

Abstract
Chinese hamster ovary (CHO) cells are widely used in the biopharmaceutical industry as a host for the production of complex pharmaceutical proteins. Thus genome engineering of CHO cells for improved product quality and yield is of great interest. Here, we demonstrate for the first time the efficacy of the CRISPR Cas9 technology in CHO cells by generating site-specific gene disruptions in COSMC and FUT8, both of which encode proteins involved in glycosylation. The tested single guide RNAs (sgRNAs) created an indel frequency up to 47.3% in COSMC, while an indel frequency up to 99.7% in FUT8 was achieved by applying lectin selection. All eight sgRNAs examined in this study resulted in relatively high indel frequencies, demonstrating that the Cas9 system is a robust and efficient genome-editing methodology in CHO cells. Deep sequencing revealed that 85% of the indels created by Cas9 resulted in frameshift mutations at the target sites, with a strong preference for single base indels. Finally, we have developed a user-friendly bioinformatics tool, named “CRISPy” for rapid identification of sgRNA target sequences in the CHO-K1 genome. The CRISPy tool identified 1,970,449 CRISPR targets divided into 27,553 genes and lists the number of off-target sites in the genome. In conclusion, the proven functionality of Cas9 to edit CHO genomes combined with our CRISPy database have the potential to accelerate genome editing and synthetic biology efforts in CHO cells. Biotechnol. Bioeng. 2014; 111: 1604–1616. © 2014 The Authors. Biotechnology and Bioengineering Published by Wiley Periodicals, Inc.
Keywords: CRISPR Cas9, genome editing, CRISPy, Chinese hamster ovary cells, database
Introduction
Chinese hamster ovary (CHO) cells are the primary factories for biopharmaceuticals, due to their capacity to correctly fold and post-translationally modify recombinant proteins compatible with humans (Jayapal et al., 2007). Genome editing and engineering are of increasing interest in this field for the purpose of increasing cellular production capabilities and improving product quality. This is facilitated by the expanding amount of data being generated for CHO cells including genomic sequences (Lewis et al., 2013; Xu et al., 2011) and other 'omics data such as transcriptomics, proteomics, and metabolomics information (Kildegaard et al., 2013). Early efforts to engineer CHO cells by gene disruptions have been performed mainly by conventional gene targeting strategies based on homologous recombination (HR) (Yamane-Ohnuki et al., 2004). However, HR-based gene targeting is rare event in mammalian cells, since non-homologous end-joining (NHEJ) occurs several orders of magnitude more frequently than HR (Sedivy and Sharp, 1989). NHEJ is an imperfect repair process that often results in insertions or deletions of DNA bases at the site of the double strand break (DSB) during repair, making NHEJ particularly applicable for generating gene disruptions. To induce site specific gene disruptions, targeting endonucleases like transcription activator-like effector nucleases (TALENs), zinc-finger nucleases (ZFNs), and meganucleases have been successfully applied to mammalian cells such as human and CHO cell lines (Galetto et al., 2009; Miller et al., 2011; Santiago et al., 2008).
More recently the RNA-guided Cas9 nuclease has proven to be a highly valuable tool for genome editing in nematodes (Waaijers et al., 2013), fruitflies (Bassett et al., 2013; Gratz et al., 2013), zebrafish (Chang et al., 2013; Hwang et al., 2013), plants (Jiang et al., 2013), mice (Wang et al., 2013; Yang et al., 2013), and human cells (Cho et al., 2013; Cong et al., 2013; Mali et al., 2013). Cas9 is the effector protein of the type II clustered regularly interspaced short palindromic repeat (CRISPR) immune system of Streptococcus pyogenes and functions as a RNA-guided endonuclease (Carroll, 2012; Jinek et al., 2012). Together with two noncoding RNAs called CRISPR-RNA (crRNA) and trans-activating crRNA (tracrRNA), Cas9 binds to and cleaves DNA in a site-specific manner. The specificity is brought about by the crRNA that basepairs to the target DNA. The target site must be adjacent to a protospacer adjacent motif (PAM) consisting of a random nucleotide and two guanines (NGG) (Jinek et al., 2012; Mali et al., 2013). The tracrRNA molecule together with crRNA functions as a scaffold onto which Cas9 binds. In recent studies, a chimeric RNA that combines the crRNA and tracrRNA termed single guide RNA (sgRNA) has been applied (Chang et al., 2013; Cong et al., 2013; DiCarlo et al., 2013; Jinek et al., 2013; Mali et al., 2013).
In order to express the small chimeric sgRNA, an RNA pol III promoter is required and in previous studies on human cells, a U6 promoter was chosen for this purpose (Cong et al., 2013; Jinek et al., 2013; Mali et al., 2013). Since the U6 promoter initiates transcription at a guanine (G), this base must be present in the 5′ end of the genomic target site sequence. The U6 promoter-dependent requirement combined with the PAM motif gives rise to the following general scheme of the genomic target site sequence: G(N)19NGG. One potential advantage of CRISPR Cas9 technology, when compared to existing methods using TALENs and ZNFs, is its relative low cost. Furthermore, in contrast to ZNFs and TALENs, time-consuming protein engineering is not required to obtain an effective endonuclease (Pennisi, 2013). Thus, the CRISPR technology is attractive for the CHO cell line engineering field and for the genome engineering and synthetic biology community at large.
In this study, we demonstrate for the first time the application of a CHO codon-optimized Cas9 for modifying the genome of CHO cells by disrupting COSMC and FUT8, which are genes encoding proteins involved in O- and N-glycosylation, respectively (Miyoshi et al., 1999; Wang et al., 2010). Deep sequencing analysis revealed a strong preference toward single nucleotide indels. Since no CRISPR bioinformatics tools are available for optimal sgRNA design in CHO cells, the web-based bioinformatics tool “CRISPy” was developed. This design tool facilitates easy and fast sgRNA selection and also incorporates information on possible off-target sites. Combining the CRISPR Cas9 technology and the CRISPy bioinformatics tool, we demonstrate efficient, fast and low cost genetic manipulation of the CHO genome.
Materials and Methods
Plasmid Construction and sgRNA Target Design
The Cas9 sequence from the S. pyogenes strain M1 GAS genome with a 3′ nuclear localization signal was codon-optimized for CHO cells, synthesized (for sequence, see Supplementary Materials and Methods) and subcloned into the mammalian expression vector pJ607-03 (DNA 2.0, Menlo Park, CA, Fig. 1A). The plasmid was then transformed into DH5α subcloning cells (Life Technologies, Paisley, U.K.). Transformant clones were selected on 100 µg/mL Ampicillin (Sigma–Aldrich, St. Louis, MO) LB plates. The chosen sgRNA target sequences are listed in Supplementary Table SI. The sgRNA expression constructs were designed by fusing tracrRNA and crRNA into a chimeric sgRNA (Jinek et al., 2012) and located immediately downstream of a U6 promoter (Chang et al., 2013). The sequences of the U6 promoter, scaffold and terminator are shown in Supplementary Materials and Methods. Initially, the sgRNA expression cassette (Fig. 1A) was synthesized as a gBlock (Integrated DNA Technologies, Leuven, Belgium) and subcloned into the pRSFDuet-1 vector (Novagen, Merck, Damstadt, Germany) using KpnI and HindIII restriction sites. This pRSFDuet-1/sgRNA expression vector was used as backbone in a PCR-based uracil-specific excision reagent (USER) cloning method (Hansen et al., 2012; Nour-Eldin et al., 2006). This method was designed to easily and rapidly change the 19 bp-long variable region (N19) of the sgRNA in order to generate our sgRNA constructs. From the pRSFDuet-1/sgRNA expression vector, a 4,221 bp-long amplicon (expression vector backbone) was generated by PCR (1×: 98°C for 2 min; 30×: 98°C for 10 s, 57°C for 30 s, 72°C for 4 min 12 s; 1×: 72°C for 5 min) using two uracil-containing primers (sgRNA Backbone_fw and sgRNA Backbone_rv, Integrated DNA Technologies, Supplementary Table SII) and the X7 DNA polymerase (Nørholm, 2010). Subsequent to Fastdigest DpnI (Thermo Fisher Scientific, Waltham, MA) treatment, the amplicon was purified from a 2% agarose TBE gel using the QIAEX II Gel Extraction Kit (Qiagen, Hilden, Germany). In parallel, 54 bp-long and 53 bp-long single stranded oligos (sense and antisense strand, respectively) comprising the variable region of the sgRNA were synthesized (TAG Copenhagen, Denmark, Supplementary Table SII). The sense and antisense single stranded oligos (100 µM) were annealed in NEBuffer4 (New England Biolabs, Ipswich, MA) by incubating the oligo mix at 95°C for 5 min in a heating block and the oligo mix was subsequently allowed to slowly cool to RT by turning off the heating block. The annealed oligos were then mixed with the gel purified expression vector backbone and treated with USER enzyme (New England Biolabs) according to manufacturer's recommendations. After USER enzyme treatment, the reaction mixture was transformed into E. coli Mach1 competent cells (Life Technologies) according to standard procedures. Transformant clones were selected on 50 µg/mL Kanamycin (Sigma–Aldrich) LB plates. All constructs were verified by sequencing and purified by NucleoBond Xtra Midi EF (Macherey-Nagel, Düren, Germany) according to manufacturer's guidelines.
Figure 1.
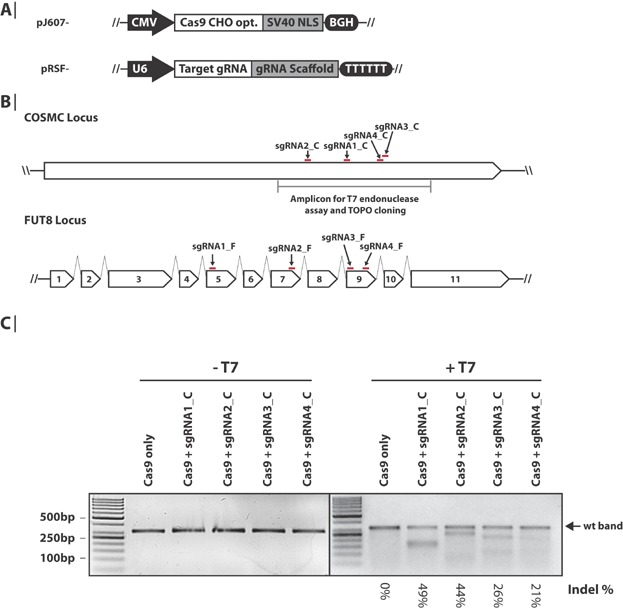
Genome editing in CHO cells by CRISPR Cas9. A: Schematics of the Cas9 and sgRNA expression cassettes. The Cas9 expression cassette consists of a CMV promoter, Cas9 ORF codon-optimized for CHO, SV40 nuclear localization sequence (NLS) and bovine growth hormone (BGH) polyadenylation signal and transcription termination sequence. The sgRNA expression cassette consists of a U6 polymerase III promoter, a target gRNA sequence, a gRNA scaffold sequence and a poly(T) termination sequence. B: Illustration of the sgRNA genomic target sites in COSMC and FUT8. Red lines denote the position of the sgRNA target sites. Introns are depicted as broken lines (not drawn to scale) and exons as arrowed boxes. C: Indel frequency in COSMC analyzed by T7 endonuclease assay. Genomic DNA was extracted from CHO-K1 cells 5 days after transfection with plasmids encoding Cas9 and sgRNA against COSMC. The PCR amplicon covering the sgRNA-target sites as shown in panel B was re-annealed to enable heteroduplex formation before treatment with T7 endonuclease where indicated. Samples were subsequently analyzed by agarose gel electrophoresis. Approximate quantification of indels (%) was peformed with ImageJ software analysis of the uncut (WT) DNA bands. For details see Supplementary Table SVII.
Cell Culture and Transfection
CHO-K1 adherent cells obtained from ATCC (#ATCC-CCL-61) were grown in CHO-K1 F-12K medium (ATCC) supplemented with 10% fetal calf serum (Life Technologies) and 1% Penicillin–Streptomycin (Sigma–Aldrich). Cells were expanded in T-75 cm2 vented cap tissue culture flasks (SARSTEDT, Nümbrecht, Germany) and experiments were performed in Advanced TC Cell Culture Multiwell plates (Greiner Bio-one, Frickenhausen, Germany). Cells were released from plastic ware using trypsin-EDTA (Sigma–Aldrich). Cells were transfected (Day 0) by the Nucleofector 2b device using the Amaxa Cell Line Nucleofector Kit V (Lonza, Basel, Switzerland) according to manufacturer's guidelines (program U-023). A total of 1 × 106 cells were transfected with 1 µg Cas9 plasmid and 1 µg sgRNA plasmid. Cells were incubated at 30°C, 5% CO2 from Day 1 to Day 2 (cold shock) and incubated at 37°C, 5% CO2 at all other times. Two days after transfection (Day 2), cells transfected with the pmaxGFP plasmid (Lonza) were used to estimate the transfection efficiency by analyzing GFP signal using a Celigo Imaging Cell Cytometer (Brooks Automation, Chelmsford, MA). The transfection efficiency was calculated as the percentage of GFP positive cells. Five days after transfection (Day 5), cells were trypsinized and pelleted (200 g, 5 min, RT). Genomic DNA was extracted from the cell pellets using QuickExtract DNA extraction solution (Epicentre, Illumina, Madison, WI) according to manufacturer's instructions and stored at −20°C.
Selection and Phenotypic Analysis of FUT8 Knockout Cells
Five days after transfection (Day 5), selection of FUT8 knockout cells was initiated by supplementing complete medium with 50 µg/mL Lens culinaris agglutinin (LCA; Vector Laboratories, Peterborough, UK) from a 5 mg/mL LCA (10 mM Hepes/NaOH, pH 8.5, 0.15 mM NaCl, 0.1 mM CaCl2) stock solution. Bright field images were taken with a Celigo Imaging Cell Cytometer (Brooks Automation). After 7 days of selection (Day 12), genomic DNA was extracted as described above. In parallel, cells were seeded in complete medium without LCA. The day after (Day 13), cells were incubated for 45 min at RT in complete medium containing 20 µg/mL fluorescein-LCA (Vector Laboratories) and two droplets NucBlue® Live ReadyProbes (Life Technologies) per mL media. Cells were washed three times with complete medium and fluorescence microscopy was performed on a LEAP instrument (Intrexon, Germantown, MD) using the two channel imaging application with the NucBlue stain as target 1 using the blue fluorescence channel and the fluorescein labeled LCA as target 2 using the green fluorescence channel.
T7 Endonuclease Assay
Genomic regions flanking the CRISPR target site for T7 endonuclease assay were amplified from the genomic DNA extracts using DreamTaq DNA polymerase (Thermo Fisher Scientific) by touchdown PCR for COSMC (95°C for 2 min; 10×: 95°C for 30 s, 69–59°C (−1°C/cycle) for 30 s, 72°C for 50 s; 20×: 95°C for 30 s, 59°C for 30 s, 72°C for 50 s; 72°C for 5 min), using PCR primers listed in Supplementary Table SII. The PCR products were subjected to a re-annealing process to enable heteroduplex formation which is sensitive to T7 digestion: 95°C for 10 min; 95–85°C ramping at −2°C/s; 85°C to 25°C at −0.25°C/s; and 25°C hold for 1 min. Re-annealed PCR products were treated with T7 endonuclease (New England Biolabs) for 30 min at 37°C. T7 digested and undigested samples were analyzed on a 3% TAE gel. The percentage of indels was estimated from analysis of the uncut (WT) gel bands with ImageJ software. For details see Supplementary Table SVII.
TOPO™ TA Cloning and Sanger Sequencing
A genomic region of 318 bp covering the four COSMC sgRNA target sites was PCR-amplified from the genomic extracts as described in the T7 endonuclease assay. PCR products were subjected to agarose gel electrophoresis and subsequently gel purified from a 1% agarose TBE gel using the QiaQuick Gel Extraction Kit (Qiagen). Purified PCR products were TOPO-cloned into the pCR4-TOPO vector using the TOPO™ TA cloning kit (Life Technologies) and subsequently transformed into E. coli Mach1 chemically competent cells (Life Technologies). Transformed Mach1 cells were then plated on LB-ampicillin agar plates and grown at 37°C overnight. Plasmids from single colony 60 µg/mL carbenicillin (Novagen, Merck) 2X YT-cultures were extracted using the Nucleospin 8/96 Plasmid kit (Macherey-Nagel). Each plasmid preparation was sequenced using the M13 forward (−20) primer (Supplementary Table SII) on an AB 3500xL Genetic Analyzer (Life Technologies) using the BigDye Terminator v3.1 cycle sequencing kit (Life Technologies).
MiSeq Library Construction and Deep Sequencing
PCR amplicons were designed to be between 150 and 200 bp long and to span the sgRNA target sequence (See Supplementary Table SII for primers and Supplementary Table SIII for amplicon sizes). Amplicons were generated from the genomic DNA extracts using Phusion Hot Start II HF Pfu polymerase (Thermo Fisher Scientific) by touch-down PCR (95°C for 7 min; 20×: 95°C for 45 s, 69°C − 59°C (−0.5°C/cycle) for 30 s, 72°C for 30 s; 35×: 95°C for 45 s, 59°C for 30 s, 72°C for 30 s; 72°C for 7 min). Amplicons were purified on 2% agarose TBE gels and bands with expected fragment sizes were excised and purified using QIAEX II Gel Extraction Kit (Qiagen). Amplicon concentration was measured on Qubit® using the dsDNA BR Assay Kit (Life Technologies). Amplicons were pooled in four for multiplexing (25 ng each, 100 ng in total). Illumina multiplexing adapters were ligated to the pooled amplicons using the TruSeq™ LT DNA Sample Preparation LT kit (Illumina) according to manufacturer's instructions. DNA concentration of the multiplexed libraries was measured with the Qubit® dsDNA BR Assay Kit, and library quality was determined with an Agilent DNA1000 Chip (Agilent Bioanalyzer 2100). Finally, multiplexed libraries were pooled and sequenced on a MiSeq Benchtop Sequencer (Illumina) using the MiSeq Reagent Kit v2 (300 cycles) according to manufacturer's protocol for a 151 bp paired-end analysis.
Deep Sequencing Data Analysis
To minimize the number of required indexes, the same index was used on different PCR products (multiplexing) and the identities of the PCR products were found in the data analysis step based on their individual PCR primer sequences. A Python script was developed to process MiSeq data resulting from the targeted re-sequencing of the Cas9 target site regions. The script performs the following tasks: (1) join paired-end reads; (2) check if resulting sequences contain correct PCR primer at both beginning and end of the sequences and discard those sequences that fail to do so; (3) compute output length of PCR product; and (4) compare PCR product length to expected PCR product length. Paired-end reads were joined using fastq-join (Aronesty 2011: http://code.google.com/p/ea-utils). Ends were checked for correct PCR primer using fastx_barcode_splitter (http://hannonlab.cshl.edu/fastx_toolkit/index.html).
Cas9 Target Finding Database and Web Interface “CRISPy”
A python script, utilizing BioPython (Cock et al., 2009), was created to search through the CHO-K1 genome obtained from http://www.chogenome.org (Hammond et al., 2012). Potential target sequences of the format G(N)19NGG were searched for in annotated exons. Each of the identified target sequences was then searched against the entire genome for potential off-targets. Only genomic sequences matching the 13 bp sequence immediately upstream of the NGG were identified as potential off-targets, and one or two mismatches were allowed. Additionally, it was tested if an off-target/mismatch was located within an exon. The database generated by the Python script was uploaded to a MySQL database and an interface based on HTML, PHP, and JavaScript was created to allow public access to the database.
Results
RNA-Guided CRISPR Cas9 Shows Targeted Endonuclease Activity in CHO
In order to test if the RNA-guided CRISPR Cas9 system could be applied for gene disruptions in CHO cells, an expression vector with a CHO codon-optimized version of Cas9 with a C-terminal SV40 nuclear localization signal under the control of a CMV promoter was constructed (Fig. 1A). To direct Cas9 to disrupt genes of interest, sgRNA expression constructs were generated using the human U6 polymerase III promoter as previously described (Mali et al., 2013) (Fig. 1A). Four sgRNAs were designed for each of the two genes; C1GALT1C1 (COSMC) encoding the C1GALT1-specific chaperone 1 and FUT8 encoding fucosyltransferase 8 (alpha-(1,6)-fucosyltransferase). COSMC is a chaperone essential for correct protein O-glycosylation (Wang et al., 2010) and FUT8 catalyzes the transfer of fucose from GDP-fucose to N-acetylglucosamine (Wilson et al., 1976). In general, it may be desirable to choose a target early in the gene in order to avoid a truncated yet partially functional protein. However, knowledge regarding alternative splicing or active sites may in some cases make a more downstream position or exon a better choice. The four sgRNA constructs for COSMC target the only exon present in the gene. This exon has previously been targeted with a ZFN in human cells (Steentoft et al., 2011). FUT8 consists of 11 exons and the FUT8 sgRNA constructs target exon 5, exon 7, and exon 9 (Fig. 1B). Exon 9 was chosen based on previously published work targeting FUT8 with a ZFN (Malphettes et al., 2010), and exons 5 and 7 were chosen to target earlier exons in the gene sequence. To compare the activity of the designed sgRNAs, adherent CHO-K1 cells were transfected transiently with the CHO codon-optimized Cas9 expression vector and each of the eight sgRNAs to introduce DSBs in the two test genes in two independent experiments (replicate 1 and 2). Initially, a T7 endonuclease assay was performed to analyze the indel frequency at the COSMC loci resulting from Cas9 guided by the four different COSMC-targeting sgRNAs (sgRNA1_C, sgRNA2_C, sgRNA3_C, and sgRNA4_C). When assayed 5 days after transfection, genomic indel events were detected for all four sgRNAs (Fig. 1C, replicate #1 is shown). Using ImageJ software analysis of the uncut gel bands, the percentage of indels generated at the COSMC loci was estimated to be between 21% and 49% (Supplementary Table SVII). The fragment sizes of the digested amplicons correspond to the expected sizes (Supplementary Table SIV).
High Indel Frequency Obtained by All Four COSMC-Targeting sgRNAs
To further assess the indel frequency achieved with the COSMC sgRNAs, TOPO cloning-based sequencing of gel-purified amplicons from the COSMC genomic site was performed (Fig. 2A, Supplementary Table SV). Consistent with the T7 endonuclease assay, Cas9 activity was observed for all four COSMC sgRNAs in two independent experiments (replicate 1 and 2). sgRNA1_C gave rise to the highest indel frequency of 48.0% and 66.7% and sgRNA4_C displayed the lowest indel frequency of 10.3% and 17.9%, in replicate 1 and 2, respectively. Based on GFP fluorescence of cells transfected with GFP-encoding plasmids, transfection efficiency was estimated to be approximately 60% and 65% for replicate 1 and 2, respectively. The indels created by the COSMC sgRNAs predominantly involved a single-base insertion of a thymine or deletions (Fig. 2B; only sgRNA1_C, replicate #1 is shown). To analyze the Cas9 activity in greater detail, deep sequencing was performed using the genomic DNA extracts from the two independent experiments (Fig. 2C). Deep sequencing data comprising between approximately 200,000–700,000 reads per sgRNA in each of the two replicates correlated well with the sequencing data obtained from TOPO cloning (between 21 and 32 sequences per sgRNA). Both sequence-based methods detected relatively high Cas9-activity for all four sgRNAs. Deep sequencing reported indel frequencies of 47.3% and 44.3% for sgRNA1_C, 45.6% and 40.2% for sgRNA2_C, 36.0% and 27.2% for sgRNA3_C and 15.2% and 13.6% for sgRNA4_C in replicate 1 and 2, respectively. Deep sequencing of control cells transfected only with Cas9-encoding plasmids showed an indel frequency of 0.1–0.2% (Supplementary Fig. S1). To examine the fidelity of both sequence-based methods, indel-containing sequences obtained from TOPO-cloning were checked using the deep sequencing data. All indels detected in the TOPO-cloning experiments were also retrieved in the deep sequencing data (data not shown).
Figure 2.
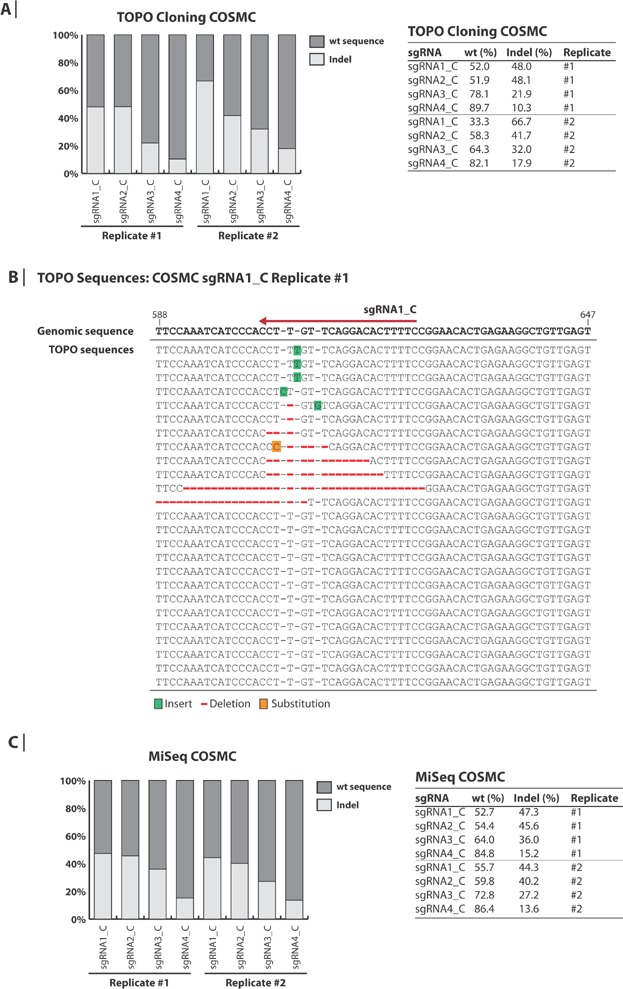
Analysis of generated indels in COSMC. A: TOPO™ TA-based sequence analysis of COSMC. Genomic DNA was extracted from CHO-K1 cells 5 days after transfection with plasmids encoding Cas9 and sgRNA against COSMC. PCR amplicons covering the sgRNA-target sites in COSMC were TOPO™ TA-cloned and Sanger sequenced. Between 21 and 32 sequences were obtained for each sgRNA. The percentages of wt and indel sequences are illustrated in the bar plot and shown in the table. B: Alignment of TOPO™ sequence traces. Genomic DNA from cells transfected with Cas9 + sgRNA1_C (replicate #1) were subjected to TOPO™ cloning as described in panel A. The sequence traces are aligned to the CHO-K1 genomic sequence. The red arrow indicates the genomic target site of sgRNA1_C. The numbers denote the position (bp) in the open reading frame of COSMC. Green, red and orange colors indicate insertions, deletions and substitutions, respectively. C: Targeted deep sequencing analysis of COSMC. The same extracted genomic DNA as described for panel A was used as template for the MiSeq analysis. Between 200,000 and 700,000 sequences were obtained for each sgRNA. The percentages of wt and indel sequences are illustrated in the bar plot and shown in the table.
Homozygous Knockout of FUT8 in CHO Cells Generated by CRISPR
The α1,6-fucosyltransferase FUT8 catalyzes the addition of fucose on IgG1 antibodies produced by CHO cells which can reduce antibody-dependent cell-mediated cytotoxicity (Niwa et al., 2004; Shields et al., 2002; Shinkawa et al., 2003). Disruption of the FUT8 gene in CHO cells is therefore attractive in order to achieve highly active and completely nonfucosylated therapeutic antibodies (Yamane-Ohnuki et al., 2004). To expand our knowledge of applying CRISPR Cas9 in CHO cells, the gene disruption efficiency of four FUT8 sgRNAs was investigated by deep sequencing. Genomic regions covering the target site of sgRNA1_F, sgRNA2_F, sgRNA3_F, and sgRNA4_F were PCR amplified and sequenced. This analysis revealed that all four sgRNAs gave rise to significant Cas9 activity with an indel frequency of 17.6% and 15.1% for sgRNA1_F, 38.7% and 31.2% for sgRNA2_F, 42.5% and 36.0% for sgRNA3_F, and 18.9% and 11.1% for sgRNA4_F in replicate 1 and 2, respectively (Fig. 3A). As previously mentioned, transfection efficiency was estimated to be approximately 60% and 65% for replicate 1 and 2, respectively. Lens culinaris agglutinin (LCA)-based selection was further used to select for FUT8-disrupted CHO cells. LCA binds fucosylated plasma membrane proteins leading to endocytosis and cell death. This enables selection for homozygous FUT8 gene disruptions, since LCA can no longer bind to cells devoid of FUT8 and these cells therefore survive (Malphettes et al., 2010). LCA-treatment was initiated 5 days after transfection and resulted in non-adherent round-shaped morphology of all control cells (Fig. 3B). However, many adherent cells were detected in the pool of cells transfected with Cas9 and the four FUT8 sgRNAs (Fig. 3B, only sgRNA3_F is shown), indicating Cas9-mediated functional knockout of FUT8 in these cells. To analyze the phenotypic change of CRISPR Cas9 mediated disruption of FUT8 on cell surface exposed α-1,6-linked fucose moieties, a lectin stain was performed (Malphettes et al., 2010; Mori et al., 2004; Yamane-Ohnuki et al., 2004). Eight days after initiation of selection, LCA selected and non-LCA selected cells transfected with and without Cas9 and sgRNAs were stained with fluorescein-labeled LCA (F-LCA) (Fig. 3C and Supplementary Fig. S2). Cells transfected with Cas9 + sgRNAs without LCA selection revealed a fraction of F-LCA negative cells, demonstrating the presence of cells with homozygous disruption of the FUT8 gene. For Cas9 + sgRNA3_F, these F-LCA negative cells constituted 29.1% of the entire population (Fig. 3D). Cells transfected with Cas9 + sgRNA3_F, which subsequently had been exposed to LCA treatment revealed that the majority of cells (98.6%) stained LCA negative (Fig. 3D). This clearly demonstrates that the LCA treatment efficiently selects for cells devoid of functional FUT8 as previously observed (Malphettes et al., 2010, Mori et al., 2004; Yamane-Ohnuki et al., 2004). Indeed, this observation was confirmed by deep sequencing, since LCA selection significantly enriched cells with FUT8 disruption to an indel frequency between 98.2% and 99.7% for the four FUT8-targeting sgRNAs (Fig. 3A).
Figure 3.
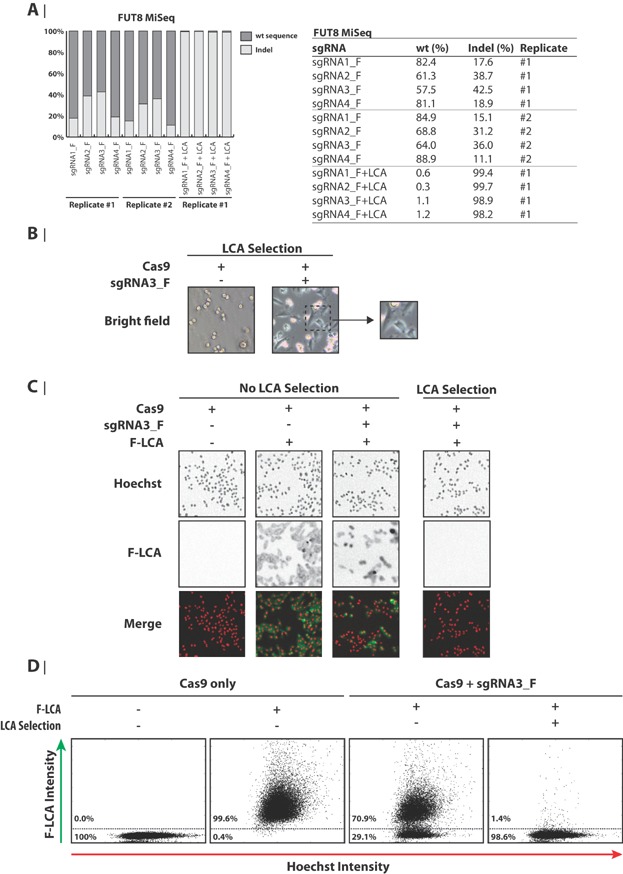
Functional and genomic knockout of FUT8 in CHO-K1. A: Targeted deep sequencing analysis of the FUT8 locus in CHO-K1 cells. Genomic DNA was extracted from CHO-K1 cells transfected with Cas9 and FUT8 sgRNAs harvested on Day 5 (no LCA selection) or Day 12 (7 days with LCA selection). The percentages of wt and indel sequences are illustrated in the bar plot and shown in the table. B: Selection of FUT8 knockout CHO-K1 cells by LCA. As indicated, cells were either transfected with only a Cas9-encoding plasmid or in combination with an sgRNA3_F-encoding plasmid. Five days after transfection (Day 5), selection with LCA was initiated. The day after (Day 6), the shown bright field images were acquired. The magnified view shows cells with normal (adherent-looking) morphology from pool of cells transfected with Cas9 and sgRNA3_F. C: Phenotypic staining of FUT8 knockout CHO-K1 cells by fluorescein-labeled LCA (F-LCA). CHO-K1 cells were treated as described for panel A. On Day 13, cells were treated with Hoechst and with F-LCA where indicated. Fluorescence microscopy images were subsequently acquired. Hoechst and F-LCA signal is depicted as red and green color, respectively, in the merged images and as grayscale in the individual images. D: Quantification of fluorescent-based phenotypic staining of FUT8 knockout CHO-K1 cells. Cells were gated based on signal intensity of Hoechst and F-LCA as shown. The percentages of F-LCA positive (FUT8 WT) and negative (phenotypic knockout of FUT8) cells are shown. LCA: Lens culinaris agglutinin.
The Majority of CRISPR-Generated Indels Is Single Base Pair Insertions
The vast amount of information obtained from deep sequencing led us to investigate further the indel sizes created by the NHEJ repair mechanism resulting from the COSMC and FUT8 sgRNAs. The frequency was calculated as an average for both independent experiments and was based on 11 × 105 reads for COSMC sgRNAs and 8 × 105 reads for FUT8 sgRNAs (Supplementary Figs. S3 and S4 and Table SVI). All targets were weighted equally with each target contributing 12.5%. The data was compiled into a single plot, displaying the frequency of specific indel sizes ranging from 37 bp deletions to 11 bp insertions within the individual targets (Fig. 4A). Surprisingly, mainly single base pair insertions were detected with a frequency of 32.8%. A high frequency of single base pair insertions was also observed in sequences obtained from TOPO cloning (Fig. 2B, only sgRNA1_C, replicate #1 is shown). Two and one base pair deletions were the second and third most frequent indel size with a frequency of 10.3% and 8.7%, respectively. Together, almost half of the identified indels (56.5%) were single or double-base pair indels. Collectively, 85% of the indels observed in this study resulted in frame shift mutations (±1 or ±2 bp) in the reading frame (Fig. 4B), which most likely leads to a loss-of-function of the target protein. This finding further underlines CRISPR Cas9 as a powerful tool to disrupt genes of interest in the CHO genome, and prompted us to develop a target design tool that facilitates identification of Cas9 targets.
Figure 4.
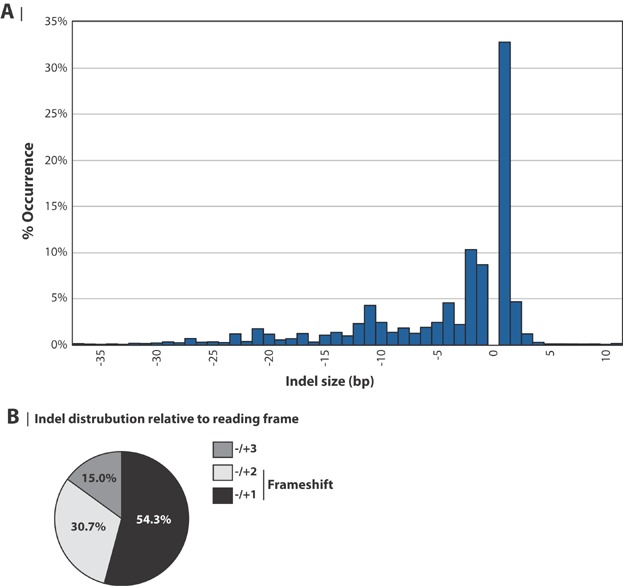
Cas9 activity in CHO results in high frequency of 1 bp indels. A: Frequency distribution of indel sizes. The size distribution of indels is based on sequences obtained from all eight sgRNAs. The indel frequency for each sgRNA is the average for both independent experiments. Normalization was performed so each of the eight sgRNA accounts for 12.5% of the data points. Only indels ranging from −37 bp (37 bp deletion) to +11 bp (11 bp insertion) are shown. B: Frameshifts generated by CRISPR Cas9. The distribution of indels generating ±1, ±2, and ±3 base pair shifts in the reading frame was calculated from data presented in panel A.
Cas9 Target Finding Tool “CRISPy” for CHO-K1
To our knowledge, there are currently three Cas9 target design tools available to the public. The “ZiFiT Targeter” (http://zifit.partners.org/ZiFiT/ChoiceMenu.aspx) identifies Cas9 targets in a given sequence. The “CRISPR Design” (http://crispr.mit.edu/) (Hsu et al., 2013), allows the user to find targets in a given sequence and then checks for off-targets in the genome of either human, mouse, zebrafish, or C. elegans. The “Cas9 guide RNA Design” tool (Ma et al., 2013) is highly similar to “CRISPR Design” with the addition of reporting content of AT (adenine and thymine) and predicting secondary RNA structure. However, neither can currently be applied to find sgRNA target sequences with off-target information in CHO genomes, nor provide pre-configured links to primer design tools.
In order to design the Cas9 system for gene knockouts, a number of tasks must be performed. Once a gene of interest has been selected, one or more targets must be identified in the exons of the gene. These targets should then be evaluated based on their position in the gene and sequence similarity to the rest of the genome. Once suitable targets have been identified, a method for monitoring creation of gene disruptions must be established. This is commonly done using PCR amplification and sequencing of the targeted genomic region. To facilitate this workflow (Fig. 5), we have developed a bioinformatics tool “CRISPy” that is freely accessible at “http://staff.biosustain.dtu.dk/laeb/crispy/.” The web interface interacts with a precompiled database of all possible Cas9 target sites in CHO-K1 genes based on the annotated CHO-K1 genome (Hammond et al., 2012; Xu et al., 2011). Every target sequence has the following format GN19NGG. For every Cas9 target sequence, we have compiled a list of off-targets of the format B13NGG (B is a nucleotide identical to the genomic sequence and N is a random nucleotide) with 0–2 mismatches. This list also shows the genomic site of each off-target for the 0 and 1 bp mismatches. Overall, this has resulted in a database with 1,970,449 targets divided into 27,553 genes.
Figure 5.

Workflow of Cas9 target finding tool: CRISPy. Screenshots of the online CRISPy tool showing the process of (1) finding a target gene, (2) selecting an exon, (3) evaluating the available targets, and (4) showing links to PCR primer designs for analysis of on- and off-target effects resulting from Cas9 + sgRNA activity. The tool is available at http://staff.biosustain.dtu.dk/laeb/crispy/.
While improvements of the CHO-K1 genome annotation are still underway, there are 21,610 coding sequences annotated as of this publication. They contain 231,866 exons. 221,353 of these (95.5%) have at least one potential Cas9 target site. Given CRISPy's 1,970,449 potential Cas9 target sites in exons, this gives an average of nine target sites per exon. If a researcher chooses one of these target sites at random, there is a risk of choosing a target site with high sequence similarity to other parts of the genome; thus producing potential off-target effects. The median number of exact DNA sequence matches elsewhere in the genome for a target site is 6. For 1 bp mismatches the number is 377 and for 2 bp mismatches the number is 4558. Off-targets are here based on the first 13 bp upstream of the PAM sequence. However, 248,777 of 1,970,449 target sites (12.6% unique sites) have zero exact off-target matches. This highlights the importance of using CRISPy to select the most specific CRISPR target site.
The user can search for a gene of interest based on the annotation available for the CHO-K1 genome at the time of database generation; for example, GeneID, gene symbol and name (Fig. 5). After clicking the gene of interest, the user will be presented with a schematic overview of the gene and targets. All exonic targets can either be listed together or on an exon-by-exon basis. Targets are listed with number of off-targets with 0, 1, or 2 bp mismatches as well as a list of genes in which one or more of the perfectly matching off-targets occur. Once one or more suitable targets have been selected, the user can click on a link to get to a target specific page with direct pre-configured links to the NCBI Primer-Blast tool (Ye et al., 2012) on which the user simply has to click the “Get Primers” button. Once the Primer-Blast tool returns results, the user can verify that the PCRs amplify the desired region by copy/pasting the target sequence into the “Find on Sequence” field, which should return at least one sequence hit (if more than one hit, then select the one at the position indicated as your PCR region). It should be noted that the pre-configured link to the Primer-Blast tool is set to generate PCR amplicons of 100–200 bp for compatibility with deep sequencing such as MiSeq. The user can alter the size on the Primer-Blast tool page if necessary. In addition there are links to the genomic sequence at either NCBI or UDEL. On the target specific page the user will also find a list of 0 or 1 bp mismatch off-targets for which there are also links to genome sequence and Primer-Blasts to facilitate easy monitoring of potential off-targets. Since the database is precompiled, there are no time-consuming computational steps.
Discussion
In this study, we demonstrate the successful application of RNA-guided CRISPR Cas9 for generating gene disruptions in CHO-K1 cells. The tested sgRNAs for COSMC created indels with a frequency between 13.6% and 47.3% according to MiSeq analysis in a pool of transfected cells with a transfection efficiency of approximately 60%. In comparison, genetic disruption frequency of 3.8% and 6.3% in CHO cells for BAK and BAX, respectively, has been observed using pre-screened ZFNs (Cost et al., 2010). With an indel frequency between 11.1% and 42.5% created at the target sites, the tested sgRNAs for FUT8 revealed an activity similar to the high efficiencies observed for COSMC. Selection pressure with LCA furthermore facilitated enrichment of cells exhibiting functional disruptions in the FUT8 gene for each of the four sgRNAs. Together, RNA-guided Cas9 activity was able to generate indels with a relatively high frequency for all eight sgRNAs examined, demonstrating that the Cas9 genome-editing methodology is robust and efficient. With these high efficiencies obtained with CRISPR Cas9 system in CHO cells, it will be worthwhile to investigate the capacity of Cas9-based multiplexing to generate multiple gene disruptions in a single round of modifications. Since Cas9-based multiplexing has successfully been performed in other mammalian cells (Cong et al., 2013; Wang et al., 2013), multiplexing using the Cas9 system may as well be a powerful technique in CHO cells.
The mutations created by the eight sgRNAs were predominantly very short indels (56.5% single or double base pair indels) with a preference for single base pair insertions. Analysis of indel sizes obtained with Cas9 in human cells revealed mainly single base pair deletions (Mali et al., 2013). The preference for small single base pair deletions was also observed in another study involving Cas9 in human cells (Wang et al., 2014). Interestingly, the preference for single base pair insertions or deletions observed in our study resulted in a high frequency of indels creating frameshifts (85%) within the open reading frame further supporting Cas9 as a highly attractive endonuclease for generating gene disruptions.
To enable high throughput automated gene disruptions in CHO, the bioinformatics tool “CRISPy” was developed to assist in identification of sgRNA target sites. The sgRNA design tool incorporates additional elements/properties not currently available elsewhere including visualization of sgRNA target sites, detailed off-target information and links to primer design tools. Since off-target indel events have been observed in previous reports on Cas9 in human cells (Fu et al., 2013; Hsu et al., 2013; Wang et al., 2014), prescreening sgRNAs for possible off-target effects represents a useful addition to the target design tool box. The CRISPy tool presented here provides upfront off-target analysis of the designed sgRNAs, enabling selection of sgRNAs with the minimal number of possible off-target sites. Furthermore, CRISPy aids researchers in primer design for targeted analysis of off-target effects. We envision incorporating new knowledge on target sequence-dependent activity of sgRNAs as it becomes available in the future.
In this study, we have been able to enrich the FUT8 knockout population by LCA selection. However, this type of selection is often unavailable and so single cell cloning will be required to obtain cells with the desired gene disruptions. This is commonly achieved through either FACS sorting or limited dilution. These clones must then be analyzed for homozygous populations through screening by fragment analysis and sequencing. However, this process can be time consuming and includes a number of challenges. With the high genome editing efficiency of the Cas9 system, the number of analyzed single cell clones sufficient for obtaining homozygous mutations is expected to be lowered considerably. Thus, the CRISPR Cas9 system holds the potential to significantly decrease the heavy workload involved in generating knockout CHO cell lines.
Our study demonstrates that design and implementation of Cas9-sgRNA-based genome engineering is straightforward and fast. Additionally, the CRISPR Cas9 system is relatively inexpensive as the Cas9 expression vector is reused and only new sgRNA constructs need to be cloned for every target sequence at the cost of a few oligonucleotides. The high efficiency, robustness, ease of use, and low costs make the CRISPR Cas9 system a highly attractive genome-editing tool for both the academic and industrial community. The introduction of the CRISPR Cas9 system in CHO cells combined with the CRISPy design tool will significantly accelerate the pace of genome editing in CHO cells and enhance the rate of CHO cell line improvement for increasing yields and quality of biopharmaceuticals.
Acknowledgments
The authors thank Jae Seong Lee and Bjørn Voldborg for valuable guidance, support, and fruitful discussions. The authors thank Patrice Menard and Sara Petersen Bjørn for technical assistance. In addition, the authors thank Anna Koza for her assistance with the MiSeq experiments. This work was supported by The Novo Nordisk Foundation.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
Supporting Information.
References
- Bassett AR, Tibbit C, Ponting CP, Liu J-L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013;4:220–228. doi: 10.1016/j.celrep.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D. A CRISPR approach to gene targeting. Mol Ther. 2012;20:1658–1660. doi: 10.1038/mt.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong J-W, Xi JJ. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013;23:465–472. doi: 10.1038/cr.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim J-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, Freyvert Y, Vafiadis A, Santiago Y, Miller JC, Rebar E, Collingwood TN, Snowden A, Gregory PD. BAK and BAX deletion using zinc-finger nucleases yields apoptosis-resistant CHO cells. Biotechnol Bioeng. 2010;105:330–340. doi: 10.1002/bit.22541. [DOI] [PubMed] [Google Scholar]
- DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013;41:4336–4343. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galetto R, Duchateau P, Pâques F. Targeted approaches for gene therapy and the emergence of engineered meganucleases. Expert Opin Biol Ther. 2009;9:1289–1303. doi: 10.1517/14712590903213669. [DOI] [PubMed] [Google Scholar]
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison JW, O'Connor-Giles KM. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194:1029–1035. doi: 10.1534/genetics.113.152710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond S, Kaplarevic M, Borth N, Betenbaugh MJ, Lee KH. Chinese hamster genome database. Biotechnol Bioeng. 2012;109:1353–1356. doi: 10.1002/bit.24374. An online resource for the CHO community at http://www.chogenome.org. [DOI] [PubMed] [Google Scholar]
- Hansen BG, Sun XE, Genee HJ, Kaas CS, Nielsen JB, Mortensen UH, Frisvad JC, Hedstrom L. Adaptive evolution of drug targets in producer and non-producer organisms. Biochem J. 2012;441:219–226. doi: 10.1042/BJ20111278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh J-RJ, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayapal KP, Wlaschin KF, Hu W-S, Yap MGS. Recombinant protein therapeutics from CHO cells—20 years and counting. Chem Eng Prog. 2007;103:40–47. [Google Scholar]
- Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013;41:1–12. doi: 10.1093/nar/gkt780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:1–9. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildegaard HF, Baycin-Hizal D, Lewis NE, Betenbaugh MJ. The emerging CHO systems biology era: Harnessing the 'omics revolution for biotechnology. Curr Opin Biotechnol. 2013;24:1–6. doi: 10.1016/j.copbio.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Lewis NE, Liu X, Li Y, Nagarajan H, Yerganian G, O'Brien E, Bordbar A, Roth AM, Rosenbloom J, Bian C, Xie M, Chen W, Li N, Baycin-Hizal D, Latif H, Forster J, Betenbaugh MJ, Famili I, Xu X, Wang J, Palsson BO. Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat Biotechnol. 2013;31:759–765. doi: 10.1038/nbt.2624. [DOI] [PubMed] [Google Scholar]
- Ma M, Ye AY, Zheng W, Kong L. A guide RNA sequence design platform for the CRISPR/Cas9 system for model organism genomes. BioMed Res Int. 2013;2013:270805. doi: 10.1155/2013/270805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malphettes L, Freyvert Y, Chang J, Liu P-Q, Chan E, Miller JC, Zhou Z, Nguyen T, Tsai C, Snowden AW, Collingwood TN, Gregory PD, Cost GJ. Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol Bioeng. 2010;106:774–783. doi: 10.1002/bit.22751. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow K, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Miyoshi E, Noda K, Yamaguchi Y, Inoue S, Ikeda Y, Wang W, Ko JH, Uozumi N, Li W, Taniguchi N. The alpha1-6-fucosyltransferase gene and its biological significance. Biochem Biophys Acta. 1999;1473(1):9–20. doi: 10.1016/s0304-4165(99)00166-x. [DOI] [PubMed] [Google Scholar]
- Mori K, Kuni-Kamochi R, Yamane-Ohnuki N, Wakitani M, Yamano K, Imai H, Kanda Y, Niwa R, Iida S, Uchida K, Shitara K, Satoh M. Engineering Chinese hamster ovary cells to maximize effector function of produced antibodies using FUT8 siRNA. Biotechnol Bioeng. 2004;88:901–908. doi: 10.1002/bit.20326. [DOI] [PubMed] [Google Scholar]
- Niwa R, Shoji-hosaka E, Sakurada M. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 2004;64:2127–2133. doi: 10.1158/0008-5472.can-03-2068. [DOI] [PubMed] [Google Scholar]
- Nour-Eldin HH, Hansen BG, Nørholm MHH, Jensen JK, Halkier BA. Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res. 2006;34:e122. doi: 10.1093/nar/gkl635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørholm M. A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. BMC Biotechnol. 2010;10:21. doi: 10.1186/1472-6750-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi E. The CRISPR craze. Science. 2013;341:833–836. doi: 10.1126/science.341.6148.833. [DOI] [PubMed] [Google Scholar]
- Santiago Y, Chan E, Liu P-Q, Orlando S, Zhang L, Urnov FD, Holmes MC, Guschin D, Waite A, Miller JC, Rebar EJ, Gregory PD, Klug A, Collingwood TN. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci USA. 2008;105:5809–5814. doi: 10.1073/pnas.0800940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedivy JM, Sharp PA. Positive genetic selection for gene disruption in mammalian cells by homologous recombination. Proc Natl Acad Sci USA. 1989;86:227–231. doi: 10.1073/pnas.86.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields RL, Lai J, Keck R, O'Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, Uchida K, Anazawa H, Satoh M, Yamasaki M, Hanai N, Shitara K. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–3473. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager KT, Kong Y, Bennett EP, Mandel U, Wandall H, Levery SB, Clausen H. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods. 2011;8:977–982. doi: 10.1038/nmeth.1731. [DOI] [PubMed] [Google Scholar]
- Waaijers S, Portegijs V, Kerver J, Lemmens BB, Tijsterman G, van den Heuvel S, Boxem M. CRISPR/Cas9-targeted mutagenesis in C. elegans. Genetics. 2013;195:1187–1191. doi: 10.1534/genetics.113.156299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ju T, Ding X, Xia B, Wang W, Xia L, He M. Cosmc is an essential chaperone for correct protein O-glycosylation. Proc Natl Acad Sci USA. 2010;107:9228–9233. doi: 10.1073/pnas.0914004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JR, Williams D, Schachter H. The control of glycoprotein synthesis: N-acetylglucosamine linkage to a mannose residue as a signal for the attachment of l-fucose to the asparagine-linked N-acetylglucosamine residue of glycopeptide from alpha1-acid glycoprotein. Biochem Biophys Res Commun. 1976;72:909–916. doi: 10.1016/s0006-291x(76)80218-5. [DOI] [PubMed] [Google Scholar]
- Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Chen W, Xie M, Wang W, Hammond S, Mikael R, Neff N, Passarelli B, Koh W, Fan HC, Gui Y, Lee KH, Betenbaugh MJ, Quake SR. The genomic sequence of the Chinese hamster ovary (CHO) K1 cell line. Nat Biotechnol. 2011;29:735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, Wakitani M, Niwa R, Sakurada M, Uchida K, Shitara K, Satoh M. Establishment of FUT8 knockout Chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng. 2004;87:614–622. doi: 10.1002/bit.20151. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.