Scholar]
- 31.Jacobi AM, Goldenberg DM, Hiepe F, Radbruch A, Burmester GR, Dorner T. Differential effects of epratuzumab on peripheral blood B cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis. 2008;67:450–7. doi: 10.1136/ard.2007.075762. [DOI] [PubMed] [Google Scholar]
- 32.Lazarus MN, Turner-Stokes T, Chavele KM, Isenberg DA, Ehrenstein MR. B-cell numbers and phenotype at clinical relapse following rituximab therapy differ in SLE patients according to anti-dsDNA antibody levels. Rheumatology (Oxford) 2012;51:1208–15. doi: 10.1093/rheumatology/ker526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, et al. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192:1453–66. doi: 10.1084/jem.192.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamore R, III, Parmar S, Patel K, Hilas O. Belimumab (benlysta): a breakthrough therapy for systemic lupus erythematosus. P T. 2012;37:212–26. [PMC free article] [PubMed] [Google Scholar]
- 35.Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune–based rheumatic diseases. Arthritis Rheum. 2001;44:1313–9. doi: 10.1002/1529-0131(200106)44:6<1313::AID-ART223>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 36.Stohl W, Metyas S, Tan SM, Cheema GS, Oamar B, Xu D, et al. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis Rheum. 2003;48:3475–86. doi: 10.1002/art.11354. [DOI] [PubMed] [Google Scholar]
- 37.Carter LM, Isenberg DA, Ehrenstein MR. Elevated serum BAFF levels are associated with rising anti–double-stranded DNA antibody levels and disease flare following B cell depletion therapy in systemic lupus erythematosus. Arthritis Rheum. 2013;65:2672–9. doi: 10.1002/art.38074. [DOI] [PubMed] [Google Scholar]
- 38.Chang SK, Arendt BK, Darce JR, Wu X, Jelinek DF. A role for BLyS in the activation of innate immune cells. Blood. 2006;108:2687–94. doi: 10.1182/blood-2005-12-017319. [DOI] [PMC free article] [PubMed] [Google Scholar]