

Abstract
Objective
The excessive deposition of extracellular matrix, including type I collagen, is a key aspect in the pathogenesis of connective tissue diseases such as systemic sclerosis (SSc; scleroderma). To further our understanding of the mechanisms governing the dysregulation of type I collagen production in SSc, we investigated the role of the activator protein 1 (AP-1) family of transcription factors in regulating COL1A2 transcription.
Methods
The expression and nuclear localization of AP-1 family members (c-Jun, JunB, JunD, Fra-1, Fra-2, and c-Fos) were examined by immunohistochemistry and Western blotting in dermal biopsy specimens and explanted skin fibroblasts from patients with diffuse cutaneous SSc and healthy controls. Gene activation was determined by assessing the interaction of transcription factors with the COL1A2 enhancer using transient transfection of reporter gene constructs, electrophoretic mobility shift assays, chromatin immunoprecipitation analysis, and RNA interference involving knockdown of individual AP-1 family members. Inhibition of fibroblast mammalian target of rapamycin (mTOR), Akt, and glycogen synthase kinase 3β (GSK-3β) signaling pathways was achieved using small-molecule pharmacologic inhibitors.
Results
Binding of JunB to the COL1A2 enhancer was observed, with its coalescence directed by activation of gene transcription through the proximal promoter. Knockdown of JunB reduced enhancer activation and COL1A2 expression in response to transforming growth factor β. In SSc dermal fibroblasts, increased mTOR/Akt signaling was associated with inactivation of GSK-3β, leading to blockade of JunB degradation and, thus, constitutively high expression of JunB.
Conclusion
In patients with SSc, the accumulation of JunB resulting from altered mTOR/Akt signaling and a failure of proteolytic degradation underpins the aberrant overexpression of type I collagen. These findings identify JunB as a potential target for antifibrotic therapy in SSc.
Systemic sclerosis (SSc; scleroderma) is a complex autoimmune disease with unknown etiology. Pathogenic processes include vascular damage, autoimmunity, and widespread fibrosis of the skin and internal organs. Transforming growth factor β (TGFβ) has been implicated in the development of many fibrotic diseases, including SSc. TGFβ is a pleiotropic mediator with a critical role in wound healing and tissue remodeling. Consequently, it is of major importance in pathologic conditions that are characterized by tissue remodeling, scarring, and fibrosis. TGFβ regulates the expression of genes that are part of, or regulate the formation of, the extracellular matrix (ECM). Type I collagen is an integral structural component of the ECM, with a major role in wound healing and connective tissue remodeling. Dysregulated or excessive production and deposition of type I collagen lead to ECM accumulation and, eventually, tissue fibrosis.
TGFβ is a potent inducer of the human procollagen type I α2 chain gene (COL1A2). We have recently described in detail the in vivo mechanism underlying the transcriptional control of COL1A2, through a complex interaction between its distal enhancer and proximal promoter, in response to TGFβ. This far-upstream enhancer (FUE) region is highly homologous to the mouse Col1a2 enhancer region (1–4), which is activated in adult mice during wound healing and fibrosis (5). Studies exploring the enhancer function have shown that TGFβ can also activate COL1A2 via a noncanonical (Smad-independent) signaling pathway requiring enhancer/promoter cooperation. This interaction appears to involve activator protein 1 (AP-1) family members and, in particular, an exchange of c-Jun for JunB in the critical AP-1 site of the enhancer, resulting in enhancer/promoter coalescence and transactivation of the transcriptional machinery bound in the promoter, by the factors bound to the enhancer. Moreover, using transgenesis, we have shown that interfering with this mechanism results in the abolition of COL1A2 expression by fibroblasts in vivo (6).
The AP-1 family of transcription factors (c-Jun, JunB, JunD, cFos, FosB, Fra-1, and Fra-2) form homodimers and heterodimers as part of a complex mode of transcriptional regulation and are induced by a large variety of cellular signals (7–12). AP-1 transcriptional regulation is known to be involved in many normal and pathogenic cellular processes (13–16). Recently, the important roles of the AP-1 family members c-Jun, c-Fos, and JunD in dermal fibrosis have been described in patients with scleroderma (17–20).
In this study, we observed that the novel TGFβ response element (TβRE) located far upstream in the enhancer of human COL1A2 was active in fibroblasts from the fibrotic lesions of patients with SSc. We also found that JunB was activated by TGFβ and detected constitutive expression of JunB in SSc dermal fibroblasts. Furthermore, inhibition of JunB resulted in the down-regulation of type I collagen expression by SSc dermal fibroblasts and reduced the ability of dermal fibroblasts to migrate into an in vitro wound. We also delineated a mechanism of JunB overexpression in SSc dermal fibroblasts, involving mammalian target of rapamycin (mTOR), Akt, and glycogen synthase kinase 3β (GSK-3β) signaling.
PATIENTS AND METHODS
Patients and cell culture
Fibroblasts were explanted from skin biopsy specimens obtained from patients with diffuse cutaneous SSc or from normal healthy control subjects, and cultures were established. The affected skin of SSc patients and skin from an equivalent location in the controls was sampled. Each of the fibroblast lines used in this study was derived from a distinct individual. The patients fulfilled the American College of Rheumatology preliminary criteria for SSc (21). The fibroblasts were used between passages 2 and 5. During TGFβ treatment, the cells were serum-starved for 8 hours. TGFβ was added to a final concentration of 2.5 ng/ml.
DNA constructs
The human collagen FUE region has been previously described by Antoniv et al (2). Details on the construction of the plasmid vectors used in this study are available upon request from the corresponding author. Briefly, construct −378pLAC in the pbgal-Basic Vector (Clontech) was derived from the −378 COL1A2/Luc plasmid (17). The genomic DNA between the enhancer and promoter was excluded from the DNA constructs.
Transient transfections
Transient transfection assays were performed using FuGene 6 transfection reagent according to the manufacturer's instructions. Briefly, various cell lines (2 × 105 cells per transfection) were cultured in vitro and transfected with 2 μg of total plasmid DNA, with pRSVLuc (0.2 μg) as a normalization control, and 6 μl of FuGene 6 reagent. The cells were harvested 72 hours posttransfection using a Dual-Light Chemiluminescent reporter gene assay system (Tropix) according to the manufacturer's instructions. All transfections were performed in triplicate within each experiment, and each experiment was repeated on more than 3 separate occasions.
Electrophoretic mobility shift assay (EMSA)
The nuclear extracts were prepared and used in EMSA analyses according to the protocol described in a Promega Gel Shift Assay Systems kit. Supershift assays were carried out using specific antibodies raised against c-Jun (sc-45x), JunB (sc-46x), and JunD (sc-74x) (all from Santa Cruz Biotechnology). Briefly, double-stranded oligonucleotides were synthesized and end-labeled using T4 kinase and 32P–γ-ATP. Labeled probes were used in binding reactions with nuclear extracts from cells that had been left untreated or treated with TGFβ. Competition was carried out with unlabeled oligonucleotides (1.75 pmoles/μl) of specific transcription factor consensus-binding sites.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis
Fibroblasts were seeded (2.5 × 105 cells per well) for 48 hours. The cells were serum-starved for 12 hours and treated with or without TGFβ (R&D Systems) at 4 ng/ml for a further 24 hours. The cells were analyzed by SDS-PAGE, followed by Western blotting. The antibodies against c-Jun, JunB, and JunD (all from Santa Cruz Biotechnology) and an anti–procollagen α1 antibody (Southern Biotechnology) were used.
RNA inhibition using small interfering RNA (siRNA)
Human JunB siRNA target sequences were obtained from Dharmacon (JunB siGenome SMARTpool reagent) and used according to the manufacturer's instructions. Transfection into explanted human dermal fibroblasts was achieved using Oligofectamine transfection reagent (Invitrogen) according to the manufacturer's instructions. The siRNA were transfected at concentrations of 10 nM and 25 nM. Scrambled control siRNA (K-002800; Dharmacon) were also used and transfected at a concentration of 25 nM.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using a commercially available kit (Upstate Biotechnology), according to the manufacturer's instructions, on primary human dermal fibroblasts that were left untreated or treated with TGFβ. Briefly, DNA–protein complexes were immunoprecipitated with antibodies to RNA polymerase II, c-Jun, JunB, or JunD (Santa Cruz Biotechnology). Polymerase chain reaction amplifications were carried out with the appropriate primers spanning the human COL1A2 promoter and enhancer. These primers are specific to human DNA, as previously reported (6).
Immunohistochemistry
Dermal biopsy specimens obtained from patients with diffuse cutaneous SSc or healthy controls were postfixed in neutral buffered formalin and paraffin embedded. Sections (3 μm) were cut, dewaxed, and incubated with H2O2(3%) at room temperature for 10 minutes in the dark. When required, antigen retrieval was performed using hot citrate buffer (0.01M, pH 6.0) for 10 minutes. The sections were incubated in blocking serum (20%) for 10 minutes and washed with phosphate buffered saline. Sections were incubated with primary antibodies for procollagen (Abcam) or for c-Jun, JunB, or JunD (all from Santa Cruz Biotechnology). Specificity of staining was confirmed by assessing control dermal biopsy sections incubated with isotype-matched irrelevant control antibodies. Sections were incubated with secondary biotinylated antibodies, and then with Vectastain avidin–biotin complex conjugated with horseradish peroxidase (Vector Laboratories) and visualized using 3,3′-diaminobenzidine. Sections were viewed and photographed on an Axioskop microscope and Carl Zeiss digital camera using Axiovision software.
Immunofluorescence
Colocalization between cell-specific antigens was investigated using 2-, 3-, and 4-color immunofluorescence labeling. Cells were grown in 8-well chamber slides, fixed with ice-cold methanol for 2 minutes, blocked with normal serum, and incubated with primary antibodies for 1 hour at room temperature. Primary antibodies were raised in different species in each experiment, and appropriate secondary antibodies conjugated to Alexa Fluor fluorophores (Life Technologies) were used. Vectashield mounting medium containing DAPI (Vector Laboratories) was used to mount the slides. Cells were viewed and photographed on an Axioskop Z fluorescence microscope with an Axiocam digital camera in combination with Axiovision software (Carl Zeiss).
Deconvolution of Z-stacks was used to assess nuclear/cytoplasmic immunostaining. Analysis of staining of the nuclei was carried out using ImageJ software. Four separate fields of view were imaged in triplicate. The total number of nuclei stained with DAPI was calculated using the software and verified by eye. The total number of fluorescent cells or nuclei was then determined using the software, with verification by eye. The mean ± SEM values for nuclear/cytoplasmic staining were calculated, and Student's t-test was applied for comparisons of the data between groups.
Migration assays
Dermal fibroblasts were either left untreated, treated with scrambled control siRNA, or treated with specific siRNA for JunB, prior to migration experiments. Migration into a scratch wound was performed using a 96-well floating-pin transfer device with a pin diameter of 1.58 mm. The pin array was placed in the top corner of a well, pushed down into the 96-well plate to engage all pins, and then pulled toward the user. Plates were returned to the tissue-culture incubator for 48 hours, and migration was assessed in the presence of mitomycin C (5 μg/ml; Sigma). Images of the wounds were obtained at 24 hours of culture, and the extent of wound closure was assessed (using the Wimasis Image Analysis platform, available at http://www.wimasis.com), with results expressed as the percentage of wounded area covered.
Inhibition of mTOR, Akt, and GSK-3β signaling
Normal and SSc dermal fibroblasts were serum-starved overnight and treated with an inhibitor of mTOR (20 nM rapamycin; Sigma), phospho-Akt (50 nM GSK690693; Seleckchem), GSK-3β (inhibitor VII, 500 nM AR-A014418; Merck-Calbiochem), and proteasome (500 nM MG-132; Merck-Calbiochem).
Statistical analysis
Results are expressed as the mean ± SEM. Student's unpaired t-tests were used for comparison between 2 groups. P values less than 0.05 were considered statistically significant.
RESULTS
Type I collagen expression in SSc skin
We have previously shown that overexpression of type I collagen in the skin tissue of adult patients is regulated at the transcriptional level by the COL1A2-FUE, which drives expression in a spatial and temporal manner (5). More recently, we demonstrated that this complex regulatory region contains an AP-1–dependent TβRE (6).
The TβRE sequence of the human COL1A2-FUE (shown in the schematic representation in Figure 1A) is located between 19.2 kb and 20.0 kb upstream of the transcriptional origin in a previously identified region of open chromatin, footprint 2 (F2) (2). Fine-mapping of the region identified a putative AP-1 site that was shown to mediate the TGFβ response (6).
Figure 1.

Structure and function of the far-upstream enhancer (FUE) of human COL1A2. A, Schematic representation of the FUE genomic location shows 3 open chromatin areas (footprint 1 [F1]–F3) along with known DNase hypersensitive sites (arrowheads), the critical activator protein 1 (AP-1) site in F2, and the position of the proximal promoter (PP). B and C, LacZ reporter gene constructs driven by the thymidine kinase (TK) promoter alone or fused to the COL1A2-FUE (−21.1/18.8pLAC-TK) (B) or LacZ reporter gene constructs driven by the COL1A2 proximal promoter (C) were transfected into dermal fibroblasts from normal healthy controls or patients with systemic sclerosis (SSc) (n = 3 per group), and response to treatment with transforming growth factor β (TGFβ; 4 ng/ml) (black bars) or without TGFβ (gray-shaded bars) was measured. In B, cotransfection of a Smad7 expression vector was also included. In C, a mutated COL1A2-FUE (−21.1/18.8pLAC-δAP-1-20.03) with the AP-1 site abolished (red “x”) was also included. Bars show the mean ± SEM of quadruplicate experiments. Statistical significance was assessed using Student's t-test. D, Intracellular type I procollagen expression was assessed by immunohistochemical staining of skin sections from healthy control subjects or patients with active diffuse SSc. Insets show IgG controls.
Reporter gene constructs containing the wild-type human COL1A2 enhancer region and the heterologous thymidine kinase (TK) promoter, lacking a Smad-dependent TβRE (21.1/18.8pLAC-TK), were used to assess the TβRE in the FUE region. These constructs were cotransfected with Smad7 expression vectors into normal or SSc dermal fibroblasts, and the response to TGFβ treatment was assessed (Figure 1B). The results indicated that the COL1A2-FUE was activated by TGFβ in both normal and scleroderma fibroblasts. The addition of Smad7 had no effect, thus indicating that the activation of the upstream TβRE is independent of Smad. Moreover, TGFβ treatment was not required for induction of COL1A2-FUE activity in SSc fibroblasts, suggesting that the factors induced by TGFβ were constitutively present.
To examine the role of the AP-1 site, we used reporter gene constructs containing the wild-type human COL1A2 enhancer region fused to the natural COL1A2 proximal promoter (21.1/18.8pLAC) or a mutant construct in which the TβRE/AP-1 site was deleted (21.1/18.8pLAC-δAP-1-20.03). In both normal and SSc fibroblasts, COL1A2-FUE activity was abolished in the presence of the AP-1 mutation, resulting in attenuated reporter gene activity upon the addition of TGFβ. Importantly, the higher basal reporter gene activity in SSc cells was also effectively abolished in the presence of the mutant construct (Figure 1C).
Consistent with these results, immunohistochemical staining of skin sections for intracellular procollagen revealed an overexpression of intracellular type I procollagen in the SSc dermis compared to the normal dermis (Figure 1D). The differences were particularly pronounced in fibroblasts located in the suprabasal layer (Figure 1D).
Role of type I collagen enhancer TβRE in scleroderma dermal fibroblasts
We carried out EMSAs of nuclear extracts from normal and SSc dermal fibroblasts using 32P-labeled oligonucleotide probes of the COL1A2 wild-type sequence containing the putative AP-1 TβRE (Figure 1A) after the cells had been cultured in the presence or absence of TGFβ (2 ng/ml). The EMSA profile differed between normal and SSc nuclear extracts (Figure 2A). The SSc gel-shift pattern resembled that of nuclear extracts from TGFβ-treated normal fibroblasts (Figure 2A). A single-shifted band of diminished intensity was observed in untreated normal nuclear extracts, compared to 4 shifted bands in SSc nuclear extracts and TGFβ-treated normal nuclear extracts. The band specificity was tested with the addition of unlabeled oligonucleotide (cold competitor), which competes with the 32P-labeled probe. When an AP-1 mutant probe was used, no shifted bands were apparent in either normal or SSc nuclear extracts.
Figure 2.

Binding of JunB and c-Jun to the AP-1 site of the COL1A2 enhancer. A, Electrophoretic mobility shift assays (EMSAs) were carried out using a probe for the COL1A2-FUE AP-1 site. Nuclear extracts from dermal fibroblasts of normal healthy controls and patients with SSc (n = 3 per group) treated with or without TGFβ (2 ng/ml) were incubated with 32P-labeled probe and analyzed by EMSA. Control experiments using unlabeled (cold) oligonucleotide (oligo) probe as a competitor or a 32P-labeled probe containing a mutated AP-1 site were included. B, Supershift EMSA was performed to confirm the identity of the transcription factors binding the AP-1 probe. Nuclear extracts (n = 3 per group) were preincubated with non-neutralizing antibodies against c-Jun, JunB, or JunD prior to adding the labeled probe. C, Chromatin immunoprecipitation assays were carried out with specific antibodies (+IgG) against RNA polymerase II (Pol II), c-Jun, or JunB, using DNA from normal and SSc dermal fibroblasts (n = 3 per group) treated with or without TGFβ (2 ng/ml), with expression assessed in the F2 and proximal promoter sites. Nonspecific antibody (+IgG Con) and nonimmunoprecipitated DNA (input) were included as controls. D, Schematic representations show a putative mechanism for enhanced COL1A2 expression in response to TGFβ in SSc fibroblasts, mediated by c-Jun and JunB and involving the FUE AP-1 site, compared to basal COL1A2 expression in normal fibroblasts. See Figure 1 for other definitions.
Supershift assays using specific antibodies (Figure 2B) revealed that normal nuclear extracts bound c-Jun and, to a lesser extent, JunD. In SSc, c-Jun binding was greatly reduced, and JunB, which was previously absent in normal nuclear extracts, accounted for 2 of the shifted bands. JunD binding was more pronounced in SSc extracts (Figure 2B). These gel-shift findings suggest that in SSc dermal fibroblasts or in normal nuclear extracts treated with TGFβ, JunB binding replaces c-Jun binding on the AP-1 site in the COL1A2 upstream enhancer.
These results were confirmed using ChIP assays, which demonstrated the association of the F2 region in the FUE with the proximal promoter and the transcriptional machinery. ChIP was conducted on explanted normal or SSc fibroblasts that were left untreated or treated with TGFβ and then incubated with specific antibodies against RNA polymerase II, c-Jun, and JunB. As expected, RNA polymerase II coprecipitated with the proximal promoter in both normal and SSc cells with or without TGFβ treatment (Figure 2C). RNA polymerase II also coprecipitated with the F2 enhancer sequence, despite not being able to bind the enhancer directly, thereby demonstrating the interaction between the transcriptional machinery in the promoter with the factors bound to the F2 region. Thus, the data confirm the coalescence of the enhancer/promoter.
In normal fibroblasts, in the absence of TGFβ, c-Jun appeared to be mediating this interaction, whereas there was no JunB binding on the F2 enhancer sequence (Figure 2C). TGFβ treatment resulted in a decrease in c-Jun binding and an increase in JunB binding (Figure 2C). In SSc fibroblasts, c-Jun binding was greatly diminished on both the enhancer and the proximal promoter (Figure 2C). In contrast, JunB appeared to bind the F2 region in the absence of TGFβ treatment and was further increased upon the addition of TGFβ (Figure 2C).
Based on these findings, a simplified model of c-Jun/JunB regulation can be put forward (Figure 2D), in which basal expression of COL1A2 occurs in normal fibroblasts as a result of c-Jun being bound to the AP-1 site (−20.0 kb) in the F2 region. However, in SSc dermal fibroblasts or in TGFβ-treated normal fibroblasts, the AP-1 site is occupied by JunB, resulting in a stronger interaction of factors bound to the FUE with the transcriptional machinery in the proximal promoter, and ultimately leading to higher expression of COL1A2.
Expression levels of AP-1 family members in scleroderma
We tested the expression levels of AP-1 family members in dermal fibroblasts explanted from biopsy specimens that were obtained from a cohort of patients with active diffuse cutaneous SSc. Western blot analysis revealed that protein levels of c-Jun, JunD, cFos, Fra-1, and Fra-2 were the same in both the control and SSc cohorts, and only the level of JunB differed. JunB was absent in the control cohort (Figure 3A). The time course of TGFβ treatment of normal fibroblasts showed that JunB was the only AP-1 family member that was induced by TGFβ treatment over 24 hours (Figure 3B).
Figure 3.

Protein expression levels of AP-1 family members in dermal fibroblasts from healthy controls and patients with diffuse SSc. A, Expression of c-Jun, JunB, JunD, c-Fos, Fra-1, and Fra-2 in normal and SSc fibroblasts (n = 10 per group) was assessed by Western blotting. Five representative samples are shown, with GAPDH used as the loading control. B, Western blotting was used to assess a normal fibroblast line for protein expression at various times over a 24-hour period after serum starvation and addition of TGFβ (2 ng/ml). C and D, Top, Three-color immunofluorescence staining was used to assess the cellular location of c-Jun (C) or JunB (D) in normal and SSc dermal fibroblasts (n = 3 per group; representative results shown). DAPI was used to stain nuclei, and phalloidin (Phal) was used to stain actin filaments. Bottom, The number of cells showing nuclear or cytoplasmic staining was assessed and expressed as a percentage of the total number of cells. Bars show the mean ± SEM aggregate results from 2 independent experiments. Statistical significance was assessed using Student's t-test. See Figure 1 for other definitions.
We examined the intracellular distribution of c-Jun and JunB in normal and SSc dermal fibroblasts (Figures 3C and D). In normal fibroblasts, c-Jun expression was mainly nuclear, and JunB expression was low. In SSc fibroblasts, cytoplasmic c-Jun expression was observed in >90% of cells (Figure 3C), and concomitantly, >90% of the SSc cells showed nuclear expression of JunB (Figure 3D).
We determined the expression and distribution of JunB, c-Jun, and JunD in sections of dermal biopsy specimens obtained from healthy controls and patients with diffuse cutaneous SSc. The expression of c-Jun was observed in the dermis of both normal control subjects and patients with diffuse cutaneous SSc (Figure 4B). Similarly, nuclear expression of JunD was observed in dermal fibroblasts and around vessels in the dermis of both normal subjects and patients with diffuse cutaneous SSc (Figure 4C). However, JunB expression was absent from the dermis of normal subjects, whereas in SSc patients, JunB was highly expressed in dermal fibroblasts and around blood vessels (Figure 4A).
Figure 4.

Protein expression of activator protein 1 family members in skin biopsy specimens from healthy controls and patients with diffuse systemic sclerosis (SSc). Expression of JunB (A), c-Jun (B), and JunD (C) was determined by immunohistochemistry in skin biopsy specimens (n = 5 per group) using specific antibodies, visualized using 3,3′-diaminobenzidine (brown staining) and counterstained with hematoxylin. Images are shown at low (top panels) and high (bottom panels) magnification. Representative images from 1 patient are shown. Insets, Isotype control antibodies used at the same concentration as the primary antibodies. Arrows indicate positive expression. Bars = 100 μM.
Inhibition of JunB in SSc
Having established the differential expression of JunB, we tested the functional role of JunB in normal and scleroderma dermal fibroblasts. We used siRNA to target JunB in dermal fibroblasts from healthy controls and patients with diffuse cutaneous SSc (Figure 5A). Normal fibroblasts expressed only low levels of JunB, except when treated with TGFβ. After treatment with TGFβ, normal fibroblasts that were either left untreated or transfected with scrambled control siRNA expressed JunB and type I collagen. However, when JunB was inhibited by specific siRNA for JunB in normal fibroblasts, there was a dose-dependent decrease in the expression of type I collagen and connective tissue growth factor (CTGF; CCN2) (Figure 5A).
Figure 5.

Effect of JunB down-regulation by small interfering RNA (siRNA) on the expression of fibrotic markers and cell migration. A, JunB siRNA (siJunB) was used at concentrations of 10 nM and 25 nM, and scrambled control siRNA (siControl) was used at 25 nM. The levels of JunB, c-Jun, and JunD, as well as the fibrotic markers type I collagen (Col1) and connective tissue growth factor (CCN2), in cells from normal healthy controls and patients with SSc (n = 3 per group) with or without treatment with TGFβ were assessed by Western blotting. Six different cell lines were used; representative images are shown. B, Migration of normal and SSc fibroblasts was assessed by in vitro scratch wound assay. Top, Fibroblasts from normal healthy controls and patients with SSc (n = 3 per group) were either left untreated or treated with TGFβ and transfected with siJunB (20 nM) or siControl (20 nM). Six different cell lines were used; representative images are shown. Each assay was carried out in triplicate. Bottom, The percentage of the scratch area covered by migrating fibroblasts was measured after 24 hours. Bars show the mean ± SEM. Statistical significance was assessed using Student's t-test. See Figure 1 for other definitions.
When fibroblasts from patients with diffuse cutaneous SSc were transfected with scrambled control siRNA, expression of JunB and type I collagen was observed. SSc fibroblasts also expressed JunB regardless of whether TGFβ was added. Inhibition of JunB using specific siRNA for JunB resulted in a dose-dependent decrease in type I collagen and CTGF expression.
Expression of c-Jun and JunD was unaffected in both normal and scleroderma fibroblasts in which JunB had been inhibited. This suggests that JunB is involved in the activation of fibroblasts and in the establishment of the fibrotic phenotype.
We also assessed the effect of JunB inhibition on fibroblast migration in a scratch wound assay (Figure 5B). Scratch wounds were generated on a fibroblast monolayer using a scratch tool that creates equal wound areas allowing for cell migration. Normal fibroblasts that were activated with TGFβ and then transfected with scrambled control siRNA (25 nM) covered a mean ± SEM 92.2 ± 1.98% of the scratch area after 48 hours. Inhibition of JunB with siRNA for JunB (25 nM) in normal fibroblasts resulted in only 26.6 ± 4.5% coverage after 48 hours. Scleroderma fibroblasts that were not treated with TGFβ but were transfected with scrambled control siRNA covered 91.25 ± 3.35% of the scratch wound area, whereas when the cells were transfected with siRNA for JunB, only 20.75 ± 1.34% of the scratch area was covered after 48 hours. Similar results were obtained with SSc fibroblasts that were treated with TGFβ and then transfected with scrambled control siRNA (96.75 ± 2.1% area coverage) or with siRNA for JunB (19.25 ± 1.72% area coverage). Interestingly, untreated normal fibroblasts, when transfected with scrambled control siRNA, covered 70.6 ± 4.1% of the scratch area after 48 hours, whereas those transfected with siRNA for JunB covered 47.8 ± 3.3% of the scratch area. Since the expression of JunB in normal fibroblasts grown in confluent monolayers was low, one can speculate that this discrepancy could be attributed to a localized increase in JunB expression within cells adjacent to the scratch wound, where contact inhibition is abolished and the migration process is initiated.
Mechanism of JunB overexpression in SSc
Although JunB protein was overexpressed, JunB messenger RNA levels were not significantly different between normal and SSc fibroblasts (Figure 6A). We hypothesized that JunB may be regulated by a posttranslational mechanism. Previous studies in patients with cancer have shown that JunB is regulated by GSK-3β phosphorylation, leading to JunB degradation via the proteasome (22,23). Inhibiting GSK-3β phosphorylation using the specific inhibitor AR-A14418 increased the level of JunB protein in normal fibroblasts, making it roughly equal to the level observed in SSc fibroblasts (Figure 6B). Treatment with the proteasome inhibitor MG-132 for 3 hours produced similar results. Interestingly, neither inhibitor appeared to influence the levels of c-Jun (Figure 6B) or JunD (results not shown).
Figure 6.
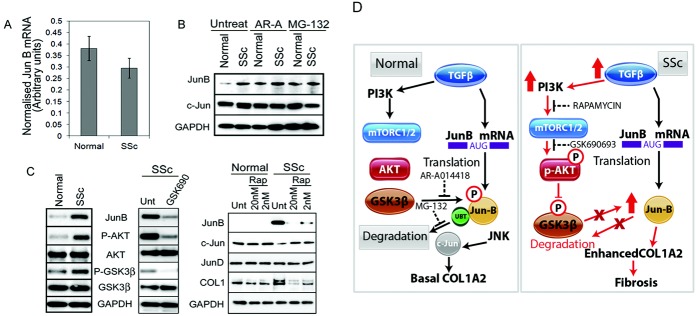
Pathways that lead to JunB overexpression in patients with SSc. A, JunB mRNA levels were assessed in untreated fibroblasts from normal healthy controls and patients with diffuse SSc. Bars show the mean ± SEM (n = 6 per group). B, Fibroblasts from normal healthy controls and patients with diffuse SSc (n = 3 per group) were serum-starved and then left untreated or treated for 3 hours with the specific glycogen synthase kinase 3β (GSK-3β) inhibitor AR-A14418 (AR-A) or with the proteasome inhibitor MG-132. Expression of JunB and c-Jun was assessed by Western blotting. C, The role of mammalian target of rapamycin (mTOR)/Akt signaling in JunB expression was assessed by Western blotting. Left, Western blot showing the difference in Akt and GSK-3β phosphorylation between serum-starved SSc and normal fibroblasts. Middle, Western blot of serum-starved SSc fibroblasts left untreated (Unt) or treated with TGFβ (2 ng/μl) for 16 hours, prior to removal of TGFβ and treatment with the specific Akt inhibitor GSK690693 (50 μM) for 3 hours. Right, Western blot of serum-starved normal or SSc fibroblasts left untreated or treated with 2 different concentrations of rapamycin (Rap) for 6 hours, prior to harvesting each protein. All Western blots are representative of at least 3 independent experiments. D, Schematic diagrams show the regulation of JunB levels by TGFβ, mTOR, Akt, and GSK-3β in normal and SSc fibroblasts. PI3K = phosphatidylinositol 3-kinase; AUG = adenosine, uridine, and guanosine; P = phosphorylated; UBT = ubiquitinated (see Figure 1 for other definitions).
Phospho-Akt, which is known to phosphorylate and inactivate GSK-3β, was up-regulated in SSc fibroblasts. We found that the levels of both phospho-Akt and phospho–GSK-3β were higher in SSc fibroblasts compared to normal fibroblasts (Figure 6C).
To test the involvement of Akt phosphorylation, we stimulated SSc fibroblasts with TGFβ in the presence or absence of 50 nM GSK690693 (Akt inhibitor). After treatment of SSc fibroblasts with GSK690693, we observed reduced phosphorylation of both Akt and GSK-3β and lower expression of JunB (Figure 6C).
Since Akt and GSK-3β are both downstream of the mTOR pathway, we also utilized rapamycin to investigate the role of mTOR in the accumulation of JunB in SSc fibroblasts. Treatment of SSc fibroblasts with rapamycin resulted in decreased levels of JunB and type I collagen protein expression, in a dose-dependent manner. Rapamycin treatment had no effect on the levels of c-Jun and JunD (Figure 6C).
We also investigated the effect of rapamycin on the cellular localization of JunB, c-Jun, and JunD in normal and SSc fibroblasts (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38897/abstract). In normal fibroblasts, in which there was no JunB expression and c-Jun was mainly located in the nucleus, rapamycin treatment had no effect on the phenotype of the cells (low levels of type I procollagen and α-smooth muscle actin [α-SMA]). However, in SSc fibroblasts, in which the cells displayed a fibrotic phenotype (high levels of type I procollagen and α-SMA) and there was high nuclear expression of JunB and cytoplasmic expression of c-Jun, rapamycin treatment resulted in a normalizing effect, whereby the nuclear levels of c-Jun increased, the nuclear levels of JunB decreased, and both type I collagen and α-SMA were down-regulated. Taken together, these data show that in SSc dermal fibroblasts, activated mTOR induces the accumulation of JunB by inducing the phosphorylation of Akt, which in turn phosphorylates and deactivates GSK-3β, thereby preventing the degradation of JunB by the proteasome. In contrast, in normal fibroblasts, GSK-3β is active and targets JunB for proteasomal degradation (Figure 6D).
DISCUSSION
The excessive deposition of type I collagen is a common pathologic feature of all fibrotic diseases. In SSc, collagen deposition in the dermis and the resultant fibrosis is an important contributor to the chronic disease process, leading to manifestations such as ulcers, reduced hand function, and a significant decrease in quality of life.
In this study, we show that the expression and deposition of type I collagen is potently regulated by the COL1A2-FUE, which interacts with the proximal promoter of the gene to initiate transcription. We previously showed that this regulatory element is required in order to reactivate COL1A2 expression in adult dermal tissue during processes of wound healing, repair, or pathologic development (5). We have now determined that within this enhancer lies a small regulatory sequence containing an AP-1 consensus site, which is essential for enhancer function and controls the overexpression of COL1A2 by scleroderma fibroblasts. Previously, we described a mechanism in which TGFβ increases COL1A2 expression by activating JunB expression, which leads to exchange of c-Jun for JunB, thus stabilizing and enhancing promoter/enhancer coalescence (6). The significance of these study findings is that it uncovers the unifying mechanism underlying the function of AP-1 family members in the control of collagen expression.
Although both JunB and c-Jun can bind the AP-1–FUE site and direct collagen expression, JunB mediates enhanced expression, whereas c-Jun is responsible for basal expression levels. In SSc dermal fibroblasts, constitutive JunB expression is responsible for enhanced collagen expression. Other investigators have reported that AP-1 members such as JunD, c-Jun, and Fra-2 are important in scleroderma dermal fibrosis (17–20,24). These studies are consistent with our findings. Collagen gene regulation is complex and involves several pathways, some of which are AP-1 dependent, that are likely to have an impact on each other. Indeed, although COL1A1 and COL1A2 are on separate chromosomes, they are coordinately expressed via mechanisms not fully understood. Herein we have focused on the COL1A2 enhancer/proximal promoter interface, finding that although c-Jun occupied the COL1A2 enhancer TβRE under normal conditions, it was JunB that was critical in activating and enhancing type I collagen expression in SSc dermal fibroblasts.
We also found that specific inhibition of JunB by siRNA resulted in decreased type I collagen expression and a decreased ability of SSc dermal fibroblasts to migrate into a scratch wound, thus demonstrating the importance of JunB for many normal dermal fibroblast functions. We therefore suggest that JunB orchestrates the activation of the COL1A2 enhancer. Consistent with previous reports, it is likely that other AP-1 family members are also necessary for FUE activity and form part of the regulatory complex that assembles in the enhancer, but their action is conditional, requiring the presence of JunB, which is the critical regulatory component.
JunB was originally discovered for its role in cancer as an early growth-response gene that inhibited cell proliferation and transformation and antagonized c-Jun activity. However, the findings from recent reports suggest that JunB has oncogenic activities, such as promoting proliferation in cutaneous lymphomas (25), and it can cooperate with c-Jun in the development of fibrosarcoma (26).
In this study, we identified aberrant proteasomal targeting, rather than direct transcriptional activation, as the reason behind the constitutive JunB expression in scleroderma skin. Indeed, there are several examples of JunB regulation by the ubiquitin–proteasome pathway in the literature (22,27,28). GSK-3β phosphorylation of JunB is therefore likely to be one of the critical events determining the difference in collagen deposition between normal and SSc fibroblasts. Our results show that Akt activity plays an important role by inactivating GSK-3β through phosphorylation.
Consistent with previous reports highlighting the role of the mTOR pathway in type I collagen regulation (29) and in animal models of scleroderma (30), we found that rapamycin was effective at reducing JunB expression and collagen production in SSc fibroblasts. The therapeutic potential of rapamycin was recently confirmed in human patients. A small, randomized, single-blind pilot study of rapamycin in patients with early diffuse SSc demonstrated that rapamycin significantly improved both the modified Rodnan skin thickness score and the patient's global assessment of health (31).
In conclusion, the results of this study demonstrate that type I collagen overexpression in scleroderma skin is regulated, in part, by the persistent expression of JunB, which occupies the TβRE in the COL1A2 enhancer. The constitutive up-regulation of JunB expression in SSc fibroblasts appears to be attributable to aberrant signaling of GSK-3β through the mTOR/Akt pathway, leading to failure of the mechanism that normally targets JunB for proteasomal degradation. Inhibition of Akt or mTOR results in decreased levels of JunB and type I collagen and a normalization of the fibrotic phenotype of scleroderma fibroblasts in vitro, whereas inhibition of GSK-3β results in constitutive expression of JunB in normal fibroblasts. In summary, these findings add yet another layer of complexity in the regulation of type I collagen by AP-1 transcription factors and fibrosis (18–20,24), reinforcing the view that these pathways have translational utility for the treatment of SSc.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Ponticos had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Ponticos, Papaioannou, Holmes, Denton, Bou-Gharios, Abraham.
Acquisition of data. Ponticos, Papaioannou, Xu, Khan, Denton, Bou-Gharios, Abraham.
Analysis and interpretation of data. Ponticos, Papaioannou, Denton, Bou-Gharios, Abraham.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Figure 1.
REFERENCES
- 1.De Val S, Ponticos M, Antoniv TT, Wells DJ, Abraham D, Partridge T, et al. Identification of the key regions within the mouse pro-α2(I) collagen gene far-upstream enhancer. J Biol Chem. 2002;277:9286–92. doi: 10.1074/jbc.M111040200. [DOI] [PubMed] [Google Scholar]
- 2.Antoniv TT, De Val S, Wells D, Denton CP, Rabe C, de Crombrugghe B, et al. Characterization of an evolutionarily conserved far-upstream enhancer in the human α2(I) collagen (COL1A2) gene. J Biol Chem. 2001;276:21754–64. doi: 10.1074/jbc.M101397200. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka S, Antoniv TT, Liu K, Wang L, Wells DJ, Ramirez F, et al. Cooperativity between far upstream enhancer and proximal promoter elements of the human α2(I) collagen (COL1A2) gene instructs tissue specificity in transgenic mice. J Biol Chem. 2004;279:56024–31. doi: 10.1074/jbc.M411406200. [DOI] [PubMed] [Google Scholar]
- 4.De Crombrugghe B, Vuorio T, Karsenty G, Maity S, Rutheshouser EC, Goldberg H. Transcriptional control mechanisms for the expression of type I collagen genes. Ann Rheum Dis. 1991;50(Suppl 4):872–6. doi: 10.1136/ard.50.suppl_4.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ponticos M, Abraham D, Alexakis C, Lu QL, Black C, Partridge T, et al. Col1a2 enhancer regulates collagen activity during development and in adult tissue repair. Matrix Biol. 2004;22:619–28. doi: 10.1016/j.matbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Ponticos M, Harvey C, Ikeda T, Abraham D, Bou-Gharios G. JunB mediates enhancer/promoter activity of COL1A2 following TGF-β induction. Nucleic Acids Res. 2009;37:5378–89. doi: 10.1093/nar/gkp544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–57. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 8.Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001;20:2413–23. doi: 10.1038/sj.onc.1204380. [DOI] [PubMed] [Google Scholar]
- 9.Angel P, Szabowski A. Function of AP-1 target genes in mesenchymal-epithelial cross-talk in skin. Biochem Pharmacol. 2002;64:949–56. doi: 10.1016/s0006-2952(02)01158-9. [DOI] [PubMed] [Google Scholar]
- 10.Brenner DA, O'Hara M, Angel P, Chojkier M, Karin M. Prolonged activation of jun and collagenase genes by tumour necrosis factor-α. Nature. 1989;337:661–3. doi: 10.1038/337661a0. [DOI] [PubMed] [Google Scholar]
- 11.George SJ, Lloyd CT, Angelini GD, Newby AC, Baker AH. Inhibition of late vein graft neointima formation in human and porcine models by adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-3. Circulation. 2000;101:296–304. doi: 10.1161/01.cir.101.3.296. [DOI] [PubMed] [Google Scholar]
- 12.Verrecchia F, Tacheau C, Schorpp-Kistner M, Angel P, Mauviel A. Induction of the AP-1 members c-Jun and JunB by TGF-β/Smad suppresses early Smad-driven gene activation. Oncogene. 2001;20:2205–11. doi: 10.1038/sj.onc.1204347. [DOI] [PubMed] [Google Scholar]
- 13.Gurzov EN, Bakiri L, Alfaro JM, Wagner EF, Izquierdo M. Targeting c-Jun and JunB proteins as potential anticancer cell therapy. Oncogene. 2008;27:641–52. doi: 10.1038/sj.onc.1210690. [DOI] [PubMed] [Google Scholar]
- 14.Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann Rheum Dis. 2011;70(Suppl 1):i109–12. doi: 10.1136/ard.2010.140533. [DOI] [PubMed] [Google Scholar]
- 15.Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein U, Tian J, et al. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13:607–19. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verrecchia F, Wagner EF, Mauviel A. Distinct involvement of the Jun-N-terminal kinase and NF-κB pathways in the repression of the human COL1A2 gene by TNF-α. EMBO Rep. 2002;3:1069–74. doi: 10.1093/embo-reports/kvf219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avouac J, Palumbo K, Tomcik M, Zerr P, Dees C, Horn A, et al. Inhibition of activator protein 1 signaling abrogates transforming growth factor β–mediated activation of fibroblasts and prevents experimental fibrosis. Arthritis Rheum. 2012;64:1642–52. doi: 10.1002/art.33501. [DOI] [PubMed] [Google Scholar]
- 18.Palumbo K, Zerr P, Tomcik M, Vollath S, Dees C, Akhmetshina A, et al. The transcription factor JunD mediates transforming growth factor β-induced fibroblast activation and fibrosis in systemic sclerosis. Ann Rheum Dis. 2011;70:1320–6. doi: 10.1136/ard.2010.148296. [DOI] [PubMed] [Google Scholar]
- 19.Reich N, Tomcik M, Zerr P, Lang V, Dees C, Avouac J, et al. Jun N-terminal kinase as a potential molecular target for prevention and treatment of dermal fibrosis. Ann Rheum Dis. 2012;71:737–45. doi: 10.1136/annrheumdis-2011-200412. [DOI] [PubMed] [Google Scholar]
- 20.Reich N, Maurer B, Akhmetshina A, Venalis P, Dees C, Zerr P, et al. The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum. 2010;62:280–90. doi: 10.1002/art.25056. [DOI] [PubMed] [Google Scholar]
- 21.Subcommittee for Scleroderma Criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Preliminary criteria for the classification of systemic sclerosis (scleroderma) Arthritis Rheum. 1980;23:581–90. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 22.Perez-Benavente B, Garcia JL, Rodriguez MS, Pineda-Lucena A, Piechaczyk M, Font de Mora J, et al. GSK3-SCFFBXW7 targets JunB for degradation in G2 to preserve chromatid cohesion before anaphase. Oncogene. 2013;32:2189–99. doi: 10.1038/onc.2012.235. [DOI] [PubMed] [Google Scholar]
- 23.Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science. 2004;306:271–5. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- 24.Maurer B, Reich N, Juengel A, Kriegsmann J, Gay RE, Schett G, et al. Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann Rheum Dis. 2012;71:1382–7. doi: 10.1136/annrheumdis-2011-200940. [DOI] [PubMed] [Google Scholar]
- 25.Mao X, Orchard G, Lillington DM, Child FJ, Vonderheid EC, Nowell PC, et al. BCL2 and JUNB abnormalities in primary cutaneous lymphomas. Br J Dermatol. 2004;151:546–56. doi: 10.1111/j.1365-2133.2004.06106.x. [DOI] [PubMed] [Google Scholar]
- 26.Bossy-Wetzel E, Bravo R, Hanahan D. Transcription factors junB and c-jun are selectively up-regulated and functionally implicated in fibrosarcoma development. Genes Dev. 1992;6:2340–51. doi: 10.1101/gad.6.12a.2340. [DOI] [PubMed] [Google Scholar]
- 27.Gao M, Karin M. Regulating the regulators: control of protein ubiquitination and ubiquitin-like modifications by extracellular stimuli. Mol Cell. 2005;19:581–93. doi: 10.1016/j.molcel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Benavente B, Farras R. Regulation of GSK3β-FBXW7-JUNB axis. Oncotarget. 2013;4:956–7. doi: 10.18632/oncotarget.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shegogue D, Trojanowska M. Mammalian target of rapamycin positively regulates collagen type I production via a phosphatidylinositol 3-kinase-independent pathway. J Biol Chem. 2004;279:23166–75. doi: 10.1074/jbc.M401238200. [DOI] [PubMed] [Google Scholar]
- 30.Yoshizaki A, Yanaba K, Yoshizaki A, Iwata Y, Komura K, Ogawa F, et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 2010;62:2476–87. doi: 10.1002/art.27498. [DOI] [PubMed] [Google Scholar]
- 31.Su TI, Khanna D, Furst DE, Danovitch G, Burger C, Maranian P, et al. Rapamycin versus methotrexate in early diffuse systemic sclerosis: results from a randomized, single-blind pilot study. Arthritis Rheum. 2009;60:3821–30. doi: 10.1002/art.24986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1.