

Abstract
The heart is highly energy dependent with most of its energy provided by mitochondrial oxidative phosphorylation. Mitochondria also play a role in many other essential cellular processes including metabolite synthesis and calcium storage. Therefore, maintaining a functional population of mitochondria is critical for cardiac function. Efficient degradation and replacement of dysfunctional mitochondria ensures cell survival, particularly in terminally differentiated cells such as cardiac myocytes. Mitochondria are eliminated via mitochondrial autophagy or mitophagy. In the heart, mitophagy is an essential housekeeping process and required for cardiac homeostasis. Reduced autophagy and accumulation of impaired mitochondria have been linked to progression of heart failure and aging. In this review, we discuss the pathways that regulate mitophagy in cells and highlight the cardioprotective role of mitophagy in response to stress and aging. We also discuss the therapeutic potential of targeting mitophagy and directions for future investigation.
Keywords: autophagy, mitophagy, mitochondria, parkin, BNIP3, FUNDC1
the heart is highly energy dependent, and the majority of its energy is provided by mitochondrial oxidative phosphorylation. Mitochondria also play a role in many other cellular processes including metabolite synthesis and calcium storage (41). Hence, maintaining a functional population of mitochondria is critical for cardiac function. Damaged mitochondria produce less ATP, generate greater amounts of reactive oxygen species (ROS), and can activate apoptosis and/or necrosis. Thus efficient removal of dysfunctional mitochondria and biogenesis of functional mitochondria are critical for the maintenance of cellular homeostasis. This is of particular importance in cells such as cardiac myocytes, which are terminally differentiated. The mechanism that the cell uses to eliminate damaged mitochondria is known as mitochondrial autophagy or mitophagy, and involves selective sequestering of the organelle inside an autophagosome and subsequent fusion with a lysosome where degradation occurs (98). Autophagy was initially considered to be a nonselective bulk degradation pathway, especially during stress such as starvation. However, given the critical role of mitochondria in myocytes, it seems unlikely that degradation of mitochondria is a random process, especially during energy deficient conditions such as starvation. This could lead to the degradation of too many mitochondria and compromise cellular survival. A recent study examined changes in the cellular proteome during starvation and found that cytosolic proteins, multiprotein complexes, and organelles show distinct patterns of degradation (46). This study reported that cytosolic proteins are degraded early during starvation, whereas mitochondria are degraded at a much later time point. This suggests that the degradation of proteins and organelles is a selective process during starvation.
Mitophagy is an essential housekeeping process that is required to maintain cardiac homeostasis. Studies suggest that mitophagy is important in eliminating impaired mitochondria both under baseline conditions and in response to stress (4, 9, 18, 32, 50, 82). Studies have linked impaired mitochondrial function and reduced autophagy to progression of heart failure and age-related cardiac pathologies (35, 61). Recent evidence also suggests a specific role for mediators of mitophagy in eliciting cardioprotective benefits (18, 32, 50, 82). However, the exact nature of the relationship between mitophagy and cardioprotection is still under investigation. In this review, we discuss our current understanding of the pathways that regulate mitophagy in cells and the cardioprotective role of mitophagy in response to stress and aging. We also highlight the therapeutic potential of targeting mitophagy, as well as future directions for investigation.
Activation of Autophagy
When mitochondria become damaged or functionally impaired, there is an increase in the number of autophagosomes in the cell. This induction of autophagy occurs in discrete steps, and each step is regulated by a number of autophagy-related (Atg) proteins (Fig. 1) (44). A class III PI3K complex, composed of BECLIN 1(Atg6)/VPS34/VPS15, is responsible for nucleation of the isolation membrane (also known as the phagophore). The phagophore then elongates and two ubiquitin-like conjugation systems, ATG12-ATG5 and ATG8/light chain 3 (LC3), contribute to this step. Once this is complete, the mature LC3-II remains associated with the autophagosome membrane, where it interacts with specific adapter proteins or receptors that mark the mitochondria for degradation (52). The marked mitochondrion is then fully engulfed by the autophagosome. The final step involves autophagosome-lysosome fusion, during which acid hydrolase enzymes degrade the cellular content (44).
Fig. 1.
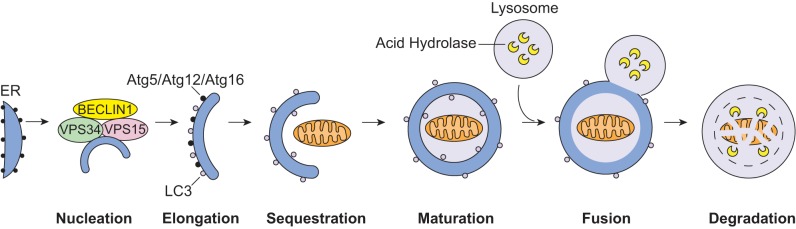
Overview of mitochondrial autophagy. Mitochondrial autophagy begins with nucleation step by the BECLIN 1/VPS34/VPS15 complex, which initiates formation of the autophagosome. Next, ATG5/ATG12/ATG16 and light chain 3 (LC3) are involved in elongating the membrane. The autophagosome then fuses around a mitochondrion, sequestering it inside the mature double membrane vesicle. Finally, the autophagosome fuses with a lysosome and the mitochondrion is degraded by lysosomal hydrolases. ER, endoplasmic reticulum.
Targeting of Mitochondria to Autophagosomes
To ensure that healthy mitochondria are not sequestered and degraded by the autophagy-lysosomal pathway, dysfunctional mitochondria are selectively labeled or selected for degradation. To date, two different mechanisms of selective mitophagy have been described: phosphatase and tensin homolog induced putative kinase 1 (PINK1)/Parkin- and mitochondrial receptor-mediated mitophagy.
PINK1/Parkin-Mediated Mitophagy
The PINK1/Parkin pathway is involved in marking dysfunctional mitochondria for clearance by autophagy (Fig. 2A) (67). The serine/threonine kinase PINK1 is normally imported into healthy mitochondria by the translocase of the outer membrane (TOM) complex, where it is actively degraded by mitochondrial processing peptidase and presenilin-associated rhomboid-like protease (37). However, when a mitochondrion loses its membrane potential, import of PINK1 is abrogated. This leads to accumulation of PINK1 on the outer mitochondrial membrane and recruitment of the E3 ubiquitin ligase Parkin (37). The recruitment and activation of Parkin by PINK1 involve several steps. First, PINK1 phosphorylates mitofusin 2 (MFN2), which then acts as a mitochondrial receptor for Parkin (9). PINK1 must also phosphorylate ubiquitin to fully activate the E3 ubiquitin ligase activity of Parkin (40, 45). Activated Parkin is then responsible for ubiquitinating a number of proteins on the outer mitochondrial membrane. Parkin can mediate nonclassical K63-linked ubiquitination, which targets proteins for degradation by the autophagic-lysosomal pathway (21). The ubiquitin on these substrates serve as a signal for degradation. Adaptor proteins such as p62/SQSTM1 bind to the ubiquitinated proteins via its ubiquitin associated domain and to LC3 on the autophagosome (38). Parkin can also mediate the classical, proteasomal degradation-associated, K48-linked ubiquitination of mitochondrial proteins (52).
Fig. 2.

Mitophagy pathways. A: PINK1/Parkin-mediated mitophagy is initiated upon accumulation of PINK1 on the outer membrane of depolarized mitochondria. PINK1 then phosphorylates mitofusin 2 (MFN2), which leads to recruitment of Parkin. PINK1 also phosphorylates ubiquitin, and activated Parkin ubiquitinates its substrates. The p62 adaptor protein binds to ubiquitinated proteins and LC3 on the autophagosome. B: BNIP3 and NIX act as mitochondrial receptors and directly bind to LC3 to induce mitophagy. C: dephosphorylation of FUNDC1 by PGAM5 allows FUNDC1 to directly interact with LC3 to induce mitophagy.
Mitochondrial proteins that are ubiquitinated by Parkin include MFN1/2, MIRO, Hexokinase I, and voltage-dependent anion channel (VDAC)1 (23, 70, 72, 88, 95). MFN1/2 and MIRO are shared PINK1 and Parkin substrates. They are phosphorylated by PINK1 before their ubiquitination by Parkin (88, 95). Interestingly, these substrates are degraded by the proteasome, which assists with the mitophagy process. Parkin-mediated degradation of the fusion proteins MFN1/2 maintains mitochondria in a fragmented state, which facilitates mitophagy (88). Moreover, MIRO is a component of the complex that anchors kinesin to the mitochondria and Parkin-mediated degradation of MIRO leads to release of the damaged mitochondria from the tubulin network (95). The importance of MIRO in regulating mitophagy in myocytes still needs to be investigated. These studies suggest that UPS and autophagy pathways coordinate to clear dysfunctional mitochondria.
The importance of specific Parkin substrates in mitophagy is still unclear and controversial. For instance, silencing of VDAC1 with siRNA in HeLa cells reduces Parkin mitochondrial translocation and clearance (23). Similarly, Sun et al. (85) showed that all three VDAC proteins (1, 2, and 3) can recruit Parkin to damaged mitochondria and that their loss results in impaired mitophagy. However, a contrasting study found that VDAC1 is dispensable for mitophagy, and fibroblasts lacking both VDAC1 and VDAC3 eliminate mitochondria as efficiently as wild-type fibroblasts (66). This suggests that there is a redundancy between the different substrates to ensure degradation. Although only a few mitochondrial substrates have been identified to date, it is very likely that other still unidentified mitochondrial substrates of Parkin exist. Future efforts should focus on identifying novel Parkin substrates on mitochondria and elucidate their roles in mitophagy.
Cells have evolved mechanisms that negatively regulate Parkin-mediated mitophagy. For instance, the mitochondrial deubiquitinase USP30 opposes Parkin-mediated mitophagy by removing ubiquitin from Parkin substrates (5). Additionally, the anti-apoptotic B-cell lymphoma (BCL)-2 proteins, such as B-cell lymphoma-extra large (BCL-XL) and myeloid cell leukemia-1 (MCL-1), abrogate Parkin-mediated mitophagy by inhibiting Parkin translocation to depolarized mitochondria (28). MCL-1 and BCL-XL directly interact with Parkin in HeLa cells, and this interaction is increased after CCCP treatment (28). In contrast, Thomas et al. (90) found that PINK1/Parkin-mediated mitophagy is impaired in MCL-1-deficient hearts where deletion of MCL-1 in adult myocytes results in reduced PINK1 and accumulation of Parkin in the cytosol. Although these studies indicate that the anti-apoptotic BCL-2 proteins can regulate Parkin-mediated mitophagy, several questions need to be resolved. First, it is puzzling why anti-apoptotic proteins inhibit clearance of depolarized mitochondria since these organelles can activate apoptosis. The study by Hollville et al. (28) did not assess whether inhibiting Parkin-mediated clearance of damaged mitochondria has an effect on cell death. Also, it is unclear how loss of MCL-1 impairs the PINK1/Parkin pathway. Loss of MCL-1 leads to development of rapid heart failure, and it is unknown whether the impaired PINK1/Parkin pathway is a cause or consequence of the heart failure. Clearly, additional studies are needed to elucidate how the anti-apoptotic proteins regulate mitophagy in cells.
Mitochondrial Receptor-Mediated Mitophagy
BNIP3 and NIX.
Another pathway involved in mitophagy occurs through the BCL-2-related proteins BNIP3 and BNIP3L/NIX. These atypical BH3-only proteins are well known activators of cell death. BNIP3 activates BAX/BAK in the outer mitochondrial membrane and causes opening of the mitochondrial permeability transition pore (49, 76, 94). NIX activates cell death via the mitochondrial apoptotic pathway (99) and induces necrotic cell death by perturbing endoplasmic reticulum/sarcoplasmic reticulum calcium stores (17). Interestingly, recent studies have identified an additional function for these two proteins where they act as autophagy receptors on mitochondria in cells. For instance, BNIP3 promotes mitophagy in various cells, including cardiomyocytes (26, 59, 74, 91). NIX is required for mitochondrial elimination in maturing reticulocytes (78, 81). Both BNIP3 and NIX localize to the outer mitochondrial membrane where they act as receptors for targeting autophagosomes to mitochondria (Fig. 2B) (27, 69). Using their conserved LC3-interacting region motifs, they can directly bind to LC3/γ-aminobutyric acid receptor-associated protein (GABARAP) on the autophagosome (27, 69), eliminating the need for adaptor proteins. Because BNIP3/NIX and PINK1/Parkin play roles in mitophagy, this raises the question of whether they participate in the same pathway to clear mitochondria. However, the signals that activate the two pathways appear to be different. Whereas Parkin-mediated mitophagy requires loss of mitochondrial membrane potential (67), BNIP3 promotes mitophagy even when mitochondria retain their membrane potential (77). However, BNIP3 overexpression also induces translocation of Parkin to mitochondria in cardiac myocytes and Parkin-deficient myocytes exhibit reduced autophagy in response to BNIP3 overexpression (53). Additionally, NIX-deficient mouse embryonic fibroblasts have reduced Parkin translocation in response to CCCP treatment (15). Although these studies indicate potential coordination between BNIP3/NIX and the PINK1/Parkin pathways to clear mitochondria, the role and regulation of this potential crosstalk need to be investigated. In addition, in vivo mouse studies indicate that BNIP3/NIX is important for the normal turnover of mitochondria (18). Although it is clear that both BNIP3 and NIX are dual regulators of cell death and mitophagy, when and how they switch between these two opposing functions are unclear. It is also unknown whether other BH3-only proteins can function as autophagy receptors.
FUNDC1.
FUNDC1 is an outer mitochondrial membrane protein that has been implicated in hypoxia-mediated mitophagy in mammalian cells (55). Similar to BNIP3 and NIX, FUNDC1 acts as a receptor and mediates mitophagy through its interaction with LC3 through its LIR motif (55). In contrast to BNIP3 and NIX, FUNDC1 does not have any pro-death activity. A recent study reported that the PGAM5 phosphatase dephosphorylates FUNDC1 during hypoxia or FCCP treatment, which promotes the interaction of FUNDC1 with LC3 and mitophagy (Fig. 2C) (8). Not surprisingly, new evidence show a link between the PGAM/FUNDC1 and the PINK1/Parkin pathways. Both PINK1 and Parkin are involved in familial Parkinson's disease (14), and PGAM5-deficient mice develop a Parkinson's disease phenotype (57). A recent study discovered that PGAM5 is required for stabilization of PINK1 on damaged mitochondria and that loss of PGAM5 abrogates PINK1-mediated mitophagy (57). In addition, both PGAM5 and BNIP3 play roles in hypoxia-mediated mitophagy (8, 91), raising the possibility that there is also cooperation between BNIP3 and PGAM5/FUNDC1 in regulating mitochondrial clearance in response to hypoxia.
Cardiolipin.
Cardiolipin is present in inner mitochondrial membranes, where it is essential for optimal function of numerous enzymes involved in mitochondrial metabolism (33). Recently, it was reported that there is a significant redistribution of cardiolipin from the inner mitochondrial membrane to the outer mitochondrial surface during mitophagy (11). This study also reported that LC3 binds to cardiolipin on damaged mitochondria and that prevention of this interaction results in inhibition of mitochondrial delivery to autophagosomes (11). Thus it is possible that the redistribution of cardiolipin also acts as a signal for elimination of damaged mitochondria. These studies were conducted in neurons, and it will be necessary to determine whether cardiolipin regulates mitophagy in cardiac myocytes. It will also be interesting to investigate whether cardiolipin plays a role in activating other autophagy receptors or recruiting Parkin to mitochondria.
Overall, these studies demonstrate that multiple redundant pathways exist in cells to ensure clearance of dysfunctional mitochondria. These studies also indicate an intriguing link between different mediators of mitophagy, suggesting that these pathways may be more connected than previously thought. How these pathways coordinate mitophagy in vivo under baseline conditions and during stress still needs to be investigated.
Mitochondrial Dynamics Regulate Mitophagy
Mitochondrial dynamics have been implicated in regulating mitophagy. Studies have demonstrated that mitochondrial fission facilitates mitophagy (36, 39, 53, 92), whereas fusion protects against mitochondrial clearance (24, 75). Fission occurs in an asymmetric manner, which allows for segregation and degradation of only damaged mitochondrial components (92). The dynamin-like GTPase dynamin-related protein 1 (DRP1) plays a critical role in mediating mitochondrial fission and mitophagy. Deletion of DRP1 in cardiac myocytes results in mitochondrial elongation due to unopposed fusion, inhibition of mitophagy, and mitochondrial dysfunction, which lead to cardiac dysfunction and increased susceptibility to myocardial ischemia-reperfusion injury (36, 39). Mitochondrial fusion is regulated by Mitofusin 1 and 2 (MFN1 and -2) in the outer mitochondrial membrane and optic atrophy 1 in the inner mitochondrial membrane (12, 79). Fusion of mitochondria protects them from clearance by autophagosomes (24, 75). Interestingly, there is cross talk between the mitophagy and mitochondrial dynamics pathways. For instance, overexpression of BNIP3 leads to recruitment of DRP1 to mitochondria and induction of mitophagy (53). In addition, Parkin-mediated ubiquitination of MFN1/2 promotes their degradation by the proteasome (22, 72). This shift in the balance of fission/fusion proteins ensures that mitochondria are maintained in a fragmented state to allow mitophagy to proceed. Exactly how mitochondria coordinate with proteins involved in regulating their shape and clearance still needs further investigation.
Damaged Mitochondria Signals to Induce Autophagy
In addition to being marked for degradation, dysfunctional mitochondria must signal to the autophagy machinery to initiate the formation of additional autophagosomes. There are multiple ways this can be accomplished (Fig. 3). First, BH3-only proteins such as BAD, BNIP3, and NIX can directly activate autophagy by disrupting the BCL-2-BECLIN 1 interaction (3, 60). The anti-apoptotic BCL-2 proteins negatively regulate autophagy through their interaction with BECLIN 1 (60, 71), and the release of BECLIN 1 allows autophagy to be initiated (Fig. 3A). Similarly, Bax interacting factor-1 (Bif-1) is a positive regulator of Parkin-mediated mitophagy (86). This study found that Parkin translocation to damaged mitochondria is intact in the absence of Bif-1. Instead, Bif-1-deficiency results in accumulation of immature autophagosomes in cells, suggesting that Bif-1 promotes the maturation of autophagosomes downstream of Parkin. In contrast, two other BH3-only proteins have been reported to have the opposite effect on autophagy. BIM and BMF localize to the cytoskeleton under baseline conditions where they inhibit autophagy (13, 58). BMF interacts with and stabilizes the BCL-2-BECLIN1 complex (13), whereas BIM sequesters BECLIN1 and prevents it from initiating autophagy (58). Thus this suggests that the regulation of autophagy by the BH3-only proteins might depend on their subcellular localization.
Fig. 3.

Induction of autophagy by damaged mitochondria. A: BH3-only proteins directly induce autophagy by disrupting the BCL-2/BECLIN1 complex to release BECLIN1. The BECLIN1/VPS34/VPS15 complex can then induce autophagy. B: damaged mitochondria produce less ATP production, which activates the energy sensor AMPK. AMPK then activates Unc-51-like kinase (ULK)1, which activates the BECLIN1/VPS34/VPS15 complex. C: damaged mitochondria produce reactive oxygen species (ROS) that inhibit mammalian target of rapamycin (mTOR). Inhibition of mTOR leads to activation of autophagy. D: opening of the mitochondrial permeability transition pore (mPTP) results in influx of solutes and water into the mitochondrial matrix, which leads to disruption of the proton gradient and oxidative phosphorylation. It also causes depolarization and mitochondrial swelling that can activate autophagy.
Additionally, when mitochondrial function is compromised, they produce less ATP, which leads to activation of AMPK and subsequent initiation of autophagy (Fig. 3B) (100). AMPK phosphorylates and activates the Unc-51-Like Kinases (ULK), which, in turn, activates the BECLIN 1/VPS34/VPS15 complex and initiation of autophagy (42). ULK exists as two different isoforms: ULK1 and ULK2. Both can induce autophagy and have compensatory functions (10). However, recent studies indicate that ULK1 plays a specific role in regulating mitophagy (51, 96). In addition to activating the BECLIN 1/VPS34/VPS15 complex, ULK1 translocates to mitochondria where it phosphorylates and activates the mitophagy receptor FUNDC1 (96). Interestingly, both ULK1- and NIX-deficient erythrocytes are unable to clear their mitochondria via autophagy (51, 81), suggesting that they act in the same pathway. It will be interesting to explore whether ULK1 is an upstream regulator of NIX and whether they coordinate to regulate mitophagy in other tissues including the heart.
ROS is a byproduct of oxidative phosphorylation, and myocytes have a great antioxidant capacity to neutralize the ROS. However, excess ROS from damaged mitochondria act as an important signal for mitophagy (Fig. 3C) (80, 83). Song et al. (83) recently reported that suppression of mitochondrial ROS in transgenic mice overexpressing a mitochondrial targeted catalase leads to impaired mitophagy in the myocardium. ROS can activate autophagy by inhibiting mammalian target of rapamycin (mTOR), a negative regulator of autophagy (1), or by activating BNIP3 (31, 48). Thus it is likely that a specific threshold for ROS levels exists in myocytes, and once that threshold has been reached, it signals activation of autophagy and mitophagy via inhibition of mTOR and activation of BNIP3.
Another mitophagy signal involves the mitochondrial permeability transition pore (mPTP) (Fig. 3D). Opening of this pore causes an influx of solutes and water and leads to swelling of the inner membrane and disruption of mitochondrial membrane potential (2). Opening of the mPTP has also been shown to induce mitophagy during nutrient deprivation (7, 19). Cyclophilin D (CypD) is an essential component of the mPTP, and CypD-deficient myocytes fail to induce autophagy during starvation (7). In contrast, myocytes overexpressing CypD have enhanced autophagy during starvation, suggesting that CypD and the mPTP are critical in the initiation of autophagy during starvation. Interestingly, BNIP3 induces autophagy and mitophagy independently of the mPTP (74). This suggests that the mPTP is responsible for activating mitophagy under certain conditions such as starvation. It is currently unknown if the mPTP plays a role in PINK1/Parkin-mediated mitophagy.
AMBRA1 (activating molecule in Beclin1-regulated autophagy) is a critical regulator of autophagy initiation and interacts with BECLIN 1 to initiate formation of the phagophore (20). It has been proposed that there is a reserve of AMBRA1 at the mitochondria under normal conditions, which locally activate the BECLIN1/VPS34/VPS15 complex at the mitochondria in response to stress. This facilitates formation of the autophagosome near or at the depolarized mitochondria (93). AMBRA1 also interacts with Parkin at the mitochondria, and the interaction between Parkin and AMBRA1 is increased during prolonged mitochondrial depolarization (93). Although AMBRA1 is not required for translocation of Parkin to depolarized mitochondria, it is critical for subsequent mitochondrial clearance (84). Interestingly, AMBRA1 can also interact directly with LC3 through its LIR (LC3 interacting region) motif, and this interaction is critical for Parkin-dependent mitophagy (84). Surprisingly, specific targeting of AMRA1 to mitochondria leads to Parkin- and p62-independent mitophagy, suggesting that AMBRA1 can also act as a mitophagy receptor.
Alternative Pathways of Mitophagy
Although traditional autophagy is dependent on Atg5 to form autophagosomes, recent studies have described an alternative Atg5-independent form of autophagy (68). This alternative form of autophagy is independent of Atg5 and Atg7 but, similar to autophagy, requires ULK1 and BECLIN 1 (Fig. 4A) (68). Unlike in traditional autophagy, the formation of autophagosomes occurs in a Rab9-dependent manner with vesicles derived from the trans-Golgi (68). This alternative autophagy pathway plays a role in clearing mitochondria during erythrocyte differentiation (29). This indicates that the Rab9-dependent alternative autophagy can also contribute to mitochondrial degradation. Additional studies are needed to determine the functional importance of this pathway in the myocardium, as well as its role in disease. It will also be necessary to decipher during what conditions traditional versus alternative autophagy are activated.
Fig. 4.

Alternative pathways of mitochondrial clearance. A: ULK1 activates alternative autophagy via the BECLIN1/VPS34/VPS15 complex. The autophagosome membrane is derived from the trans-Golgi and is dependent on Rab9. The autophagosome sequesters the mitochondrion and fuses with a lysosome. B: microautophagy involves direct engulfment of cytosolic cargo into lysosomes. The lysosome forms an invagination to envelope the cargo. The internalized vesicle and its content are then degraded inside the lysosome by acid hydrolases.
Microautophagy is another less known form of autophagy that occurs in mammalian cells. In this process, proteins and organelles are directly internalized into lysosomes through invaginations of the lysosomal membrane (Fig. 4B). Microautophagy has primarily been studied in yeast, but there is evidence that it also takes place in mammalian cells (65, 87). Nonselective microautophagy results in the degradation of randomly sequestered soluble intracellular material. This process has been observed in both yeast and mammalian cells (64). Selective microautophagy has only been described in yeast and involves the direct sequestration of organelles such a mitochondria, nucleus, mitochondria, and peroxisomes into lysosomes (54). Currently, very little is known about the molecular mechanisms underlying microautophagy in mammalian cells. Its functional role in mammalian cells is not well understood, and it is unknown if alterations in this play a role in disease.
Mitophagy and Cardioprotection
The half-life of mitochondria in the myocardium has been reported to range from days to weeks (43, 63). Thus mitochondrial degradation is critical for cardiac homeostasis, and interfering with this process leads to accumulation of dysfunctional mitochondria and cardiac dysfunction (4, 18, 32, 47, 50). Many studies have reported that autophagy is increased in the myocardium in response to stress, and this is initially a protective response activated by the cell (25, 62, 89). To date, few studies have focused on the specific role of mitophagy in the heart, but emerging evidence support a protective role for mitophagy in response to stress. Increased mitophagy was initially described in myocytes overexpressing BNIP3 and in hearts subjected to ex vivo I/R (26). Subsequent studies using knockout mouse models have confirmed the importance of mitophagy in cardioprotection. For instance, PINK1-deficiency increased the susceptibility of the heart to I/R injury ex vivo (53). These mice also develop heart failure more rapidly in response to pressure overload than wild-type mice (4). Parkin-deficient mice accumulate dysfunctional mitochondria after a myocardial infarction, which leads to increased mortality (50). Parkin-deficient mice are also more susceptible to doxorubicin-mediated cardiotoxicity (32). In addition, Parkin plays a role in ischemic preconditioning, and Parkin null mice are resistant to preconditioning (34). This suggests that brief episodes of ischemia induce Parkin-mediated clearance of unstable mitochondria that are likely to cause damage during a prolonged ischemic episode. These studies clearly demonstrate the important cardioprotective role of mitophagy in the cardiovascular system, and enhancing mitophagy might represent a promising future therapeutic target.
In contrast, other studies have found that augmented autophagy can promote loss of myocytes (56, 62, 101). Although enhanced autophagy is protective during ischemia, it switches to a detrimental role during reperfusion (62). Increased autophagy is also detrimental in a model of pressure overload (101), confirming that chronic and excessive activation of autophagy can be dangerous to myocytes. Studies assessing the effect of chronic and excessive mitophagy in the myocardium are lacking, but excessive degradation of mitochondria in myocytes will be harmful. Hence, it will be important to determine the threshold of mitophagy in myocytes. It is likely that the levels and extent of both general autophagy and mitophagy determine whether it will be detrimental or beneficial to the cell. Excessive and/or prolonged induction of autophagy may be detrimental by causing overclearing of organelles and critical proteins. Thus additional research is necessary to determine the dynamics that regulate this transition between autophagy-induced protection and cell death in the heart.
Recently, a study reported that Parkin-mediated mitophagy is protective in pancreatic β-cell function in diabetes (30). Diabetic cardiomyopathy is associated with mitochondrial dysfunction (6), and it is possible that impaired autophagy and mitophagy may contribute to the pathology. Interestingly, Xu et al. (97) reported that general autophagy, as well as PINK1 and Parkin protein levels, are significantly reduced in Type 1 diabetic hearts. Interestingly, this study noted that Rab9 is increased and that it colocalizes with mitochondria, suggesting that alternative mitophagy is increased to compensate for reduced traditional mitophagy. This implies that alternative mitophagy is independent of PINK1 and Parkin, a concept that needs to be further explored.
Mitophagy in Aging
There is a reduction in mitochondrial DNA (mtDNA) quality and mitochondrial function with age in most tissues (61). Unfortunately, autophagy is also reduced with age (35, 61). Parkin-mediated mitophagy is also reduced in senescent cells (32). Thus reduced autophagy and mitophagy in the aging myocardium could contribute to accumulation of dysfunctional mitochondria. In support of this, mice deficient in Parkin accumulate abnormal mitochondria in the myocardium at a much earlier age than wild-type and exhibit enhanced age-dependent accumulation of mtDNA deletion mutations and oxidative damage (32, 47). In contrast, overexpression of Parkin in myocytes increases mitochondrial turnover and delays age-related cardiac abnormalities (32). Additionally, increasing general autophagy in the heart systemically by overexpressing the critical autophagy protein Atg5 also delayed aging (73). These studies demonstrate the important cardioprotective role of autophagy and mitophagy in aging. However, although upregulation of mitophagy appears to be promising in combating the aging process, more research is necessary to fully understand its specific role in clearing mitochondria.
Therapeutic Potential of Mitophagy
Despite major advances in the treatment of cardiovascular diseases, there is still a need to find improved therapies. A future potential therapy includes targeting of autophagy, since many studies have reported that enhancing autophagy is cardioprotective. However, a major challenge is to find safe and efficient targets in the autophagy pathway. Many of the regulators of autophagy, such as mTOR and AMPK, also regulate other critical cellular processes, and modulating general autophagy via these pathways can produce unwanted off-target effects. There is also a concern that activating general autophagy will not result in the selective removal of damaged mitochondria.
Thus an alternate and novel approach would be to selectively target mitophagy to clear impaired mitochondria without increasing the general nonselective autophagy. This would help avoid chronic nonspecific cellular clearance and minimize off-target effects. Data from Parkin transgenic mice indicate that enhancing levels of Parkin in the heart have no adverse effects under normal conditions and the increased rate of mitochondrial turnover delays aging (32). However, it still needs to be investigated whether myocardial ischemia or pressure overload will be protective or result in excessive clearance in these mice. Although BNIP3 and NIX also promote mitophagy, they are more risky targets due to their pro-death activity. In fact, BNIP3 and NIX cardiac-specific transgenic mice have increased apoptosis, which leads to cardiac dysfunction (16, 99). Although current studies suggest that enhancing mitophagy is cardioprotective, a more in-depth characterization of the pathways involved is necessary to identify optimal therapeutic targets.
Conclusion and Future Directions
Our current knowledge of mitophagy in the heart is very limited, and a number of unresolved questions remain. For instance, studies suggest that enhanced mitophagy can protect against stress that acutely damages mitochondria. However, whether mitophagy will be cardioprotective in other chronic diseases such as diabetic cardiomyopathy, pressure overload, or late-onset anthracycline-mediated cardiotoxicity still needs to be investigated. Similarly, the threshold for mitophagy in myocytes is currently unknown, and it is unclear how much mitochondria can be cleared before the cells become energy deficient and activate cell death. Additionally, mitochondria that have been removed must also be replaced through biogenesis, and very little is known about how these two processes are connected. For optimal cardioprotection, future therapies should aim to increase mitophagy while simultaneously augmenting mitochondrial biogenesis to prevent mitochondrial depletion and cell death. In conclusion, although3 the majority of current studies support the concept that mitophagy is cardioprotective, additional studies are necessary before this pathway can be targeted therapeutically.
GRANTS
This work was supported in part by National Institutes of Health Grants HL-087023, HL-101217, HL-085577 (to Å. Gustafsson), HL-67724, HL-91469, HL-102738, HL-112330, AG-23039 (to J. Sadoshima), and T32HL-007444 (to A. Moyzis). This work was also supported by the Fondation Leducq Transatlantic Networks of Excellence (to J. Sadoshima) and an American Heart Association Established Investigator Award 14EIA18970095 (to Å. Gustafsson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.G.M. prepared figures; A.G.M. and Å.B.G. drafted manuscript; A.G.M. and Å.B.G. edited and revised manuscript; A.G.M., J.S., and Å.B.G. approved final version of manuscript.
REFERENCES
- 1.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA 107: 4153–4158, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baines CP. The cardiac mitochondrion: nexus of stress. Annu Rev Physiol 72: 61–80, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29: 2570–2581, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA 108: 9572–9577, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510: 370–375, 2014. [DOI] [PubMed] [Google Scholar]
- 6.Blake R, Trounce IA. Mitochondrial dysfunction and complications associated with diabetes. Biochim Biophys Acta 1840: 1404–1412, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 6: 462–472, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, Duan L, Wang X, Liu L, Liu X, Shen Y, Zhu Y, Chen Q. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell 54: 362–377, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340: 471–475, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci USA 108: 11121–11126, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu CT, Bayir H, Kagan VE. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy 10: 376–378, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101: 15927–15932, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Contreras AU, Mebratu Y, Delgado M, Montano G, Hu CA, Ryter SW, Choi AM, Lin Y, Xiang J, Chand H, Tesfaigzi Y. Deacetylation of p53 induces autophagy by suppressing Bmf expression. J Cell Biol 201: 427–437, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deas E, Wood NW, Plun-Favreau H. Mitophagy and Parkinson's disease: the PINK1-parkin link. Biochim Biophys Acta 1813: 623–633, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 285: 27879–27890, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 117: 2825–2833, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, Molkentin JD, Kranias EG, Dorn GW 2nd. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest 119: 203–212, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorn GW., 2nd Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 3: 374–383, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 15: 2286–2287, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature 447: 1121–1125, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Galan JM, Haguenauer-Tsapis R. Ubiquitin lys63 is involved in ubiquitination of a yeast plasma membrane protein. EMBO J 16: 5847–5854, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19: 4861–4870, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 281: 29776–29787, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 14: 146–157, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287: 19094–19104, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell 55: 451–466, 2014. [DOI] [PubMed] [Google Scholar]
- 29.Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, Shimizu S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun 5: 4004, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, Iwai-Kanai E, Ikeda K, Ueyama T, Ogata T, Matoba S. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc Natl Acad Sci USA 111: 3116–3121, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, Ikeda K, Ueyama T, Okigaki M, Matsubara H. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol 52: 175–184, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun 4: 2308, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Houtkooper RH, Vaz FM. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol Life Sci 65: 2493–2506, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 6: e20975, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikeda Y, Sciarretta S, Nagarajan N, Rubattu S, Volpe M, Frati G, Sadoshima J. New insights into the role of mitochondrial dynamics and autophagy during oxidative stress and aging in the heart. Oxid Med Cell Longev 2014: 210934, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. In press. [DOI] [PubMed] [Google Scholar]
- 37.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 7: 279–296, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawsn VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33: 2798–2813, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. In press. [DOI] [PubMed] [Google Scholar]
- 42.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP, Ping P. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics 11: 1586–1594, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol 21: 336–345, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166, 2014. [DOI] [PubMed] [Google Scholar]
- 46.Kristensen AR, Schandorff S, Hoyer-Hansen M, Nielsen MO, Jaattela M, Dengjel J, Andersen JS. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics 7: 2419–2428, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Kubli DA, Quinsay MN, Gustafsson AB. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol 6: e24511, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol 295: H2025–H2031, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J 405: 407–415, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288: 915–926, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112: 1493–1502, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8: 1986–1990, 2009. [DOI] [PubMed] [Google Scholar]
- 53.Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301: H1924–H1931, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69: 1125–1136, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185, 2012. [DOI] [PubMed] [Google Scholar]
- 56.Lu L, Wu W, Yan J, Li X, Yu H, Yu X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int J Cardiol 134: 82–90, 2009. [DOI] [PubMed] [Google Scholar]
- 57.Lu W, Karuppagounder SS, Springer DA, Allen MD, Zheng L, Chao B, Zhang Y, Dawson VL, Dawson TM, Lenardo M. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson's-like movement disorder. Nat Commun 5: 4930, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, Rubinsztein DC. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell 47: 359–370, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 8: 297–309, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. Embo J 26: 2527–2539, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Heart Circ Physiol 305: H459–H476, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100: 914–922, 2007. [DOI] [PubMed] [Google Scholar]
- 63.Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem 246: 2425–2429, 1971. [PubMed] [Google Scholar]
- 64.Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7: 673–682, 2011. [DOI] [PubMed] [Google Scholar]
- 65.Mortimore GE, Wert JJ Jr, Adams CE. Modulation of the amino acid control of hepatic protein degradation by caloric deprivation. Two modes of alanine co-regulation. J Biol Chem 263: 19545–19551, 1988. [PubMed] [Google Scholar]
- 66.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461: 654–658, 2009. [DOI] [PubMed] [Google Scholar]
- 69.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Okatsu K, Iemura S, Koyano F, Go E, Kimura M, Natsume T, Tanaka K, Matsuda N. Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem Biophys Res Commun 428: 197–202, 2012. [DOI] [PubMed] [Google Scholar]
- 71.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122: 927–939, 2005. [DOI] [PubMed] [Google Scholar]
- 72.Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 5: e10054, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 4: 2300, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 6: 17–24, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108: 10190–10195, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 91: 226–231, 2002. [DOI] [PubMed] [Google Scholar]
- 77.Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson AB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 18: 721–731, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114: 867–874, 2001. [DOI] [PubMed] [Google Scholar]
- 80.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26: 1749–1760, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104: 19500–19505, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Siddall HK, Yellon DM, Ong SB, Mukherjee UA, Burke N, Hall AR, Angelova PR, Ludtmann MH, Deas E, Davidson SM, Mocanu MM, Hausenloy DJ. Loss of PINK1 increases the heart's vulnerability to ischemia-reperfusion injury. PLoS One 8: e62400, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW 2nd. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res 115: 348–353, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M, Cecconi F. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem 287: 40652–40660, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takahashi Y, Hori T, Cooper TK, Liao J, Desai N, Serfass JM, Young MM, Park S, Izu Y, Wang HG. Bif-1 haploinsufficiency promotes chromosomal instability and accelerates Myc-driven lymphomagenesis via suppression of mitophagy. Blood 121: 1622–1632, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takikita S, Myerowitz R, Zaal K, Raben N, Plotz PH. Murine muscle cell models for Pompe disease and their use in studying therapeutic approaches. Mol Genet Metab 96: 208–217, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci USA 105: 9745–9750, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson AB. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 27: 1365–1377, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 27: 6229–6242, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J 27: 433–446, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci 31: 10249–10261, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vande VC, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol 20: 5454–5468, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147: 893–906, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, Zhu Y, Zhang X, Li L, Zhang L, Sui S, Zhao B, Feng D. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15: 566–575, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 288: 18077–18092, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22: 124–131, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN, Dorn GW 2nd. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med 8: 725–730, 2002. [DOI] [PubMed] [Google Scholar]
- 100.Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res 111: 800–814, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117: 1782–1793, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]