

Abstract
Inhibition of β-adrenergic receptor (β-AR) signaling is one of the most common therapeutic approaches for patients with arrhythmias. Adenylyl cyclase (AC) is the key enzyme responsible for transducing β-AR stimulation to increases in cAMP. The two major AC isoforms in the heart are types 5 and 6. Therefore, it is surprising that prior studies on overexpression of AC5 and AC6 in transgenic (Tg) mice have not examined mediation of arrhythmogenesis. Our goal was to examine the proarrhythmic substrate in AC5Tg hearts. Intracellular calcium ion (Ca2+i) was imaged in fluo-4 AM-loaded ventricular myocytes. The sarcoplasmic reticulum (SR) Ca2+ content, fractional Ca2+ release, and twitch Ca2+ transient were significantly higher in the AC5Tg vs. wild-type (WT) myocytes, indicating Ca2+ overload in AC5Tg myocytes. Action potential (AP) duration was significantly longer in AC5Tg than in WT myocytes. Additionally, AC5Tg myocytes developed spontaneous Ca2+ waves in a larger fraction compared with WT myocytes, particularly when cells were exposed to isoproterenol. The Ca2+ waves further induced afterdepolarizations and triggered APs. AC5Tg hearts had increased level of SERCA2a, oxidized Ca2+/calmodulin-dependent protein kinase II (CaMKII), and phosphorylation of ryanodine receptors (RyR) at the CaMKII site, especially after isoproterenol treatment. This was consistent with higher reactive oxygen species production in AC5Tg myocytes after isoproterenol treatment. Isoproterenol induced more severe arrhythmias in AC5Tg than in WT mice. We conclude that AC5 overexpression promotes arrhythmogenesis, by inducing SR Ca2+ overload and hyperactivation of RyR (phosphorylation by CaMKII), which in turn induces spontaneous Ca2+ waves and afterdepolarizations.
Keywords: adenylyl cyclase type 5, overexpression, calcium handling, electrophysiology, arrhythmias
enhanced β-adrenergic receptor (β-AR) signaling, by increasing adenylyl cyclase (AC) activity and cAMP, is one of the most common mechanisms for arrhythmias (15, 26), and conversely, inhibition of β-AR signaling is one of the most common therapeutic approaches for patients with arrhythmias (8). There are two major AC isoforms in the heart (type 5 and type 6, AC5 and AC6) and despite the fact that AC is the key enzyme responsible for transducing β-AR stimulation to increase cAMP, little has been studied on how overexpressing either AC5 or AC6 affects cardiac arrhythmias. Interestingly the prior literature on this topic has been controversial, showing that these two isoforms do not always regulate the heart identically. For example, some studies showed that AC6 overexpression protects the heart against myocardial infarction (20), whereas other studies showed it failed to protect the heart from chronic pressure overload (11). In addition, some studies have shown that AC5 overexpression rescued the cardiomyopathy induced by Gαq expression (17), but not the one induced by β1-AR (22). Furthermore, AC5 inhibition has been shown to be cardioprotective (16, 25), while AC5 overexpression to be deleterious (14).
The goal of the present investigation was to determine the role of AC5 signaling in arrhythmogenesis by using a transgenic mouse with AC5 overexpression (AC5Tg). We showed that cardiac-specific overexpression of AC5 is detrimental to cardiac tissue by directly causing Ca2+i overload and oxidative stress, thus generating a proarrhythmic substrate.
MATERIALS AND METHODS
Mouse models.
Generation of cardiomyocyte-restricted AC5Tg mice has been described in detail previously (12). Mice of either sex were used at 2–5 mo of age. Animals used in this study were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). This study was approved by the Animal Care and Use Committee at Rutgers-New Jersey Medical School.
Cell isolation.
Ventricular myocytes were enzymatically isolated from the left ventricles of adult mouse hearts. After being anesthetized with isoflurane in a covered beaker, the hearts were removed from the mice and perfused retrogradely in Langendorff fashion at 37°C with nominally Ca2+-free Tyrode's solution containing ∼1 mg/ml collagenase (type II; Worthington) and 0.1 mg/ml protease (type XIV, Sigma) for 13–15 min. After the enzyme solution was washed out, the heart was removed from the perfusion apparatus and swirled in a culture dish. The Ca2+ concentration was slowly increased to 1.0 mM, and the cells were stored at room temperature until they were ready for use. The cells were used within 8 h of isolation. All single cell electrophysiological experiments were performed at 35–37°C.
Patch-clamp methods.
The myocytes were patch-clamped using the whole cell configuration of the patch-clamp technique in the current-clamp or the voltage-clamp mode. To record APs, patch pipettes (2–5 MΩ) were filled with an internal solution containing (in mM) 110 K+-aspartate, 30 KCl, 5 NaCl, 10 HEPES, 0.1 EGTA, 5 Mg-ATP, 5 Na2-creatine phosphate, and 0.05 cAMP (pH 7.2, adjusted with KOH). The myocytes were superfused with normal Tyrode's solution containing (in mM) 136 NaCl, 5.4 KCl, 0.33 Na2PO4, 1.0 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4, adjusted with NaOH). Action potentials (APs) were elicited with 2-ms, 2- to 4-nA square pulses at various pacing cycle lengths (PCLs).
To record L-type Ca2+ current (ICa,L), patch pipettes were filled with internal solution containing (in mM) 110 Cs-aspartate, 30 CsCl, 5 NaCl, 10 HEPES, 0.1 EGTA, 5 MgATP, 5 Na2-phosphocreatine, pH 7.2 with CsOH. The cells were perfused with a modified Tyrode's solution containing (in mM) 120 NaCl, 20 TEACl, 0.33 Na2PO4, 1.0 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4 adjusted with NaOH. The myocytes were stimulated at a PCL of 6 s with a double-pulse protocol. Following a 100-ms prepulse to −40 mV from the holding potential of −80 mV (to inactivate Na+ current and T-type Ca2+ current), ICa,L was elicited by a subsequent test depolarization step to test potentials (−40 to +50 mV at a 10-mV step) for 300 ms. For total outward K+ current recording, the pipette and superfusion solutions were the same as those for AP recording. Tetrodotoxin (10 μM) and CdCl2 (0.5 mM) were added into the Tyrode's solution to inhibit INa and ICa,L. K+ currents were elicited from a holding potential of −80 mV by a series of 400-ms test pulses from −40 to +40 mV in 10-mV increments. The inward Na+/Ca2+ exchange current (INCX) was recorded by a rapid application of 10 mM caffeine (∼2-s duration). The application of caffeine was always preceded by a set of ten 200-ms conditioning depolarization pulses from −40 to 0 mV to ensure a consistent degree of SR Ca2+ loading. A holding potential of −40 mV was maintained during caffeine application. Electrical signals were measured with an Axopatch 700A patch-clamp amplifier, controlled by a personal computer using Digidata 1322A interface, driven by pCLAMP10 software.
Ca2+i measurement.
Myocytes were loaded with the Ca2+ indicator fluo-4 by incubating them for ∼30 min in bath solution containing 4 μM fluo-4 AM (Molecular Probes), after which the cells were washed and placed in a heated chamber on a Nikon Eclipse TE200 inverted microscope. Ca2+i fluorescence [excitation/emission wavelengths (EX/EM): 485/530 nm] was recorded using an AndorIxon charge-coupled device camera (Andor Technology) operating at ∼50 fps. Fluorescence intensities were measured as the ratio of fluorescence (F) over the basal diastolic fluorescence (F0).
Measurement of mitochondrial ROS production.
Changes in mitochondrial superoxide production were monitored using MitoSOX Red (Invitrogen/Molecular Probes). Isolated cardiomyocytes were loaded with MitoSOX Red (5 μM) for 15 min at 37°C followed by washout. MitoSOX Red fluorescence (EX/EM: 485/585 nm) was monitored using a Nikon Eclipse TE200 inverted microscope and recorded using an Ixon Charge-Coupled Device camera (Andor Technology) operating at ∼50 fps. The fluorescence images were obtained at the start, and after 40 min in the absence or presence of 1 μM isoproterenol.
Western blot analysis.
Total protein extract from heart tissues (or isolated myocytes) were prepared in a buffer containing protease, kinase, and phosphatase inhibitors. Specific antibodies were used to detect expression or activation of differential proteins. The films were densitometrically evaluated utilizing “Quantity One” software (Bio-Rad Laboratories). Antiserum for oxidized CaMKII was a generous gift from Dr. Mark Anderson (Univ. of Iowa). Other antibodies were from Thermo Fisher Scientific [ryanodine receptor (RyR), dihydropyridine receptor (DHPR)], Santa Cruz Biotech (PLN, β1-AR), or Abcam (P-RyR, Pan-cadherin).
Electrocardiogram.
Standard lead II ECG signals were recorded in anesthetized mice (pentobarbital 0.04 mg/g) by using bipolar limb leads through a differential amplifier (model DP301, Warner Instruments). The signals were filtered between 0.1 and 100 Hz, displayed, and recorded to computer by using AxoScope 10 through MiniDigi 1A converter (Molecular Device).
Arrhythmia scoring system.
The definitions and point values of various arrhythmic events were as follows (7, 10, 27): 0 point: no arrhythmia; 1 point: premature ventricular contraction (PVC); 2 point: paired PVC; 3 point: bigeminal or trigeminal PVC or nonsustained ventricular tachycardia (VT) (≥ 3, but <10 consecutive PVC); 4 point: sustained VT (≥ 10 consecutive PVC); 5 point: ventricular fibrillation (VF).
Statistics.
Results are expressed as means ± SE. Individual groups were compared by two-tailed Student's t-test or Fisher's exact tests as indicated in the text. Results were considered statistically significant if the P value was <0.05.
RESULTS
Characterization of AP and Ca2+ handling properties in AC5Tg mice at baseline.
To determine whether the overexpression of AC5 affects the cellular electrical and Ca2+ handling properties, we compared APs, Ca2+ transients (CaT), and SR Ca2+ load in isolated ventricular myocytes. As shown in Fig. 1, an apparent late plateau was observed after the rapid repolarization phase in AC5Tg myocytes, causing prolongation of the AP duration (APD) at 90% repolarization (APD90) (WT 46.7 ± 6.9 vs. AC5Tg 65.2 ± 7.6 ms, P < 0.05), while there was no change in the APD at 50% repolarization (APD50) (Fig. 1, A and B). In some cells, we also observed early afterdepolarizations (EADs) in AC5Tg myocytes (Fig. 1C, left). Blocking Na+/Ca2+ exchange current (INCX) by replacing Na+ with Li+ in the Tyrode's perfusate eliminated EADs and shortened the APD90 (Fig. 1C), suggesting the involvement of a high Ca2+ load and forward-mode INCX in the APD prolongation in AC5Tg myocytes. Consistent with this assumption, the twitch CaT (F/F0, WT 2.4 ± 0.1 vs. AC5Tg 3.4 ± 0.1), the SR Ca2+ content (F/F0, WT 3.4 ± 0.2 vs. AC5Tg 4.2 ± 0.2), and the fractional Ca2+ release (WT 68.3 ± 3.4% vs. AC5Tg 84.0 ± 2.3 %) were significantly increased, and the 50% duration of twitch CaT (T50) (WT 227.3 ± 14.2 vs. AC5Tg 127.3 ± 11.1 ms) was shortened in AC5Tg myocytes (Fig. 2).
Fig. 1.
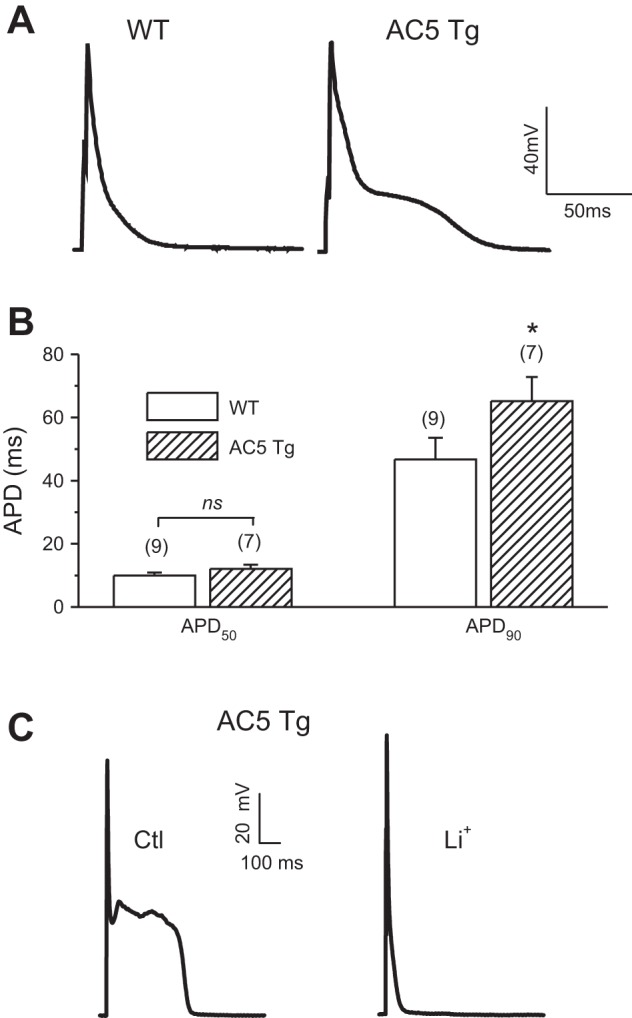
Prolonged action potential (AP) durations (APDs) in adenylyl cyclase type 5 transgenic (AC5Tg) myocytes. A: representative AP recordings from ventricular myocytes of wild-type (WT) and AC5Tg mice. B: summary of APD at 50% repolarization (APD50) and APD at 90% repolarization (APD90) values. *P < 0.05 compared with WT control myocytes. Cell number is indicated above each bar (from 3–5 mice each). C: effect of Na+-free, Li+-containing Tyrode's solution on the AP properties in AC5Tg, suggesting the contribution of Ca2+ overload and Na+/Ca2+ exchange current (INCX) on the prolonged APD and early afterdepolarizations (EADs).
Fig. 2.

Increased sarcoplasmic reticulum (SR) Ca2+ contents and intracellular Ca2+ (Ca2+i) transient amplitudes in AC5Tg myocytes. A: twitch Ca2+ transient (CaT) and SR Ca2+ content measured as the height of caffeine (10 mM)-induced Ca2+ transient in a WT ventricular myocyte. B: same as A, except in an AC5Tg ventricular myocyte. C–F: summarized data for SR Ca2+ contents, twitch CaT, fractional SR Ca2+ release, and 50% duration of twitch CaT in WT and AC5Tg ventricular myocytes. Cell number is indicated above each bar (from 3–5 mice each). *P < 0.05, **P < 0.01 compared with WT control myocytes.
Alterations in membrane currents in AC5Tg myocytes.
To obtain the direct evidence for potential upregulation of INCX, we measured SR Ca2+ release and INCX in response to caffeine (10 mM) application in fluo-4-loaded myocytes. We found significant increases in the amplitudes of both caffeine-induced Ca2+ transients (SR Ca2+ content) and peak INCX current density in response to caffeine in AC5Tg myocytes compared with WT controls (Fig. 3, A–D). These results were consistent with AP morphology changes as shown in Fig. 1A. Since INCX current is dependent on SR Ca2+ release, we further measured the ratio of peak INCX density against corresponding peak F/F0 [INCX/(F/F0)] as described previously (18). The normalized INCX exhibited the same magnitude in WT (0.39 ± 0.05) and in AC5Tg (0.40 ± 0.06). These results suggest that the enhanced INCX in AC5Tg is due to the elevation of Ca2+i loading rather than increased NCX expression.
Fig. 3.

Alterations in membrane currents in AC5Tg myocytes. A and B: caffeine (Caff; 10 mM)-induced Ca2+ transients (i.e., SR Ca2+ contents) and INCX in AC5Tg (n = 6) vs. WT myocytes (n = 6). C and D: simultaneously recorded INCX in the same cells as in A and B. E and F: representative current traces of L-type Ca2+ current (ICa,L) and current-voltage (I-V) relations recorded from AC5Tg vs. WT myocytes. G and H: representative current traces of outward potassium current (IK) and I-V relations for both peak (Ipeak) and steady-state currents (Iss) recorded from AC5Tg (n = 6) vs. WT myocytes (n = 6). *P < 0.05 compared with WT control myocytes. Myocytes used in the observations were from 3–5 mice in each group.
The ICa,L and potassium current (IK) are the other two major current components determining the AP duration. We therefore evaluated the current density level of ICa,L and total IK. As shown in Fig. 3, E and F, the ICa,L current density was increased in the AC5Tg myocytes at various holding potentials. However, the outward IK,total densities were not altered in the AC5Tg myocytes compared with WT (Fig. 3, G and H).
Alterations in other Ca2+ handling proteins in AC5Tg myocytes.
To determine if the alterations in Ca2+ handling properties are mediated by changes in expression level or function of any Ca2+ handling proteins, we next carried out Western blotting analyses. As shown in Fig. 4A, the expression levels of both SR Ca2+ ATPase (SERCA2a) and phospholamban (PLN) were upregulated (SERCA2a, from 1.00 ± 0.08 to 2.94 ± 0.19; PLN, from 1.00 ± 0.13 to 3.11 ± 0.25, n = 3, P < 0.05, respectively) in AC5Tg hearts, while no alteration was found in total calsequestrin (CSQ) expression (Fig. 4A). On the other hand, as shown in Fig. 4B, we did not observe any significant alteration in the expression level of β1-AR, L-type calcium channel (DHPR), and Na+/Ca2+ exchanger type 1 (NCX1) in the membrane preparation of AC5Tg vs. WT hearts.
Fig. 4.

Western blot analysis of Ca2+ handling proteins in AC5Tg hearts. A: expression levels of SR Ca2+ ATPase (SERCA2a), phospholamban (PLN), and calsequestrin (CSQ) in AC5Tg vs. WT. Representative blot bands (Aa) and summarized bar graph (Ab) are shown. CSQ shows no difference and serves as an internal control. Data were normalized to WT on the same gel (n = 3). B: expression levels of β1-AR, DHPR, and Na+/Ca2+ exchanger type 1 (NCX1) from membrane preparation of AC5Tg vs. WT hearts. Representative blot bands (Ba) and summarized bar graph (Bb) are shown. Pan-cadherin serves as an internal control. Data were normalized to WT on the same gel (n = 4). Equal sample loading was also confirmed by Ponceau S staining of the Western blot membrane. C: total ryanodine receptor (RyR) expression and phosphorylation of RyR (pRyR) at position Ser2808 (PKA site) and Ser2814 (CaMKII site) at baseline (vehicle injection, n = 5 in WT and n = 6 in AC5Tg) and after isoproterenol treatment (1 μM) for 40 min (n = 4) (D). Representative blot bands (Ca and Da) and summarized bar graph (Cb and Db) are shown in each panel. Data were normalized to WT on the same gel. ns, nonsignificant; *P < 0.05, **P < 0.01 compared with WT.
In addition, no differences were observed in total RyR2 expression levels between AC5Tg and WT hearts at either baseline or 40 min after isoproterenol injection (Fig. 4, C and D). Since the phosphorylation status of RyR2 is closely related to hyperactivity of RyR2 and intracellular (arrhythmogenic) Ca2+ waves, we also compared the phosphorylation levels of RyR2 at protein kinase A (PKA) (S2808) and CaMKII (S2814) sites by using phosphospecific antibodies. As shown in Fig. 4, C and D, the level of phosphorylation at the CaMKII site was significantly higher in AC5Tg hearts compared with WT at both baseline and 40 min after isoproterenol injection, while there was no difference in the levels of phosphorylation at the PKA site in AC5Tg vs. WT. This may be caused by a higher CaMKII activity in AC5Tg hearts than in WT hearts.
We next evaluated the expression and activation level of CaMKII. As shown in Fig. 5, the total expression of CaMKII remained the same. While the phosphorylated form of CaMKII (ratio) was also at the same level in WT and AC5Tg at either baseline or after isoproterenol treatment, the level of oxidized forms of CaMKII was higher in AC5Tg hearts compared with WT at both baseline and after isoproterenol treatment. In addition, the oxidation level of CAMKII in AC5Tg was further upregulated by isoproterenol treatment (P < 0.05).
Fig. 5.

Western blot analysis of CaMKII activation in AC5Tg hearts. A: Western blot bands of total, phosphorylated (P-CaMKII), and oxidized CaMKII (O-CaMKII) in AC5Tg vs. WT hearts at baseline (BL) and after isoproterenol treatment (ISO; 1 μM). Ba: summarized bar graph showing that phosphorylated (at Thr286) CaMKII levels remained the same in all the groups. Bb: summarized bar graph showing that oxidized CaMKII levels were significant higher in AC5Tg hearts, especially after isoproterenol treatment. Heart number is indicated above each bar. *P < 0.05 compared with WT; #P < 0.05 compared with AC5TG baseline.
Enhanced ROS production under β-AR stimulation in AC5Tg myocytes.
Previous studies have shown that β-AR stimulation increases oxidative stress level (ROS production) in the heart (1, 30), and ROS play an important role in cardiac arrhythmogenesis (23, 28). Our recent study has also revealed that manganese superoxide dismutase (MnSOD) is downregulated in AC5Tg mice at baseline and in response to chronic β-AR stimulation (14). Next, we compared the tolerance to oxidative stress in AC5Tg vs. WT mouse myocytes exposed to acute isoproterenol treatment. As shown in Fig. 6, MitoSox-Red fluorescence (an indicator of mitochondrial superoxide level) was measured and compared between WT and AC5Tg myocytes. While treatment with isoproterenol (1 μM, for 40 min) increased MitoSox-Red fluorescence in both WT and AC5Tg myocytes, the magnitude of enhancement was more striking in AC5Tg than in WT. This result suggests that the AC5Tg cardiomyocytes may be less tolerant to oxidative stress induced by acute β-AR stimulation. We assume that isoproterenol-promoted ROS production is likely to account for the augmentation of SR Ca2+ leak and generation of Ca2+ waves during β-AR stimulation via combined phosphorylation and oxidation of RyRs.
Fig. 6.

Reactive oxygen species (ROS) production in AC5Tg myocytes. A: representative images showing MitoSOX Red-loaded WT and AC5Tg cardiomyocytes under control conditions and after exposure to 1 μM isoproterenol for 40 min. B: summary of MitoSOX Red fluorescence in WT and AC5Tg cardiomyocytes in the absence (vehicle) or presence of 1 μM isoproterenol (Iso). Cell number is indicated above each bar (from 3–5 mice each). *P < 0.05, **P < 0.01.
Ca2+ overload and increased spontaneous Ca2+ release in AC5Tg myocytes.
The aforementioned alterations in electrophysiology and Ca2+ handling in AC5Tg myocytes present a proarrhythmic substrate. We next tried to obtain direct evidence by evaluating the incidence of spontaneous Ca2+ wave and afterdepolarizations. As shown in Fig. 7, another important finding was that AC5Tg myocytes frequently developed Ca2+ aftertransients (oscillations) following each paced beat (Fig. 7, A and B) after stimulation with isoproterenol. These spontaneous premature Ca2+ release events occurred in a significantly larger fraction of AC5Tg cells compared with WT myocytes even under basal conditions, but the incidence was even more striking when cells were exposed to isoproterenol (42.9% vs. 61.9%) (Fig. 7C). The spontaneous Ca2+ waves produced early (EADs) and/or delayed afterdepolarizations (DADs) and triggered APs (Fig. 7E). These data strongly suggest that SR Ca2+ overload and spontaneous Ca2+ release-induced EADs and DADs may facilitate the incidence of arrhythmias in AC5Tg mice in vivo.
Fig. 7.

Spontaneous Ca2+ waves and triggered activities (TAs) in AC5Tg myocytes. A: representative Ca2+ fluorescence recordings in the absence (Ctl) and presence of isoproterenol (1 μM) in a WT myocyte. B: representative Ca2+ fluorescence recordings in the absence (Ctl) and presence of isoproterenol (1 μM) in an AC5Tg myocyte. Prominent Ca2+ aftertransients (asterisks) were observed in AC5Tg myocytes after isoproterenol challenge. C: incidence rate of Ca2+ oscillations and waves in WT and AC5Tg myocytes in the presence and absence of 1 μM isoproterenol. Myocytes used in observations were from 3–5 mice in each group. E: simultaneous line-scan image along the long axis of the myocyte (Ea), Ca2+i transients (Eb), and APs (Ec) from an AC5Tg myocyte in the presence of isoproterenol (1 μM). Aberrant spontaneous Ca2+ waves (indicated by the arrows in Ea, and w's in Eb) causing delayed afterdepolarizations (DADs) (d) and triggered beats (*) in Eb and Ec. Pacing marks are indicated by the arrows. D: same as E, except from a WT myocyte. Neither apparent Ca2+ waves nor triggered activities were observed in this specific cell.
Higher susceptibility to isoproterenol-induced arrhythmias in AC5Tg mice.
In the following experiments, we assessed the effect of catecholamine challenge (isoproterenol injection, 6 mg/kg ip) in anesthetized AC5Tg vs. WT mice. Representative ECGs (lead II) recorded from AC5Tg and WT are shown in Fig. 8. We observed that only AC5Tg mice exhibited frequent PVCs and polymorphic VTs after isoproterenol treatment (Fig. 8, B, i and ii). The arrhythmia score was significantly higher in AC5Tg than WT (Fig. 8C). These data suggest that AC5Tg mice are more susceptible to catecholamine challenge-induced arrhythmias. It should be noted that the time courses for isoproterenol-induced heart-rate increase (within 1 min) and occurrence of VT/VF (peak at 20–40 min after injection) are significantly different, which is consistent with the notion that different signaling pathways may be involved in these two influences.
Fig. 8.
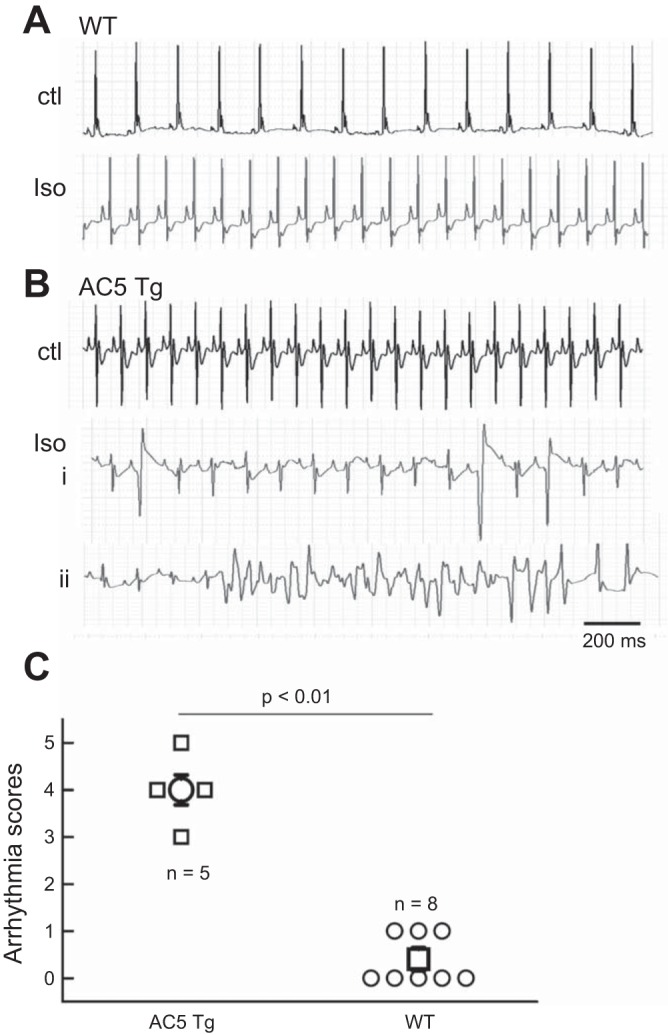
Isoproterenol-induced polymorphic ventricular tachycardias (VTs) in AC5Tg mice. Surface ECG (lead II) signals recorded from an anesthetized WT mouse (A) and an AC5Tg mouse (B). Representative traces before isoproterenol injection (Ctl) and 30 min after isoproterenol injection are shown in each case. Multiple PVCs (i) and polymorphic VT (ii) were observed after Iso injection. C: summarized arrhythmia scores in AC5Tg and WT. The significant comparison between two groups was conducted with Fisher's exact tests (P < 0.01).
DISCUSSION
AC5 and AC6 are the two major AC isoforms in the heart, responsible for the generation of cAMP in response to β-AR stimulation. Considering this and the fact that β-AR stimulation is a major mechanism mediating arrhythmogenesis, it is surprising that no prior study on AC5Tg or AC6Tg models have studied their role in arrhythmogenesis, which is the central feature of the present investigation. In the present study, we examined regulation of the electrophysiological and Ca2+ handling properties and arrhythmic propensity in the AC5Tg cardiomyocytes. The major findings are 1) the AC5Tg cardiomyocytes are characterized by Ca2+i overload, i.e., increased SR Ca2+ content and intracellular CaT; 2) isoproterenol treatment causes more SR spontaneous Ca2+ release/Ca2+ waves in AC5Tg than in WT. This subsequently accounts for higher predisposition to arrhythmias in AC5Tg mice in vivo; 3) the molecular basis for these functional changes may be due to the increased levels of expression and activity of Ca2+ handling proteins, such as SERCA, ICa,L, and NCX in AC5Tg myocytes; and 4) exaggerated ROS generation, oxidation of CaMKII, and phosphorylation of RyR2 also contribute to the higher arrhythmic susceptibility in AC5Tg mice.
Prior studies in AC5Tg and AC6Tg mice, although not examining arrhythmogenesis, have examined Ca2+ regulation. A recent study by Timofeyev et al. (21) reported that AC5 and AC6 exhibit distinct subcellular compartmentalization and response differently on regulation of ICa,L in ventricular myocytes. Their results suggest that the AC5 isoform is mainly localized in the t-tubular region and shows less effect on ICa,L regulation by β-AR stimulation due to restriction by phosphodiesterase. However, our present data demonstrate that AC5 does play a critical role in the regulation of Ca2+ handling and arrhythmogenesis, when it is overexpressed. This discrepancy may be caused by the secondary remodeling of antioxidative ability (14) and Ca2+ handling proteins in AC5Tg mice. Another possibility may be that AC5 present in other subcellular regions, where it is coupled to other cellular signaling pathways, such as regulation of RyR activity and/or mitochondrial function (ROS production), bypasses ICa,L.
Electrophysiological basis for the arrhythmogenic substrate in AC5Tg cardiomyocytes.
It has been well demonstrated that afterdepolarizations and TAs are among the main mechanisms responsible for cardiac arrhythmias. Both EADs and DADs are arrhythmogenic when they reach the threshold and initiate ectopic cardiac beats. Our recent study has shown that both ICa,L and INCX are important inward currents that account for EAD generation under different pathological conditions; while reactivation of ICa,L plays a predominant role in EAD genesis under oxidative stress, spontaneous calcium waves and subsequent INCX are a predominant cause for EADs under the Ca2+ overload condition (31). Our present data suggest that the prolongations of APD and EADs recorded from AC5Tg myocytes are most likely due to the increase in both ICa,L and INCX (inward currents) while the outward IK remains unchanged (Fig. 3). Furthermore, the spontaneous Ca2+ waves and subsequent INCX (or Iti) contribute to the genesis of EADs and DADs. Since there was no alteration of DHPR expression (Fig. 4B), we assume that the increase of ICa,L in AC5Tg myocytes was mediated by PKA-dependent regulation. This assumption fits with the leftward shift in the I-V relation of ICa,L activation as shown in Fig. 3F.
Remodeling of Ca2+ handling proteins in AC5Tg mouse heart.
Alteration of Ca2+ handling occurs under various pathological conditions such as ischemia, hypertrophy, and heart failure (2, 5, 19, 29). Our results have shown that the Ca2+ handling properties are also changed in AC5Tg ventricular myocytes. The increased twitch CaT amplitudes and Ca2+ contents in the AC5Tg myocytes (Fig. 2) suggest that the AC5 overexpression increases the propensity for Ca2+ overload. It is known that PLN binds to SERCA2a and inhibits SR Ca2+ uptake by SERCA2a in ventricular myocytes. We found that in AC5Tg myocytes, both SERCA and PLN levels were high (Fig. 4A) and the overall effect on SR Ca2+ uptake is enhanced, as indicated by the shorter CaT duration (T50, Fig. 2, B and F) and higher SR Ca2+ contents. This result suggests that SERCA upregulation plays a major role in increasing SR Ca2+ uptake, while increased PLN level shows less inhibitory effect.
While the total expression of RyR remained unchanged in AC5Tg hearts, the activation of RyR for both Ca2+-induced Ca2+ release (CICR) and diastolic spontaneous Ca2+ leak may be enhanced by CaMKII phosphorylation in AC5Tg hearts, since the activation of CaMKII was upregulated, especially after isoproterenol treatment. Thus, more spontaneous Ca2+ waves occurred in AC5Tg myocytes under both baseline and isoproterenol stimulation (Fig. 7). Therefore the overall alteration of Ca2+ handling properties in AC5Tg myocytes seem to be due to the combination of direct activation of Ca2+ handling proteins (e.g., L-type Ca2+ channels) by AC5-cAMP-PKA and compensatory changes in Ca2+ handling protein expression (e.g., SERCA and PLN).
Ca2+ overload occurs when there is a large increase in intracellular Ca2+ levels that cannot be handled by the cell (i.e., beyond the limits of the ability of cardiac cells to handle an increased calcium load; 24). Ca2+ overload may occur in the sarcoplasmic reticulum, mitochondria, or the cytoplasm. When Ca2+ overload happens it results in abnormal cell function, such as arrhythmias and heart failure. This seems to be the case in AC5Tg myocytes, especially during stimulation with isoproterenol, when more spontaneous Ca2+ waves, EADs, DADs, and arrhythmias were observed at the single cell level and in vivo as evidenced by the ECG recordings.
While we observed that ICa,L was increased in AC5Tg myocytes, most likely due to PKA-dependent activation, RyR2 phosphorylation level at S2808 (PKA site) did not show changes at either baseline or after isoproterenol treatment. We do not have a ready explanation for the differential effects of PKA on ICa,L and RyR phosphorylation. One possibility is that the stimulatory effect of the overexpressed AC5 may be compartmentalized and masked by the action of PDE, which would restrict local activation of PKA and PKA-dependent phosphorylation of RyR2.
Increased oxidative stress in AC5Tg cardiomyocytes.
Our recent studies have shown that the protein expression levels of MnSOD are significantly reduced (by 38%) in AC5Tg hearts, suggesting that AC5Tg hearts are more susceptible to oxidative stress (14). Recent studies have shown that β-AR stimulation promotes ROS production from mitochondria (1), which may account for Ca2+ wave generation (3). Our present results are consistent with these observations and further indicate that both Ca2+ overload and higher ROS levels are required for the higher propensity of arrhythmias in AC5Tg mice.
Interestingly, the time course for heart rate increase (fight or flight response) was very different from that for the arrhythmia generation (pathological response) after isoproterenol stimulation, suggesting different mechanisms may exist for these two responses. While the classical stimulatory G protein-AC-cAMP-PKA pathway likely contributes to the heart rate increase and acute Ca2+ overload (ICa,L activation and CaT enhancement), increased arrhythmogenicity may be mediated by the synergistic effect of Ca2+ overload and higher ROS production-CaMKII activation-RyR release. This notion makes sense since it has been suggested that both Ca2+ overload and reduction of SR threshold for spontaneous Ca2+ release are required for a higher incidence of arrhythmias (3, 9). Further mechanistic studies are needed to assess the synergistic roles of differential signaling pathways in causing arrhythmias in AC5Tg hearts. An alternative explanation may involve potential vagal reflex following isoproterenol injection. It has been well known that EAD generation is slow rate dependent. Meanwhile, DADs may be overdriven by fast rate. Therefore, when the heart rate is dramatically increased shortly after isoproterenol injection, the arrhythmogenic effect may be masked. However, when the strong vagal reflex brings heart rate down, the arrhythmogenic aspects of isoproterenol administration are likely to be revealed.
Our previous studies have shown that the cAMP synthesis ability is significantly enhanced in AC5Tg heart (∼20-fold higher) compared with WT (14). Isoproterenol treatment (chronic infusion for 7 days at a dose of 60 mg·kg−1·day−1) promotes the cAMP synthesis rate by ∼22-fold in WT, and interestingly it can further enhance it by ∼2-fold in AC5Tg hearts (13). Combined with our present study, these data imply that isoproterenol-induced arrhythmias in AC5Tg hearts require both PKA (ICa,L activation) and ROS-CaMKII (RyR2 activation) pathways.
Sympathetic stress and arrhythmias.
The sympathetic nervous system is activated both in response to physiological stress, e.g., exercise, and pathological stress, e.g., heart failure. Severe arrhythmias (VTs/VFs) and sudden cardiac deaths have been linked to sympathetic stimulation in several familial/genetic arrhythmias, such as catecholaminergic polymorphic ventricular tachycardia (CPVT) and long QT syndrome (LQTS) type II. In this study, we found that arrhythmias only occur in AC5Tg mice after isoproterenol injection, and the mechanism resembles that for CPVT. Although no structural changes (e.g., point mutations) seem to be involved, AC5Tg cardiomyocytes exhibited spontaneous RyR Ca2+ releases (Ca2+ waves) and EAD/DAD generation at the single cell level after isoproterenol treatment. PVCs and polymorphic VTs were generated as a result of isoproterenol injection in AC5Tg mice. Therefore, it seems likely that a CPVT phenotype can be also induced by a functional upregulation of multiple Ca2+ handling proteins under sympathetic stress conditions. In addition, elevated psychosocial/mental stress (with higher sympathetic tone) favors the occurrence of cardiac arrhythmia and sudden death (4, 6). Our present study may also provide new insights into its underlying molecular and cellular mechanism(s).
Conclusions.
Our results showed that AC5 overexpression exerts a proarrhythmic effect, by inducing SR Ca2+ overload, increased oxidative stress, spontaneous Ca2+ release/Ca2+i waves, and triggered activities, suggesting a novel genetic model for isoproterenol-induced arrhythmias. Since cardiac arrhythmias are a major cause of morbidity and mortality in patients with heart disease, modification of AC signaling in general, and reduction in AC5, in particular, may provide a novel therapeutic approach for arrhythmias induced by increased β-AR signaling.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL-97979, 5P01-AG-027211, 5R21-HL-097264, 1R01-HL-102472, 5R01-HL-033107, 5T32-HL-069752, 5R01-HL-095888, 5P01-HL-069020, 5R01-HL-091781, R01-HL-106511, R01-HL-093481, and 1R01-HL-119464 and by the National Natural Science Foundation of China Grants 81170597 and 81470510.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Z.Z., G.J.B., H.W., N.F., R.G., X.S., L.Y., and L.-H.X. performed experiments; Z.Z., G.J.B., H.W., N.F., R.G., X.S., L.Y., and L.-H.X. analyzed data; Z.Z., G.J.B., H.W., L.Y., S.F.V., and L.-H.X. interpreted results of experiments; Z.Z., G.J.B., H.W., N.F., R.G., X.S., L.Y., and L.-H.X. prepared figures; Z.Z. and L.-H.X. drafted manuscript; Z.Z., G.J.B., H.W., N.F., R.G., X.S., L.Y., D.E.V., S.F.V., and L.-H.X. approved final version of manuscript; D.E.V., S.F.V., and L.-H.X. edited and revised manuscript; L.-H.X. conception and design of research.
REFERENCES
- 1.Andersson DC, Fauconnier J, Yamada T, Lacampagne A, Zhang SJ, Katz A, Westerblad H. Mitochondrial production of reactive oxygen species contributes to the beta-adrenergic stimulation of mouse cardiomycytes. J Physiol 589: 1791–1801, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bito V, Heinzel FR, Biesmans L, Antoons G, Sipido KR. Crosstalk between L-type Ca2+ channels and the sarcoplasmic reticulum: alterations during cardiac remodelling. Cardiovasc Res 77: 315–324, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during beta-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol 590: 3291–3304, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brodsky MA, Sato DA, Iseri LT, Wolff LJ, Allen BJ. Ventricular tachyarrhythmia associated with psychological stress. The role of the sympathetic nervous system. JAMA 257: 2064–2067, 1987. [PubMed] [Google Scholar]
- 5.Clusin WT. Calcium and cardiac arrhythmias: DADs, EADs, and alternans. Crit Rev Clin Lab Sci 40: 337–375, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Critchley HD, Taggart P, Sutton PM, Holdright DR, Batchvarov V, Hnatkova K, Malik M, Dolan RJ. Mental stress and sudden cardiac death: asymmetric midbrain activity as a linking mechanism. Brain 128: 75–85, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Curtis MJ, Walker MJ. Quantification of arrhythmias using scoring systems: an examination of seven scores in an in vivo model of regional myocardial ischaemia. Cardiovasc Res 22: 656–665, 1988. [DOI] [PubMed] [Google Scholar]
- 8.Darbar D, Roden DM. Future of antiarrhythmic drugs. Curr Opin Cardiol 21: 361–367, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Eisner DA, Kashimura T, O'Neill SC, Venetucci LA, Trafford AW. What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J Mol Cell Cardiol 46: 474–481, 2009. [DOI] [PubMed] [Google Scholar]
- 10.Gao J, Zhang L, Wang Y, Lu B, Cui H, Fu W, Wang H, Yu Y, Yu X. Antiarrhythmic effect of acupuncture pretreatment in rats subjected to simulative global ischemia and reperfusion—involvement of adenylate cyclase, protein kinase A, and L-type Ca2+ channel. J Physiol Sci 58: 389–396, 2008. [DOI] [PubMed] [Google Scholar]
- 11.Guellich A, Gao S, Hong C, Yan L, Wagner TE, Dhar SK, Ghaleh B, Hittinger L, Iwatsubo K, Ishikawa Y, Vatner SF, Vatner DE. Effects of cardiac overexpression of type 6 adenylyl cyclase affects on the response to chronic pressure overload. Am J Physiol Heart Circ Physiol 299: H707–H712, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu CL, Chandra R, Ge H, Pain J, Yan L, Babu G, Depre C, Iwatsubo K, Ishikawa Y, Sadoshima J, Vatner SF, Vatner DE. Adenylyl cyclase type 5 protein expression during cardiac development and stress. Am J Physiol Heart Circ Physiol 297: H1776–H1782, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwatsubo K, Bravo C, Uechi M, Baljinnyam E, Nakamura T, Umemura M, Lai L, Gao S, Yan L, Zhao X, Park M, Qiu H, Okumura S, Iwatsubo M, Vatner DE, Vatner SF, Ishikawa Y. Prevention of heart failure in mice by an antiviral agent that inhibits type 5 cardiac adenylyl cyclase. Am J Physiol Heart Circ Physiol 302: H2622–H2628, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai L, Yan L, Gao S, Hu CL, Ge H, Davidow A, Park M, Bravo C, Iwatsubo K, Ishikawa Y, Auwerx J, Sinclair DA, Vatner SF, Vatner DE. Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of MnSOD via the SIRT1/FoxO3a pathway. Circulation 127: 1692–1701, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nam GB, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation 111: 2727–2733, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okumura S, Takagi G, Kawabe J, Yang G, Lee MC, Hong C, Liu J, Vatner DE, Sadoshima J, Vatner SF, Ishikawa Y. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci USA 100: 9986–9990, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrashevskaya N, Gaume BR, Mihlbachler KA, Dorn GW 2nd, Liggett SB. Bitransgenesis with beta(2)-adrenergic receptors or adenylyl cyclase fails to improve beta(1)-adrenergic receptor cardiomyopathy. Clin Transl Sci 1: 221–227, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan H, Mitchell S, Vainoriene M, Lou Q, Xie LH, Ren S, Goldhaber JI, Wang Y. Gi alpha 1-mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathy. Circulation 116: 596–605, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest 115: 2305–2315, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WY, Clopton P, Hammond HK. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation 114: 388–396, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Timofeyev V, Myers RE, Kim HJ, Woltz RL, Sirish P, Heiserman JP, Li N, Singapuri A, Tang T, Yarov-Yarovoy V, Yamoah EN, Hammond HK, Chiamvimonvat N. Adenylyl cyclase subtype-specific compartmentalization: differential regulation of L-type Ca2+ current in ventricular myocytes. Circ Res 112: 1567–1576, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timofeyev V, Porter CA, Tuteja D, Qiu H, Li N, Tang T, Singapuri A, Han PL, Lopez JE, Hammond HK, Chiamvimonvat N. Disruption of adenylyl cyclase type V does not rescue the phenotype of cardiac-specific overexpression of Galphaq protein-induced cardiomyopathy. Am J Physiol Heart Circ Physiol 299: H1459–H1467, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomaselli GF, Barth AS. Sudden cardio arrest: oxidative stress irritates the heart. Nat Med 16: 648–649, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Vassalle M, Lin CI. Calcium overload and cardiac function. J Biomed Sci 11: 542–565, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Vatner SF, Park M, Yan L, Lee GJ, Lai L, Iwatsubo K, Ishikawa Y, Pessin J, Vatner DE. Adenylyl cyclase type 5 in cardiac disease, metabolism, and aging. Am J Physiol Heart Circ Physiol 305: H1–H8, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res 100: 105–111, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 106: 1288–1293, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res 104: 79–86, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest 115: 556–564, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Yao L, Nagai Y, Fujisawa Y, Miyatake A, Abe Y. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc Res 65: 230–238, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Z, Wen H, Fefelova N, Allen C, Baba A, Matsuda T, Xie LH. Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes: predominant by Ca waves or Ca currents? Am J Physiol Heart Circ Physiol 302: H1636–H1644, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]