

Abstract
Monitoring and control of primary cell cultures is challenging as they are heterogenous and dynamically complex systems. Feedback signaling proteins produced from off-target cell populations can accumulate, inhibiting the production of the desired cell populations. Although culture strategies have been developed to reduce feedback inhibition, they are typically optimized for a narrow range of process parameters and do not allow for a dynamically regulated response. Here we describe the development of a microbead-based process control system for the monitoring and control of endogenously produced signaling factors. This system uses quantum dot barcoded microbeads to assay endogenously produced signaling proteins in the culture media, allowing for the dynamic manipulation of protein concentrations. This monitoring system was incorporated into a fed-batch bioreactor to regulate the accumulation of TGF-β1 in an umbilical cord blood cell expansion system. By maintaining the concentration of TGF-β1 below an upper threshold throughout the culture, we demonstrate enhanced ex vivo expansion of hematopoietic progenitor cells at higher input cell densities and over longer culture periods. This study demonstrates the potential of a fully automated and integrated real-time control strategy in stem cell culture systems, and provides a powerful strategy to achieve highly regulated and intensified in vitro cell manufacturing systems. Biotechnol. Bioeng. 2014;111: 1258–1264. © 2013 The Authors Biotechnology and Bioengineering Published by Wiley Periodicals, Inc.
Keywords: process control, hematopoietic stem cells, bioreactor, soluble signaling factors, protein detection
Primary stem cell cultures are complex and heterogeneous systems. In the in vitro culture of human hematopoietic progenitors, soluble factor-mediated feedback inhibition limits the ex vivo expansion of hematopoietic stem and progenitor cells (HSPCs) (Kirouac and Zandstra, 2006). Strategies to minimize the impact of emerging mature cell populations have provided some success towards controlling endogenous factor accumulation (Csaszar et al., 2012; Madlambayan et al., 2005; Sandstrom et al., 1995; Yang et al., 2010); however, these approaches are typically optimized for a specific set of culture conditions and do not allow for the flexibility to easily accommodate changing culture supplements or input cell compositions. One approach to overcome this limitation would be a cell culture regulation system that allows for in-line or at-line monitoring of soluble signaling factors in the culture media, thereby providing dynamic response to system variations. A major challenge in implementing control strategies for endogenously secreted proteins is their non-linear and dynamic secretion profiles (Csaszar et al., 2012) and the low concentration at which they are bioactive. As a result, dynamic regulation of signaling proteins has not yet been integrated into stem cell culture systems.
Herein, we describe the development and demonstration of a real-time control system for monitoring and controlling signaling proteins in a human HSPC expansion bioprocess using a microbead detection assay. Microbead-based biosensing platforms provide a compelling option for integrating antibody based protein detection into culture platforms as they have faster reaction kinetics and higher throughput capacity of biomolecule conjugation compared to planar detection techniques (Wilson et al., 2006). We developed a microbead sandwich antibody detection system for the rapid measurement of TGF-β1, a critical inhibitory factor of human hematopoiesis (Fortunel et al., 2000). By integrating this detection assay into a HSPC culture system (Csaszar et al., 2012), we demonstrate the ability of the strategy to expand hematopoietic progenitors at higher cell seeding densities and for longer culture times than we can achieve with alternative culture methods.
As a first step, it was investigated whether the human hematopoietic culture system would likely benefit from the incorporation of a dynamic process control system. The robust ex vivo expansion of umbilical cord blood (UCB) derived HSPCs is highly desired for hematopoietic stem cell transplantation, however, cell variability can limit the reproducibility of this expansion. To assess the variability in this system, we measured the purity of CD34+ progenitors following a lineage depletion process (commonly performed prior to seeding cells for culture) and found the frequency to be highly variable from sample to sample. This input cell variability manifests itself into differences in the kinetics and types of mature cell production, ultimately impacting the total cell expansion achieved in a non-linear manner (Fig. 1A). In a previous study, we demonstrated that experimental differences in hematopoietic cell culture outputs could be recapitulated by varying the secretion rates of endogenous regulators in silico (Kirouac et al., 2009). A large number of soluble factors are rapidly produced in HSPC culture (Fig. 1B). Globally regulating these factors with a linear fed-batch dilution scheme (which was computationally predicted to outperform a perfusion approach) allowed for the reduction of factor accumulation and the enhancement of HSPC expansion (Csaszar et al., 2012), but this approach does not account for sample-to-sample variation or changing culture parameters. Given the non-linear trajectory of both factor secretion and of cell growth, we explored alternative feeding schemes and found an exponential fed-batch feeding scheme showed a trend of outperforming other strategies when normalized to total cumulative media use over 8 days (Fig. 1C). The beneficial effects of the exponential feeding regime were particularly evident on the primitive progenitor population, as measured by long-term culture-initiating cells (LTC-ICs) (Liu et al., 2013).
Figure 1.
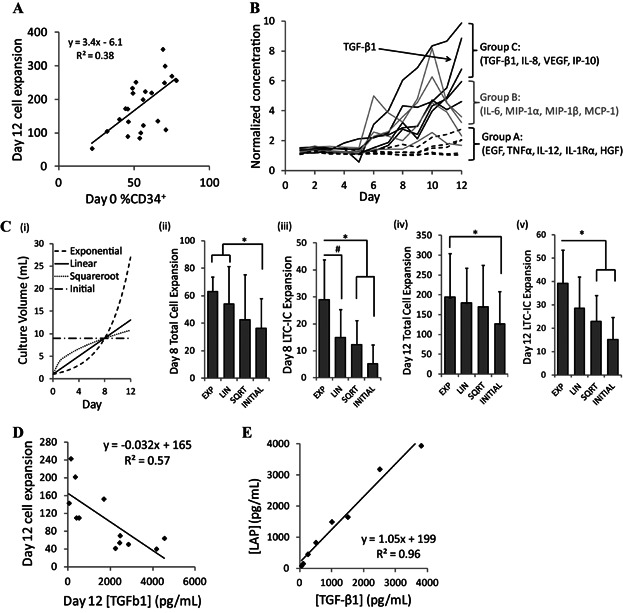
Variability in soluble factors levels and cell expansion outputs in UCB cell cultures demonstrate the need for a process control strategy. A: Variability in Day 0 cell composition (measured by % CD34+) manifests into variability in Day 12 cell expansion in a non-linear manner. B: Endogenously produced soluble signaling factors can be categorized based on the trajectory of their accumulation. Soluble factor concentrations were normalized by comparing each measured time-point to the Day 1 measured concentration (pg/mL). C: Different fed-batch feeding regimes were assessed, normalized to cumulative media usage on Day 8: (i) culture volume trajectories produced by each fed-batch feeding regime (an exponential, linear, or square root trajectory, or a initial dilution using a lower cell seeding density); (ii,iv) Total cell expansion following 8 and 12 days of culture with the different feeding strategies; (iii,v) Primitive progenitor cell expansion measured by a long term culture-initiating cell (LTC-IC) assay following 8 and 12 days of culture with the different feeding strategies. n = 5, *P < 0.05, #P < 0.1. D: Variation in TGF-β1 concentrations negatively correlate with Day 12 total cell expansion. E: TGF-β1 concentration has a strong correlation with LAP concentration, indicating that LAP can be used as a surrogate for TGF-β1 in a detection assay.
Given the apparent benefits of the exponential feeding regimen, we predicted that adjusting the media dilution scheme based on the actual measurements of soluble factors as opposed to a pre-determined algorithm would further enhance the regulation that could be achieved by providing a sample-specific dilution scheme. It was further predicted that monitoring the concentration of a sentinel endogenously produced factor would provide a good initial indication of the culture dynamics of the global secreted factor milieu and TGF-β1 was selected as an initial candidate for the development of the system. TGF-β1 is a potent inhibitor of HSPC expansion (Csaszar et al., 2012; Fortunel et al., 2000) which rapidly accumulates at high levels (>1,000 pg/mL) in this culture system in a manner that mimics the behavior of a number of secreted factors (Fig. 1B). Importantly, measured levels of TGF-β1 are negatively correlated to total cell expansion (Fig. 1D), suggesting that maintaining a low concentration of TGF-β1 should be beneficial to cell expansion. Because TGF-β1 accumulates in culture in a latent form and is difficult to measure with antibody detection without an activation step (Lyons et al., 1988), we developed the assay to monitor the latency-associated peptide (LAP) as a TGF-β1 surrogate. LAP in a propeptide that binds strongly to TGF-β1 to form the Small Latent Complex (Rifkin, 2005; Young and Murphy-Ullrich, 2004), and its concentration correlates strongly to that of TGF-β1 protein (Fig. 1E).
In order for the measurement of LAP to be used to control cell culture, it is critical for the detection assay reaction to be rapid, sensitive, and have high precision and accuracy. The reaction procedure should be simple and easy to automate for device production. We have previously developed a quantum dot (QD)-barcoded microbead platform for biosensing (Fournier-Bidoz et al., 2008; Klostranec et al., 2007). This platform is optimized for detection of genetic targets using 4-micron size beads (Gao et al., 2011; Giri et al., 2011). Microbeads doped with QDs can detect many molecules simultaneously in a single reaction chamber in less than 1 h. Here we show that a QD-magnetic nanoparticle microbead system can meet the requirements for LAP detection. The detection assay is based on a sandwich reaction; QD microbeads were conjugated with capture antibodies of LAP via EDC chemistry and the secondary probe was labeled with APC as the reporter signal readout (Fig. 2A). The optical properties of QDs were not used for detection in this study but were included to assess whether their presence would influence the detection of the target.
Figure 2.
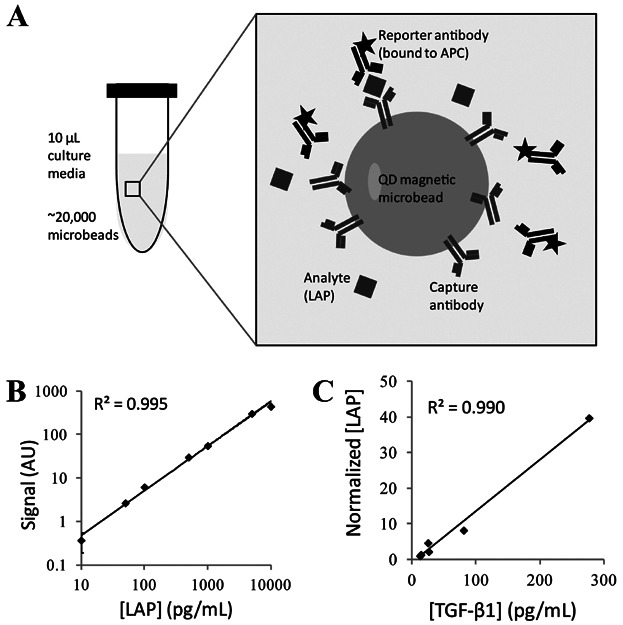
A QD microbead assay was developed and validated for the detection of LAP. A: Schematic of the QD microbead detection assay. Microbeads are conjugated with a capture antibody that binds to the corresponding analyte and the reporter antibody. By labeling the reporter antibody with a fluorescent marker, the presence of the analyte can be quantified by flow cytometry. B: Dose-response curve of LAP by QD microbead sandwich assay. LAP concentrations ranged from 10 to 10,000 pg/mL. Assay reactions were complete in 1 h and measured by flow cytometry. C: Correlation between measurements of LAP using QD microbead assay and TGF-β1 by ELISA. Culture media samples from different batches and culture periods were tested by both QD microbeads and TGF-β1 ELISA.
We evaluated the dose-response of the microbead assay using a serial concentration of LAP. Samples with known LAP concentrations were mixed with the conjugated microbeads and the resulting fluorescent signal was analyzed by flow cytometry (Fig. 2B). The results indicate that the detection limit of the assay is approximately 10 pg/mL, and the linear range of detection spans more than 3 orders of magnitude, providing a wide dynamic range for detecting protein concentrations. We then validated the accuracy of the assay by comparing the LAP measurements of conditioned media samples with the concentration of TGF-β1 measured by ELISA for the corresponding samples (Fig. 2C). The microbead detection assay correlated strongly with the ELISA measurements, suggesting that the assay can measure the concentration of LAP with high accuracy.
To demonstrate the ability of the dynamic response control system using the microbead assay, we incorporated the measurement of LAP concentration into our fed-batch expansion system (Csaszar et al., 2012). The endogenously produced concentration of LAP was measured by sampling the culture media every 12–24 h and assessing the samples with the microbead detection assay at each time-point. The sample measurement was compared to an upper LAP concentration threshold of 100 pg/mL below which it was desired to maintain the LAP levels. The difference between the measurement and the threshold was then fed into an algorithm that converted the concentration into a media dilution scheme. For this study, if the LAP concentration was measured to be >100 pg/mL, the cell culture was diluted twofold, while if the concentration was <100 pg/mL, no dilution was performed. A schematic of the “real-time control” (RTC) system is shown in Figure 3A.
Figure 3.
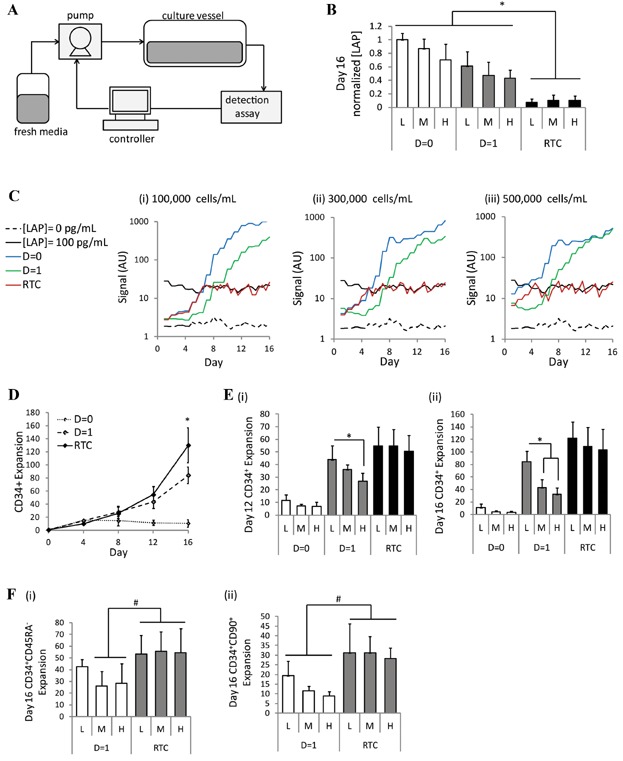
Proof-of-principle of a real-time control strategy for secreted factor concentration regulation demonstrates enhanced expansion of HSPCs. A: Schematic of process control system. A cell culture media sample is taken from the culture vessel to the microbead detection assay, where the concentration of one or more soluble factors is measured. The concentration value is fed to the controller which uses an algorithm to convert the concentration into a dilution rate. The dilution rate is fed to the pump which controls the input of fresh media into the culture vessel. B,C: LAP concentration measurements were taken every 12–24 h during a 16-day UCB culture. The “real-time control” (RTC) scheme using the detection assay was compared to a culture with no media dilution (D = 0) or a linear dilution rate of one unit volume per day (D = 1). The measurements were performed in culture with a cell seeding density of: (i) 100,000 cells/mL (L); (ii) 300,000 cells/mL (M); (iii) 500,000 cells/mL (H). [LAP] = 0 is the fresh media control and [LAP] = 100 pg/ml is recombinant LAP, diluted to 100 pg/mL in fresh media, to act as the LAP concentration threshold for the detection assay. [B] Day 16 LAP concentration, normalized to the D = 0 (L) condition (n = 3). [C] Representative plots of LAP concentrations measured every 12 h over a 16-day culture. D: CD34+ expansion was compared among the different culture strategies for cells seeded at 100,000 cells/mL. Beyond Day 12, the RTC strategy outperforms that constant D = 1 feeding scheme (n = 3). E: CD34+ expansion on: (i) Day 12 and; (ii) Day 16 for the different culture strategies at the low (L), medium (M), and high (H) cell seeding densities. F: Day 16 expansion of: (i) CD34+CD45RA− cells and; (ii) CD34+CD90+ cells with the D = 1 feeding scheme and the RTC approach for each of the low (L), medium (M), and high (H) cell seeding densities. *P < 0.05, #P < 0.1.
To test the capability of the system under different conditions, cultures were initiated at three different cell seeding densities (low (L) = 100,000 cells/mL, medium (M) = 300,000 cells/mL, high (H) = 500,000 cells/mL). Previous studies have shown that high cell densities are detrimental to primitive cell expansion when traditional feeding schemes are used due to the presence of high levels of inhibitory soluble factors (Kirouac et al., 2009). In this study, the RTC scheme maintained the LAP concentration at 100 ± 40 pg/mL for the duration of culture, regardless of the initial cell density. In contrast, the pre-set constant D = 0 (no dilution) and D = 1 (linear dilution at a constant rate of one unit volume per 24 h) feeding schemes resulted in high LAP levels (1,900–5,000 pg/mL) accumulating by Day 16 (Fig. 3B,C).
When we analyzed cell outputs, two major advantages of the control strategy were observed. We first assessed the output of cells seeded at 100,000 cells/mL. After 12 days of culture, the linear D = 1 feeding scheme led to similar CD34+ progenitor cell expansions as the RTC strategy. However, beyond Day 12, we saw enhanced expansion of the CD34+ cells with the RTC strategy, suggesting that the D = 1 scheme was not optimal past Day 12 and our progenitor expansions could be improved using the dynamic control strategy for longer term cultures (Fig. 3D). We next looked at the impact of varying cell seeding densities. We observed that with the D = 1 feeding scheme, the medium and high cell seeding densities led to lower CD34+ expansion as compared to cells seeded at the low cell density. This was evident at Day 12 and even more pronounced by Day 16. Conversely, the RTC strategy maintained consistent levels of CD34+ expansion, regardless of seeding density, at both Day 12 and Day 16 (Fig. 3E). Thus, the dynamic control strategy provides the flexibility of responding to variable input culture conditions and maintaining consistent outputs. Importantly, the ability of the RTC scheme to expand very primitive cells was also demonstrated by assessing both the CD34+CD45RA− and CD34+CD90+ populations, both of which have been shown to be very highly enriched for hematopoietic stem cells with in vivo repopulating potential (Doulatov et al., 2012), thus serving as good surrogate markers for functional HSCs. In these primitive cell populations, we again observed the ability of the RTC strategy to provide enhanced expansion beyond Day 12 at all three seeding densities, as compared to the D = 1 feeding scheme (Fig. 3F). Thus, this approach allows for highly reproducible expansion of very primitive hematopoietic cells.
This study demonstrates that using a process control scheme to regulate the feeding rate based on the measured concentration of LAP allows for enhanced regulation of HSPC culture under variable input conditions. By integrating this detection assay into an UCB culture system, we demonstrate the proof of principle of RTC of endogenously produced soluble signaling factors in a clinically relevant stem cell system. One limitation observed was that the basic control algorithm used in these studies for the RTC approach resulted in greater media volumes needed to achieve the observed progenitor cell expansion (Fig. 4A). As such, cell densities were significantly lower with the RTC approach than for the corresponding D = 1 cultures (Fig. 4B). However, it is promising that higher cell seeding densities allowed for greater relative enhancements in CD34+ cells/mL with the RTC approach (Fig. 4C). Development of more sophisticated control algorithms and adjustments to the LAP concentration threshold should allow for further process parameter optimization. For instance, the use of a proportional-integral-derivative (PID) controller would likely further minimize fluctuations of LAP levels and provide for tighter control with less media usage. The detection system also has the capability of being multiplexed to monitor either multiple secreted signaling factors (particularly ones which may have different accumulation dynamics than TGF-β1), or other endogenous or exogenous factors that have fluctuating levels in culture. The applications of QD microbeads in multiplex biosensing have been previously demonstrated (Gao et al., 2011; Giri et al., 2011), and in future studies, the regulation algorithm may involve the monitoring of several cytokines to better capture the complexity of intercellular signaling networks.
Figure 4.
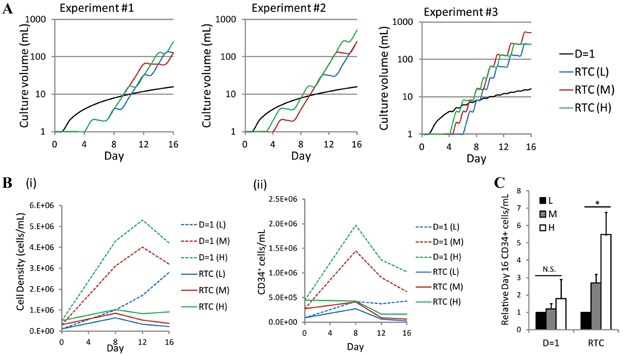
Media requirements and resulting cell densities for the different feeding schemes. A: Culture volume over time for three independent experiments and the low (L), medium (M), and high (H) cell seeding densities. B: (i) Total cell density and (ii) CD34+ cell density for cells cultured with the D = 1 feeding scheme and the RTC approach. C: Although absolute cell densities were lower for the RTC approach, higher cell seeding densities allowed for greater relative enhancements in CD34+ cells/mL with the RTC approach. *P < 0.05, N.S., not significant (P > 0.1).
The studies described here demonstrate the need for process control systems to be implemented in the control of endogenously produced signaling factors in dynamic and heterogeneous cell systems. This microbead technology will enable rapid and non-disruptive measurement of soluble factors and can be integrated into automated bioreactor systems for frequent in-line or at-line measurements. Overcoming variability in culture output is an important need within the cell manufacturing field and the routine use of in-line biosensors for signaling factor detection in stem cell cultures will be an important step for the robust and high-yield production of cell products.
Materials and Methods
Synthesis of QD Magnetic Microbeads and Conjugation of anti-LAP Antibody
QD magnetic microbeads were synthesized by the concentration-controlled flow focusing (CCFF) technique modified from a previous report (Fournier-Bidoz et al., 2008). Briefly, QDs (Cytodiagnostics, Inc., Burlington, ON, Canada) and magnetic particles were mixed with poly(styrene-co-maleic anhydride) polymer (4%, w/v, Sigma, St. Louis, MO) in chloroform. The solution was filtered and then injected into a custom-made nozzle system (Ingeniatrics, Camas, Spain). After synthesis, the microbeads were hardened by overnight stirring. For conjugation, 10 million microbeads were incubated with EDC (1 mg/mL in MES buffer, 50 mM, pH 6.0) for 15 min. Forty microliter of anti-LAP capture antibody (0.5 mg/mL in PBS buffer, R&D Systems, Minneapolis, MN) was added to the beads and incubated for 4 h at room temperature with constant rotation.
Cytokine Sandwich Assay
Each assay reaction contained 1 µL of capture antibody-conjugated beads (approximately 20,000 beads), 0.5 µL of biotin-labeled reporter antibody (50 µg/mL), and 10 µL of culture media sample. The reaction was carried out at 37° for 40 min. Hundred microliter of APC-labeled streptavidin solution was added and incubated for another 20 min. The QD magnetic microbeads were then pulled together by a magnet to wash and analyzed by flow cytometry.
Umbilical Cord Blood Cell Processing and In Vitro Culture
UCB samples were collected from consenting donors according to ethically-approved procedures at Mt Sinai hospital (Toronto, ON, Canada). Lineage negative (Lin−) progenitor cells were isolated from the mononuclear cell fraction as described (Kirouac et al., 2009). Lin- cells were seeded in 1 mL at an initial density of 1 × 105 cells/mL (unless otherwise indicated) in IMDM serum free media with 20% BIT serum substitute (Stemcell Technologies, Vancouver, BC, Canada), 1% Glutamax (Life Technologies, Carlsbad, CA), 100 ng/mL SCF, 100 ng/mL Flt3L, 50 ng/mL TPO (all R&D Systems), and 1 µg/mL low-density lipoproteins. The syringe loaded pumping system was assembled and connected to the cell culture bag (Csaszar et al., 2009). For a dilution rate of D = 0, no media was delivered. For a dilution rate of D = 1, one unit volume of media was delivered over each 24 h period, with a pulsed delivery every 30 min.
Cell and Secreted Factor Assays
Surface marker staining was performed with conjugated human antibodies: CD34, CD45RA, and CD90 (BD Biosciences, San Jose, CA). All samples were analyzed on a FACSCanto or FACS LSR Fortessa flow cytometer (BD Biosciences). Long term culture-initiating cell (LTC-IC) assays were performed as previously described (Kirouac et al., 2009). Conditioned media samples were analyzed using the TGF-β1 Quantikine ELISA Kits (R&D Systems, Minneapolis, MN) or LAP ELISA Ready-SET-Go! kit (eBiosciences, San Diego CA), according to the manufacturer's directions. Multiplexed secreted factor measurements were made using the Human Cytokine 30-Plex panel (Invitrogen, Burlington, ON, Canada), designed for the Luminex microsphere detection platform (Luminex Co. Austin, TX).
References
- Csaszar E, Gavigan G, Ungrin M, Thérien C, Dubé P, Féthière J, Sauvageau G, Roy DC, Zandstra PW. An automated system for delivery of an unstable transcription factor to hematopoietic stem cell cultures. Biotechnol Bioeng. 2009;103:402–412. doi: 10.1002/bit.22297. [DOI] [PubMed] [Google Scholar]
- Csaszar E, Kirouac DC, Yu M, Wang W, Qiao W, Cooke MP, Boitano AE, Ito C, Zandstra PW. Rapid expansion of human hematopoietic stem cells by automated control of inhibitory feedback signaling. Cell Stem cell. 2012;10:218–229. doi: 10.1016/j.stem.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: A human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Fortunel NO, Hatzfeld A, Hatzfeld JA. Transforming growth factor-beta: Pleiotropic role in the regulation of hematopoiesis. Blood. 2000;96:2022–2036. [PubMed] [Google Scholar]
- Fournier-Bidoz S, Jennings TL, Klostranec JM, Fung W, Rhee A, Li D, Chan WCW. Facile and rapid one-step mass preparation of quantum-dot barcodes. Angewandte Chemie. 2008;47:5577–5581. doi: 10.1002/anie.200800409. [DOI] [PubMed] [Google Scholar]
- Gao Y, Stanford WL, Chan WCW. Quantum-dot-encoded microbeads for multiplexed genetic detection of non-amplified DNA samples. Small. 2011;7:137–146. doi: 10.1002/smll.201000909. [DOI] [PubMed] [Google Scholar]
- Giri S, Sykes EA, Jennings TL, Chan WCW. Rapid screening of genetic biomarkers of infectious agents using quantum dot barcodes. ACS Nano. 2011;5:1580–1587. doi: 10.1021/nn102873w. [DOI] [PubMed] [Google Scholar]
- Kirouac DC, Zandstra PW. Understanding cellular networks to improve hematopoietic stem cell expansion cultures. Curr Opin Biotechnol. 2006;17:538–547. doi: 10.1016/j.copbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Kirouac DC, Madlambayan GJ, Yu M, Sykes EA, Ito C, Zandstra PW. Cell-cell interaction networks regulate blood stem and progenitor cell fate. Mol Syst Biol. 2009;5:293. doi: 10.1038/msb.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klostranec JM, Xiang Q, Farcas Ga, Lee Ja, Rhee A, Lafferty EI, Perrault SD, Kain KC, Chan WCW. Convergence of quantum dot barcodes with microfluidics and signal processing for multiplexed high-throughput infectious disease diagnostics. Nano Lett. 2007;7:2812–2818. doi: 10.1021/nl071415m. [DOI] [PubMed] [Google Scholar]
- Liu M, Miller CL, Eaves CJ. Human long-term culture initiating cell assay. In: Helgason CD, Miller CL, editors. Basic cell culture protocols. Vol. 946. Totowa, NJ: Humana Press; 2013. pp. 241–256. [DOI] [PubMed] [Google Scholar]
- Lyons M, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol. 1988;106:1659–1665. doi: 10.1083/jcb.106.5.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlambayan GJ, Rogers I, Kirouac DC, Yamanaka N, Mazurier F, Doedens M, Casper RF, Dick JE, Zandstra PW. Dynamic changes in cellular and microenvironmental composition can be controlled to elicit in vitro human hematopoietic stem cell expansion. Exp Hematol. 2005;33:1229–1239. doi: 10.1016/j.exphem.2005.05.018. [DOI] [PubMed] [Google Scholar]
- Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: Orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- Sandstrom CE, Bender JG, Papoutsakis ET, Miller WM. Effects of CD34+ cell selection and perfusion on ex vivo expansion of peripheral blood mononuclear cells. Blood. 1995;86:958–970. [PubMed] [Google Scholar]
- Wilson R, Cossins AR, Spiller DG. Encoded microcarriers for high-throughput multiplexed detection. Angewandte Chemie. 2006;45:6104–6117. doi: 10.1002/anie.200600288. [DOI] [PubMed] [Google Scholar]
- Yang H, Robinson SN, Lu J, Decker WK, Xing D, Steiner D, Parmar S, Shah N, Champlin RE, Munsell M, Leen A, Bollard C, Simmons PJ, Shpall EJ. CD3(+) and/or CD14(+) depletion from cord blood mononuclear cells before ex vivo expansion culture improves total nucleated cell and CD34(+) cell yields. Bone Marrow Transplant. 2010;45:1000–1007. doi: 10.1038/bmt.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GD, Murphy-Ullrich JE. Molecular interactions that confer latency to transforming growth factor-beta. J Biol Chem. 2004;279:38032–38039. doi: 10.1074/jbc.M405658200. [DOI] [PubMed] [Google Scholar]