

Abstract
The physiological evidence linking the production of superoxide, hydrogen peroxide, and nitric oxide in the renal medullary thick ascending limb of Henle (mTAL) to regulation of medullary blood flow, sodium homeostasis, and long-term control of blood pressure is summarized in this review. Data obtained largely from rats indicate that experimentally induced elevations of either superoxide or hydrogen peroxide in the renal medulla result in reduction of medullary blood flow, enhanced Na+ reabsorption, and hypertension. A shift in the redox balance between nitric oxide and reactive oxygen species (ROS) is found to occur naturally in the Dahl salt-sensitive (SS) rat model, where selective reduction of ROS production in the renal medulla reduces salt-induced hypertension. Excess medullary production of ROS in SS rats emanates from the medullary thick ascending limbs of Henle [from both the mitochondria and membrane NAD(P)H oxidases] in response to increased delivery and reabsorption of excess sodium and water. There is evidence that ROS and perhaps other mediators such as ATP diffuse from the mTAL to surrounding vasa recta capillaries, resulting in medullary ischemia, which thereby contributes to hypertension.
Keywords: superoxide (O2·−), hydrogen peroxide (H2O2), nitric oxide (NO), NAD(P)H oxidase, mTAL, medullary blood flow, kidney, Dahl SS rats
reactive oxygen species (ROS) are chemically reactive molecules derived from oxygen, whose role as mediators of intracellular signaling cascades is well recognized. The following is a brief review of physiological data linking superoxide (O2·−), hydrogen peroxide (H2O2), and nitric oxide (NO) production in the medullary thick ascending limbs of Henle (mTAL) to sodium (Na+) homeostasis, the regulation of medullary blood flow (MBF), and the long-term regulation of arterial blood pressure. MBF to the renal medulla can be importantly controlled by the constriction and dilation of the descending vasa recta (VR) capillaries, which branch from the efferent arterioles of the juxtamedullary glomeruli (49, 147, 149) and whose tone is controlled by the smooth muscle-like pericytes (49, 90, 146–149). The role of NO cross talk from mTAL to surrounding VR in the renal medulla has been reviewed in detail elsewhere (19, 33) and will be discussed here largely with regard to its ROS-related counterregulatory functions. The present review will first summarize data showing that either the excess endogenous production or reduced scavenging of O2·− or H2O2 within the renal medulla results in a decrease in MBF and Na+ excretion, leading to hypertension and renal injury. Second, evidence will be summarized supporting the hypothesis that a high dietary salt intake results in increased luminal flow and delivery of NaCl to the mTAL, thereby signaling via both mechanical forces (stretch and shear) and increased Na+ transport, an increased mitochondrial and membrane NAD(P)H oxidase production of O2·− and H2O2. Third, studies will be reviewed showing that reduction of MBF leads to hypertension and renal injury when either a pharmacologically induced or a naturally occurring imbalance of the normal redox state is present in the renal medulla as is found in the Dahl salt-sensitive (SS) rat.
Role of O2·− and H2O2 in the Regulation of Medullary Blood Flow and Na+ Excretion
Medullary O2·−.
The production and interaction of O2·− and NO are acknowledged to be important determinants of vascular function in a variety of normal and pathological states (25, 70, 77, 89). Evidence that ROS could play an important role in renal dysfunction and injury related to hypertension emerged in the late 1990s (195). The major source of ROS in the kidney is NAD(P)H oxidase (61, 186, 206), but the roles of specific Nox isoforms such as Noxs 1, 2, and 4 and the mechanisms of their regulation have not been well elucidated. As discussed below, Nox2, which is expressed in high abundance in several tissues, has been the most studied. This multisubunit enzyme is composed of the membrane subunits gp91phox and p22phox and the cytosolic subunits p47phox, p67phox, p40phox, and Rac 1 or 2 (151). All of the cytosolic subunits assemble on the membrane upon activation, allowing the enzyme to generate superoxide (O2·−). However, the most abundant isoform in the kidney is recognized to be Nox4. This isoform is unique in that it releases predominantly H2O2 (9, 61, 137), which cannot scavenge nitric oxide (NO) (44). There have been, as yet, no studies to determine the role of Nox4 in a naturally occurring form of hypertension nor are there studies that have evaluated the role of Nox4 in Na+ homeostasis.
Studies focused initially on renal cortical oxidative stress, which was found to accompany hypertension in several experimental models including spontaneously hypertensive (SHR) rats, angiotensin II (ANG II)-induced hypertensive rats, renovascular hypertension, DOCA-salt hypertensive rats, and obesity-related hypertension (194). Additionally, a high dietary salt intake was found to exert a powerful influence upon the rate of renal cortical O2·− generation, as characterized by increased mRNA expression levels of the NAD(P)H subunit, gp91(phox) and p47(phox) (93). Also contributing to elevated ROS generation was the diminished expression of O2·− scavengers CuZn-SOD and (Mn)-SOD despite suppression of the renin-angiotensin system (93, 119, 186).
Intrarenal sources of ROS production were initially characterized in Sprague-Dawley (SD) rats (114) and consomic SS.13BN (206) rats, a strain with the Brown Norway chromosome 13 introgressed into the SS background shown to be relatively salt resistant (34) and commonly used as a genetically inbred control strain for physiological studies (185–187). These studies showed that NAD(P)H oxidase accounted for nearly half of all ROS production in the cortex and medulla, with mitochondria accounting for the remaining half (186, 206). The renal outer medulla exhibited the greatest levels of enzyme activity for O2·− production per gram tissue followed by the renal cortex with the least in the inner white medulla (e.g., papilla).
Evidence that medullary O2·− production could play an important role in Na+ excretion and in hypertension was revealed by studies in which the SOD inhibitor diethyldithiocarbamic acid (DETC) was infused into the renal medullary interstitial space of SD rats, both acutely (206) and chronically (114). In previous studies, we had shown hemodynamic effects (decreased MBF and hypertension) of substances infused directly into the medullary region, effects that were not exhibited when infused at the same dose systemically (108, 127). As shown in Fig. 1, acute medullary infusion of DETC produced a marked decrease (∼50%) in renal MBF (Fig. 1A) and a reduction of Na+, K+, and volume excretion (Fig. 1B) in the absence of changes in mean arterial pressure (MAP) and cortical blood flow (CBF). Conversely, the infusion of the membrane-permeable SOD mimetic tempol increased MBF and Na+ excretion by 34 and 69%, respectively, even in the presence of the NOS inhibitor Nω-nitro-l-arginine methyl ester (l-NAME) (206). It was evident from these responses that endogenously produced O2·− in the renal outer medulla was capable of exerting a tonic regulatory action on renal MBF and Na+ excretion even in the presence of ubiquitous scavenging systems.
Fig. 1.

Effect of renal medullary interstitial infusion (R.I.) of the SOD inhibitor diethyldithiocarbamic acid (DETC) on mean arterial pressure (MAP) and renal cortical and medullary laser-Doppler flow (LDF; A) and urine flow and sodium and potassium excretion (B). *P < 0.05 compared with control. Reprinted with permission from Zou et al. (206).
The consequences of sustained elevations of medullary interstitial O2·− were then studied by chronic infusion of DETC directly into the medullary interstitium of the single remaining kidney of an SD rat for a period of 5 days (114). A seen in Fig. 2, this resulted in a large reduction in MBF as determined in unanesthetized rats using implanted optical fibers and laser-Doppler flowmetry techniques. Importantly, MAP rose nearly 20 mmHg over the 5 days of DETC infusion, which was associated with nearly an eightfold increase in medullary interstitial O2·− levels determined by microdialysis (Eth/DHE ratios) at the end of the study. Notably, similar to acute experiments with DETC, no changes in cortical interstitial O2·− levels were observed following 5 days of medullary DETC infusion. Intravenous infusion of the same amounts of DETC had no effects on any of these variables, indicating that the hypertension was of primary renal medullary origin. It was concluded from this study that by reducing O2·− scavenging, the resulting sustained elevations of O2·− within the renal medulla alone could produce a sustained reduction of MBF and lead to hypertension.
Fig. 2.

Effects of chronic infusion of DETC (7.5 mg·kg−1·day, for 5 days; ●) or saline (○) into the renal medullary interstitium (r.i.) on renal medullary blood flow (n = 5–6), renal cortical blood flow, and MAP. Reprinted with permission from Makino et al. (114).
Medullary H2O2.
H2O2 has received little attention as a potentially important paracrine and/or autocrine molecule within the kidney compared with O2·−. However, H2O2 is highly reactive and relatively stable in aqueous solutions and under physiological conditions catalase can convert it into water and molecular oxygen without the cost of reduction equivalents (3, 10, 16). It has been generally thought that H2O2 could easily diffuse from the cells into the interstitial space in a manner similar to NO (100) and with a greater radius of diffusion than O2·− would serve more effectively as a paracrine signaling molecule. Although this is generally true of small and nonpolar molecules that easily cross the hydrophobic membrane lipid bilayer by simple diffusion, H2O2 has a permanent dipole moment very similar to water, and passive diffusion is similarly limited (17, 116, 169, 175). Recently, direct evidence was obtained in mammalian cells showing that aquaporin-3 (AQP3) and -8 (AQP8) but not aquaporin-1 (AQP1) can facilitate the uptake of H2O2 (125). This was demonstrated by using a highly H2O2-selective small-molecule fluorescent indicator, peroxy yellow 1 methyl ester (PY1-Me) in HEK-293 and HeLa cells. The potential physiological consequences of AQP3-regulated H2O2 permeability was determined by studies demonstrating the ability of endogenous AQP3 to amplify or diminish downstream native signaling pathways such as growth factor stimulation (125). The molecular and physiological regulation of channels able to conduct H2O2 needs to be explored in the epithelial cells of the medullary tubules since there is evidence that H2O2 may serve as a paracrine signaling molecule in the renal medulla.
A series of studies has demonstrated that H2O2, independently of O2·−, is an important participant in the regulation of MBF and Na+ excretion. A specific role for H2O2 was initially suggested by observations that the antioxidant tempol, a membrane-permeable SOD mimetic, was unable to prevent the development of hypertension induced by the chronic medullary infusion of the SOD inhibitor DETC (114). The failure of tempol to prevent hypertension in these circumstances can be explained by studies of Krishna et al. (97, 98), who showed that although tempol dismutates two O2·− molecules enabling it to act as a SOD mimetic, at high concentrations it also reacts with protonated superoxide (OOH) to produce H2O2 and oxoammonium. Evidence that this reaction was relevant to our observations was obtained when it was found that coinfusion of catalase together with tempol into the medullary interstitial space prevented the DETC-induced hypertension (115), indicating an important role for H2O2.
Studies then demonstrated that acute infusions of H2O2 into the renal medullary interstitial space of SD rats reduced MBF in a dose-related manner, responses which were inhibited by catalase (29). A dose that produced a doubling of medullary interstitial H2O2 concentrations (from 116 to 211 nM) as determined by microdialysis, reduced MBF by 31% and resulted in a marked reduction of urine flow rate (50%) and Na+ excretion (47%). The basal H2O2 concentrations of the medullary interstitial fluid (100–110 nM) of SD rats were determined to be twice as high as that found in the cortical dialysate (∼ 56 nM) (29). These basal levels of endogenously produced H2O2 within the renal medulla were found to exert a moderate tonic constrictor effect on the medullary circulation as shown by a 10% increase in MBF with medullary catalase administration associated with a reduction of medullary H2O2 levels from 116 to 43 nM. This response was also accompanied by an increase in Na+ and water excretion of 16 and 12%, respectively (29).
The consequences of chronic elevations of medullary H2O2 were determined by infusing H2O2 directly into the renal medulla of a single remaining kidney of SD rats in amounts that produced a threefold elevation of medullary interstitial H2O2 concentrations. This resulted in sustained hypertension (115). Of relevance, the medullary H2O2 concentrations achieved in these infusion studies were quite similar to those levels measured in studies in which we chronically infused DETC and tempol into the renal medulla (114). Medullary H2O2 elevations produce a salt-sensitive form of hypertension. H2O2 chronically infused in low concentrations into the medullary interstitial space of the single remaining kidney of salt-insensitive SS.13BN rats fed a 0.4% salt diet did not result in hypertension until the rats were switched to a high-salt diet (4.0% NaCl) (185). Medullary interstitial concentration of H2O2 levels determined at the end of the experiment when pressure had increased nearly 30 mmHg were 83 ± 11 nM compared with rats infused with only saline (44 nM). Together, these acute and chronic studies demonstrated that H2O2 synthesized in the renal medulla can importantly influence MBF and Na+ excretion and that moderate elevations of H2O2 result in hypertension. This led to more detailed studies to determine whether H2O2 could modulate pressure-natriuresis.
H2O2 and NO reciprocally modulate pressure-natriuresis.
The critical role of renal pressure-natriuresis in the long-term control of arterial pressure was first recognized by Arthur Guyton (68), and over the past 25 years much has been learned about the mechanisms underlying these responses (32). The prevailing hypothesis to explain the increase in Na+ and water excretion with elevations of renal perfusion pressure is based on observations that MBF is poorly autoregulated compared with cortical flow and GFR, a response that is exaggerated with volume expansion (30). As a consequence of the increase in MBF, a corresponding increase in VR capillary pressure occurs, resulting in a rise in interstitial hydrostatic fluid pressure throughout the encapsulated kidney (55). This increase in interstitial pressure in turn signals mechanisms which inhibit Na+ reabsorption by the proximal tubules. This response is at least partially mediated by an increase in 20-hydroxyeicosatetraenoic acid (20-HETE), which in turn inhibits Na+-K+-ATPase activity and an internalization of apical Na+/H+ exchangers, resulting in a greater delivery of NaCl to the mTAL and distal tubules (196, 202). Reductions in Na+ reabsorption in deeper nephron segments also occur, which are likely to be a result of washout of the medullary solute gradient and reduced passive reabsorption of sodium (30). Since this overall mechanism is triggered by the rise in MBF, much effort has been made to understand the mechanisms that regulate flow to the medulla (146).
Given the ability of elevations in H2O2 to reduce MBF and Na+ excretion and with NO having the opposite effect, the impact of increased renal perfusion pressure (RPP) upon H2O2 and NO production was determined in SD rats. H2O2 production in the renal medulla was found to be highly sensitive to acute increases in RPP. As pressures were increased in ∼30-mmHg steps between 80 and 150 mmHg, significant increases in medullary interstitial H2O2 concentrations were observed along with parallel increases in urinary excretion of H2O2 (Fig. 3A) (84). Since increases in medullary H2O2 reduce Na+ excretion, these observations indicate that H2O2 plays a modulatory role in reducing the steepness of the pressure-natriuresis relationship, serving to dampen the pressure-natriuresis response. Also, as was recognized (49, 52, 65, 80, 113), medullary NOx (NO2/NO3) and urinary excretion of NO also increased with an increase in RPP, as demonstrated in Fig. 3B. It therefore appears that both H2O2 and NO serve as important modulators of pressure-natriuresis. A shift in the balance of the production or scavenging of either of these reactive compounds would therefore be expected to alter the sensitivity (slope and intercept) of the pressure-natriuresis response. It is clear from these studies that a critical redox balance must be achieved between ROS and NO production in the renal medulla to retain a normal pressure-natriuresis relationship. If the balance is shifted toward a greater production of ROS, as observed in SS rats discussed below, a reduction in the slope of the pressure-natriuresis response would be expected with increased salt sensitivity, as has been observed (159, 161).
Fig. 3.

Responses to changes in renal perfusion pressure (RPP). Left: changes in excretion of urinary sodium (UNaV), medullary interstitial H2O2, and urinary excretion of H2O2 (UH2O2V) following a change in RPP. Right: changes in excretion of UNaV, medullary interstitial NO2/NO3 (NOx), and urinary excretion of NO2/NO3 (UNOxV) following a change in RPP. Values are means ± SE. *Significant change from the lowest pressure (P < 0.05). **Significant change from both the lowest and the intermediate pressure (P < 0.05). Regraphed from results presented in Jin et al. (84).
Some investigators have ascribed a more fundamental role to NO in pressure-natriuresis and propose it as the cause of this phenomena (113) since NO has been shown to reduce Na+ reabsorption (56, 106). Although the natriuretic nature of NO is undisputed, it has been found that a potent pressure-natriuresis response occurs in rats even when NOS is inhibited (67). In SD rats infused either acutely or chronically with l-NAME, although the pressure-natriuresis relationship is shifted to a higher operating point, the rise in RPP is accompanied by increased Na+ excretion (67). These observations indicate that NO is an important modulator of pressure-natriuresis, but not the fundamental mechanism driving the relationship between RPP and Na+ excretion.
Consequences of Increased mTAL Luminal Flow and Na+ Delivery on Epithelial Cell O2·−, H2O2, and NO Production
O2·− and NO production.
The effects of increased luminal flow and Na+ delivery on ROS production in the mTAL is important to understand since the tubules appear to be the major source of NAD(P)H oxidase. A large proportion (25–30%) of the filtered Na+ is absorbed by the mTAL, a function that is critical for body Na+ homeostasis and survival. The active transport of Na+ in the mTAL results in high levels of mitochondrial oxygen utilization further enabled by levels of mitochondrial cell density, which are among the highest in the body, including the heart (7, 20, 45, 94). Since ROS levels within mitochondria are 5- to 10-fold higher than other cytosolic and nuclear compartments, mTAL mitochondria would be expected to have developed strong systems to protect from the highly damaging effects of oxidant stress. However, this appears not to be the case and this vulnerability is made even more challenging due to the limited supply of blood flow to the outer medulla relative to the high oxygen needs of the mTAL. The mTAL are nourished only through the descending VR that are capable of providing an amount of oxygen barely exceeding basal needs. With the outer medulla operating on the brink of hypoxia (18, 50), either a reduction of blood supply or an increased metabolic load can generate greater amounts of ROS production in the mTAL. The metabolic work of the mTAL is increased as more Na+ is delivered to the loop of Henle, an event that occurs with a rise in renal perfusion pressure or a greater filtration of NaCl as seen with high-salt feeding, hypertension, and the early stages of diabetes (134, 154, 196, 202). It is also relevant that increased delivery of NaCl to the loop of Henle driven by increased filtration is also the basis of the well-characterized tubuloglomerular feedback (TGF) responses initiated from the macula densa cells situated at the mTAL transition into the distal tubules (73).
Tubular microperfusion studies have determined that increased luminal flow within the physiological range to the mTAL provides powerful stimuli for ROS and NO production (1, 22, 32, 56, 74). These responses are mediated by mechanical factors of stretch, pressure, shear stress, and ion delivery. Although all of these increase in parallel when flow is increased, various elements of these factors mediate ROS and NO production in the mTAL in different ways (1, 21, 74, 75). It has been estimated that nearly 50% of the increase in mTAL O2·− production in response to normal increases in luminal flow can be attributed to mechanical factors and, most specifically, cellular stretch, since increases in transmural pressure or shear stress do not alone stimulate O2·− production (57, 74). Physiological increases in mTAL luminal flow (5–20 nl/min) stimulate O2·− production (e.g., rate of change in Eth/DHE) even when luminal Na+ concentration ([Na+]) is adjusted to remain unchanged as flow rate is increased (Fig. 4A) (1). However, increases in luminal [Na+] alone can also markedly affect O2·− production as seen when [Na+] was increased from 60 to 149 mM while maintaining a constant luminal flow rate (15 nl/min) (Fig. 4B) (1). This was not an osmotic effect since equivalent increases in [choline Cl] had no effect on O2·− production. When both luminal flow and [Na+] change together as is normally the case, the relationship becomes more complex as we have described (1). With a luminal [Na+] of 149 mM Na+, mTAL O2·− production was large even at very low luminal flow rates (5 nl/min) and was sustained when luminal flow rates were increased from 5 to 20 nl/min. At lower levels of luminal flow, with high concentrations of Na+, O2·− production was strongly dependent upon mTAL Na+ transport and was abolished by ouabain (1) as shown in Fig. 4C and by furosemide pretreatment (not shown) (74). When both luminal flow rates and [Na+] were high (20 nl/min and 146 mmol, respectively), ouabain significantly reduced the overall amount of O2·− being produced but the rate of increase (slope) was unchanged, indicating that mechanical factors related to the high flow rates continued to drive O2·− production. At a lower concentration of luminal Na+ (60 mM), there was no detectable rise in O2·− production throughout the period of the 5 nl/min perfusion, but a marked increase in O2·− production was measured when luminal perfusion was increased to 20 nl/min. This rate of increase in O2·− production paralleled that observed at high luminal [Na+] and was minimally influenced by ouabain treatment. Together, the results of these studies indicate that [Na+] and luminal flow are normally both important determinants of O2·− production in the mTAL, and attention must be paid to both of these variables to accurately predict the overall responses.
Fig. 4.

Response of superoxide (O2·−) production as measured by the change in the ratio of ethidium to dihydroethidium (Eth/DHE) to an increase in flow rate from 5 to 20 nl/min (A and C). Also shown is the response of both O2·− and nitric oxide (NO) production to increased luminal Na+ concentration ([Na+]) with luminal flow maintained constant at 15 nl/min (B and D). Regraphed from results presented by Abe et al. (1).
NO production in the mTAL is also stimulated by increases in luminal flow as demonstrated in several laboratories (1, 22, 144). However, the extent to which mechanical forces vs. luminal [Na+] participates in this process is unsettled. Garvin and associates (22, 144) determined that the NO responses were mediated by apical wall shear stress rather than pressure as signaled though a cGMP/PKG-dependent pathway. However, they also found that ion-free perfusates made no difference to the NO responses and concluded that [Na+] delivery or transport was not an important factor in NO production. In contrast, studies by Abe and colleagues (1), as shown in Fig. 4D, found that increases in luminal [Na+] act to suppress mTAL luminal NO production. At low concentrations of luminal Na+ (60 mM) when O2·− production was low, the rate of intracellular levels of NO production was high. When luminal [Na+] was increased to 149 mM, in the absence of any change in luminal flow rates (15 nl/min), there was a reduction in the rate of mTAL NO production. O2·− production became quite elevated at luminal [Na+] of 149 mM, which probably accounts for the associated reduction in NO production. These responses are consistent with the known interactions of O2·− and NO (79) and consistent with observations that decreases in O2·− concentrations with the SOD mimetic tempol increased NO production in the mTAL (143).
Further studies will be needed to resolve these observed differences between laboratories and to elucidate the signaling pathways involved in these responses. For example, reduction of O2·− in the presence of high levels of NO may not occur simply by scavenging since there is also formation of ONOO−. It has also been estimated that ∼20% of the net O2·− production is NO insensitive and that part of flow-induced O2·− may be generated in a compartment different from flow-stimulated NO (74).
H2O2 production in mTAL.
Given the lack of robust and selective fluorescent indicators for determining H2O2 changes within living cells, there has been limited knowledge about mTAL H2O2 production. Probes used for fluorescence imaging to a greater or lesser extent have limitations of selectivity or sensitivity. The most commonly used oxidant-sensitive dye able to penetrate cells is dihydrodichlorofluorescin (DCFH2) which is oxidized to DCF. This probe, however, does not react directly with H2O2 and is oxidized by many radical species or metal-dependent processes and the signal intensity can be changed without increases or decreases in H2O2 production. The limitations to the use of DCF have been critically analyzed and reviewed by others (13, 44, 88, 112, 184, 192).
Newer generation “non-redox” fluorescent probes are now being developed which exhibit greater stability and cellular localization in living cells (27, 41–43). One such probe is mitochondria peroxy yellow 1 (Mito-PY1) (41), which colocalizes with Mitotracker deep red and was used to assess H2O2 levels in isolated mTAL epithelial cells (142). This probe was used to determine H2O2 responses to physiological increases in luminal flow in freshly isolated, microperfused mTAL of SD rats (142). As shown in Fig. 5, as luminal perfusion of the mTAL was increased from 5 to 20 nl/min (similar to experiments shown in Fig. 4), mitochondrial H2O2 production (e.g., Mito-PY1 fluorescence) was significantly increased. These responses were abolished by inhibitors of the electron transport chain rotenone (complex 1) and antimycin A (complex 3), as shown in Fig. 5A, and by PEG-catalase (not shown). The mitochondrial H2O2 responses were not dependent on NAD(P)H oxidase since apocynin pretreatment did not alter the observed increases in mitochondrial H2O2 in response to increased luminal flow (142). The stimulus for increased mitochondria H2O2 production associated with increased luminal flow was dependent upon Na+ transport as both the Na+-K+-2Cl− cotransporter (NKCC2) inhibitor furosemide and Na+-K+-ATPase inhibitor ouabain greatly attenuated the mitochondrial H2O2 responses (Fig. 5B). Importantly, mitochondrial H2O2 was also markedly stimulated by increases in mTAL luminal [Na+] at a fixed luminal flow rate, which became especially prominent as luminal [Na+] was increased from 60 to 149 mM (142).
Fig. 5.
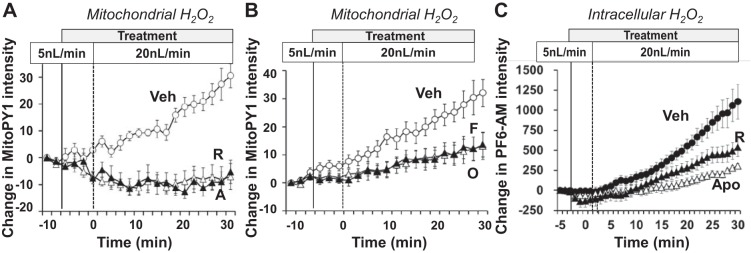
Isolated medullary thick ascending limb of Henle (mTAL) microperfusion studies in Sprague-Dawley rats under physiological conditions of increased luminal flow rate (from 5 to 20 nl/min) showing stimulation of mitochondrial H2O2 (Mito-PY1; A and B) and inhibitors of complex 1 [rotenone (R; 10 μM) and antimycin A (A; 1 μM; A)]. Furosemide (F; 100 μM) and ouabain (O; 4 mM) are shown in B. C: effects of mitochondrial H2O2 production upon whole cell H2O2 responses. The elimination of the mitochondrial component with rotenone decreased the response to increased luminal flow. In C, when apocynin (Apo; 1 mM) was given to eliminate NAD(P)H oxidase reactive oxygen species production, there was only a minimal increase in H2O2 to increased luminal flow. Regraphed from results presented in Ohsaki et al. (142).
The effects of mitochondrial H2O2 production upon whole cell (intracellular) H2O2 responses were revealed using another novel and more sensitive fluorescent probe (PF6-AM), as shown in Fig. 5C. This probe enables quantification of changes in H2O2 within the whole cell by localizing largely in the cytoplasm and in the nucleus, although entry into the mitochondria or other subcellular organelles such as endoplasmic reticulum cannot be excluded (41, 142). Large increases in PF6-AM signals were observed in response to increases in mTAL luminal flow. In the isolated mTAL pretreated with rotenone (to eliminate the mitochondrial component), a 65% reduction of the PF6-AM response to increased luminal flow was observed. That is, increased production of mitochondrial H2O2 was required to achieve maximum increases in intracellular H2O2. In studies where the mitochondria were the only probable source for changes in total cellular H2O2, as was the case in apocynin-pretreated cells, the PF6-AM signal rose only minimally in response to increased mTAL luminal perfusion (Fig. 5C).
From these observations, we have proposed that mitochondria are a key component of the signaling pathway for the overall production of cellular H2O2. Given that the membrane NAD(P)H oxidase production of ROS increases when stimulated by an increase in luminal flow or Na+ flux in the mTAL, the mitochondria appear to provide a “feed-forward” signal to membrane oxidases, resulting in an overall increase in intracellular H2O2 (142). Intercompartmental cellular signaling by ROS has been the subject of considerable interest and debate. It has been observed that vascular endothelial cells exhibit dual feed-forward loops, one from cell membrane Noxs that stimulates mitochondrial ROS, and the other from mitochondrial ROS stimulating the membrane Noxs (15, 46, 171). However, as seen in Fig. 5C, H2O2 produced from mTAL membrane NAD(P)H oxidase does not feed-forward to stimulate mitochondrial H2O2 production, which occurs even in the presence of apocynin.
Figure 6 summarizes our current working hypothesis of mitochondria-mediated signaling of membrane NAD(P)H oxidases. Illustrated is the Na+ flux-dependent stimulation of mitochondrial H2O2 production which feeds forward to stimulate the membrane Nox2 and/or Nox4. As suggested in the schematic, this could lead to a vicious cycle whereby an increase in salt diet with increased delivery of NaCl to the mTAL could produce ever increasing amounts of intracellular H2O2, leading to greater interstitial levels of H2O2. These mechanisms could be important not only in cellular energetics and renal injury, but also in the overall regulation of medullary VR blood flow, which would be consistent with what is found in Dahl salt-sensitive rats fed a high-salt diet (32), as discussed in the following sections.
Fig. 6.

Schematic of mitochondria-mediated signaling of membrane NAD(P)H oxidases.
The precise signaling pathways and mechanism(s) whereby increased luminal flow and Na+ transport in the mTAL to signal the mitochondria to produce greater amounts of ROS are unclear. Based on what is known of the kinetics of the NKCC2 cotransporter and data observed in perfused rat mTAL (66, 72, 176), it has been puzzling as to how changes in tubular [Na+] between 60 and 149 mM could affect transport rates of Na+ and thereby enhance mitochondrial H2O2 and cellular O2·− production as we have observed (142). The three NKCC2 murine isoforms when expressed in Xenopus oocytes have been found to exhibit EC50 values for Na+ ranging only from 3 to 21 mM (153). However, increases in mTAL intracellular H2O2 were prevented by furosemide inhibition of the NKCC transporter, which is difficult to reconcile with such low Km values. In vivo luminal concentrations range from 60 to 160 mM (66), which are above the purported Km saturation levels of these key transporters. The net transport rate of mTAL Na+ is exceedingly high, ranging from 87 to 870 pmol·mm−1·min−1 (20), which provides the mTAL segment with its remarkable capacity to “buffer” NaCl loads (66). The isolated mTAL even ceases to reabsorb NaCl when the luminal perfusate contains only 50 mmol/l Na+ and/or if the flow rate is very low (20). The answer to this enigma may reside in studies by Lee and McDonough (103), which discovered that an increased luminal flow and Na+ delivery resulted in rapid insertion of Na+/H+ and Na+-phosphate cotransporters in the proximal tubules (124) and NCC transporters in the distal collecting ducts (103), thereby enabling a wide transport capacity. Similar insertion of Na+ transporters could occur in the mTAL, which needs to be explored.
The processes involved in mTAL ROS production are undoubtedly complex and appear to be activated by several intracellular pathways. Recent studies have clarified one of these pathways, which involves H+ transport in the mTAL that when activated stimulates a Nox-mediated production of O2·− independently of active Na+ transport (85, 139). By utilizing SS HV1−/− mutant rats developed using zinc finger protease techniques, it was shown that the voltage-gated proton channel HV1 (37) is expressed in the mTAL membrane and contributes to O2·− production in the SS rat. HV1 was found to respond to H+ efflux (85) and to changes in intracellular Na+ (directly or indirectly). As it is believed that an increased mTAL Na+ reabsorption occurs with high-salt feeding (107, 164), it is proposed that this would result in depolarization and acidification of the mTAL, thereby promoting the opening of HV1 channels and contributing to O2·− production and oxidative stress in the outer medulla. Consistent with this hypothesis, when fed a high-salt diet, SS HV1−/− rats exhibited reduced hypertension and outer medullary renal injury and oxidative stress compared with wild-type SS rats (85).
Medullary O2·−, NO, and H2O2 as Determinants of Blood Pressure Salt Sensitivity and Renal Injury
Enhanced sensitivity of blood pressure to salt intake is present in nearly half of Americans who are afflicted with hypertension (163), including ∼75% of African American hypertensive patients (5, 177, 193), who are at substantially greater risk of developing end-stage renal disease (135). Human studies indicate that a loss of redox homeostasis and formation of excess free radicals in the kidney contribute to reduced Na+ excretory function and to the activation of proinflammatory and profibrotic pathways that exacerbate hypertension and lead to end-stage renal disease (31, 137).
Dahl SS rat model.
The SS rat, which mimics many aspects of progressive salt-sensitive human hypertension, has provided key insights and been used to identify a number of mechanisms and pathways which are believed to contribute to this form of hypertension. These include oxidative stress in the kidney (32, 95, 119, 139, 160 185,186), brain (54, 78, 82), and vasculature (48, 96, 109). Importantly, among these mechanisms is also the infiltration of immune cells into the kidney and related inflammatory responses that contribute significantly to ROS production, renal dysfunction, and hypertension (38–40, 121, 123).
SS rats exhibit a reduction of the pressure-natriuresis relationship (159, 162) even before high-salt feeding as a consequence of enhanced resorption of sodium and chloride in the mTAL (92, 161). After SS rats are fed a high-salt diet for 2 wk, they develop hypertension that cannot be reversed by returning them to a low-salt diet (159), which is associated with a reduced GFR (159, 162), which together with glomerular and fibrotic injury shift the relationship between Na+ excretion and renal perfusion pressure to yet higher levels. One of the early responses in SS rats that occurs following initiation of a high-salt diet is the reduction of MBF. As shown in Fig. 7, by day 3 of high salt, MBF in the SS rat was significantly reduced by nearly 30% but not in the salt-resistant control strain (125), indicting that medullary ischemia is a leading event contributing to tubular injury in the outer medulla. Although it has been recognized that GFR is reduced in SS rats fed a high-salt diet, until recently it has been unknown whether an early reduction of GFR was importantly involved in the initiation of the hypertension or was a consequence of the hypertension. This question has recently been resolved by a study utilizing novel techniques to determine the progression of phenotypic changes that occur in SS rats when switched from a diet containing 0.4% NaCl to that of 4.0% (35), as shown in Fig. 8. MAP determined by telemetry is seen to rise quickly during the first week, followed by a progressive rise to 160–170 mmHg by day 21 of high salt. Sequential changes in GFR were determined in these rats in the conscious state using a miniaturized device mounted on the back of the rat (165–167) enabling the determination of disappearance curves of FITC-sinistrin measured by transcutaneous excitation and real-time detection of emitted light through the skin of the dye injected from an implanted femoral venous catheter. These studies show that GFR was not significantly reduced in the SS rats until day 14 and then fell to 28% of control values by day 21 of the high-salt diet. In contrast to GFR, urine albumin excretion was clearly increased by day 7 of the high-salt diet preceding the reduction of GFR.
Fig. 7.

Effects of a high-salt diet (4%) on the changes in medullary blood flow (MBF; top), cortical blood flow (CBF; middle), and MAP (bottom) in unanesthetized Dahl S rats (left) and Dahl R rats (right). *Significant difference from control days (P < 0.05). Reprinted with permission from Miyata et al. (125).
Fig. 8.
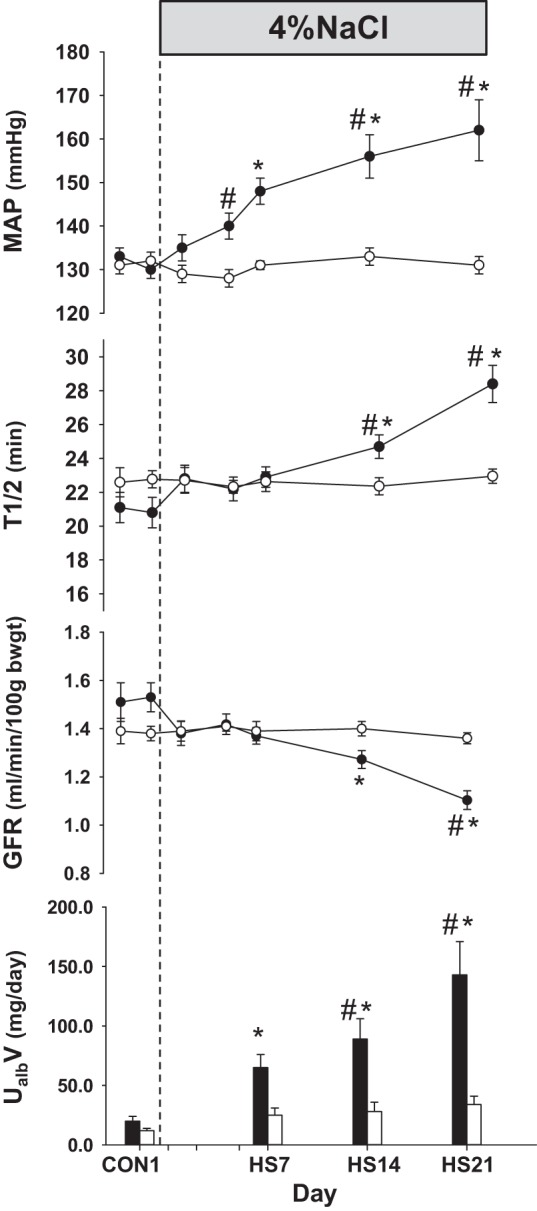
Comparison of the time control group of Dahl salt-sensitive (SS) rats (○) maintained on 0.4% salt diet with a group of SS rats fed with a high-salt diet of 4.0% (HS) for 21 days after control measurements (●). MAP, FITC-sinistrin elimination half-life (t1/2), glomerular filtration rate (GFR), and urinary excretion of albumin (UalbV) are summarized. Values are means ± SE. *P < 0.05 within-group difference from control days. #P < 0.05 between-group differences. Reprinted with permission from Cowley et al. (35).
These functional studies are consistent with recent evidence indicating that ultrastructural abnormalities such as thickening of the endoplasmic reticulum occur in the mTAL of SS rats before the development of observable histological injury (71). Additionally, we have recently found a greater increase in the number of proliferative mTAL cells in SS rats fed a high-salt diet (197). Together, these studies indicate that early structural changes also contribute to the subsequent functional changes and perhaps the alteration in the redox state of the mTAL, leading to the development of hypertension in SS rats. The extent to which pathways identified with oxidative stress in the renal cortex are responsible for the progressive reduction of GFR in SS rats remains unclear and is currently being explored. Related to this is evidence that epithelial Na channel (ENaC)-mediated sodium absorption in the cortical collecting ducts is enhanced and contributes to hypertension in SS rats (152). Inhibition of ENaC by chronic administration of benzamil significantly reduced salt-induced hypertension in SS rats. It was also found that mRNA and protein expression of ENaC subunits was significantly increased in the renal cortex of the SS rats fed a high-salt diet (87, 152). Since it was previously reported that ROS might be involved in the control of ENaC (47, 81, 190), these pathways may together act to amplify the development of salt-sensitive hypertension in SS rats. Further studies will be required to link ROS production with these tubular events in the cortex of SS rats.
Although the focus of this review is on ROS, it is important to keep in mind that the early reduction in MBF appears to be largely a consequence of the imbalance of NO and ROS found in the renal medulla of the SS rat and is exaggerated by a high-salt diet. Levels of medullary tissue NOS activity (187), and mRNA and protein levels of NOS I, II, and III (201) are all significantly lower in the outer medulla of SS rats compared with several salt-resistant rat strains. The importance of reduced NO levels in the medulla of SS rats upon salt sensitivity was clearly evident when it was found that chronic medullary infusion of l-arginine, the substrate for NOS and NO production, when infused into the single remaining kidney of SS rats completely eliminated the reduction in MBF and abolished salt-induced hypertension (125). This is consistent with the hypersensitization to vasoconstrictors discussed later in this review. The same appears to be the case in SHR compared with WKY rats in that untreated SHR exhibit significantly lower levels of endothelial and inducible NOS and a large accumulation of nitrotyrosine (2, 99, 204).
Relative concentrations and sources of medullary O2·− and H2O2 production.
Contributing importantly to the altered redox state of the renal medulla in SS rats are the elevated levels of O2·− and H2O2 production (115, 185–187) compared with salt-resistant control strains. Similar to these observations in which O2·− and H2O2 levels were assessed using microdialysis approaches in live animals, we have recently utilized another method based on the use of biosensors and assessed H2O2 levels in freshly isolated, perfused kidneys (150). These studies not only confirmed that H2O2 levels were increased in SS rats fed a high-salt diet compared with low salt but also that medullary H2O2 levels were significantly higher than in the cortex.
There are a number of factors that contribute to the elevated levels of medullary ROS. Renal medullary tissue of SS rats exhibits O2·− and H2O2 concentrations nearly twice that found in salt-resistant consomic SS.13BN rats even in a prehypertensive state while receiving a 0.4% salt diet (186, 187). SS rats excrete greater amounts of urinary 8-isoprostane levels than SS.13BN rats (133). Isolated mTAL of SS rats exhibit greater production of O2·− than mTAL of SS.13BN rats (133). The activity of several ROS-scavenging enzymes are also reduced in the outer medulla of SS rats including superoxide dismutase, glutathione peroxidase, and catalase (118, 187). NOS uncoupling occurs in the renal outer medulla after 4–6 wk of high-salt feeding as evidenced by nearly a 30% decrease in BH4 with little change of BH2 concentrations, and inhibition of O2·− production in response to the administration of the NOS inhibitor l-NAME (187).
As cited earlier in this review, a yet undetermined amount of the elevation of renal medullary ROS is related to the extent of infiltration of immune cells (121). Increased renal infiltration of T lymphocytes occurs in SS rats, which has been found to contribute to the development of the later phase of hypertension in SS rats (38). Treatment of SS rats with the immunosuppressive agent mycophenolate mofetil (MMF) was found to reduce T cell infiltration and attenuate salt-induced hypertension. However, the mechanism(s) whereby these infiltrated T cells contributes to hypertension remains unclear. It was further observed that intrarenal ANG II was inappropriately elevated in the kidneys of the SS rats fed high salt. The increased intrarenal ANG II was reduced in SS rats treated with MMF during the high-salt period. These data indicated that infiltrating immune cells participate in the production of intrarenal ANG II. The existence of a robust tubular production of ANG II of physiological importance has been established (136). We have found that mTAL of SS rats when stimulated with ANG II produce greater ROS than mTAL of salt-insensitive consomic SS.13BN rats (133). However, the relationships between ANG II and infiltrating T cells remain to be clarified. It is well recognized that ANG II can stimulate excess production of ROS in many cell types (101). The presence of ANG II in infiltrating cells of kidneys of hypertensive rats has been found using immunohistochemical and immunoblotting techniques (155, 157, 158), although it is unclear whether this ANG II is actually produced intracellularly by T cells or merely sequestered by T cells. It is therefore possible that T cells could be one of the sources of the elevations of intrarenal ANG II in the SS rat or conversely that elevations of ANG II from other intrarenal sources stimulate both ROS and ANG II production in T cells. Together, these studies indicate that ROS production from T cells interacting with the intrarenal renin-angiotensin system could be one of the many contributors to increased oxidative stress in the kidneys of SS rats.
Both membrane NAD(P)H oxidase (Noxs) and the mitochondria are important sources of O2·− production in both vascular and renal cells under physiological conditions (185). Medullary O2·− production in tissue homogenates is greater in SS rats, and the NAD(P)H oxidase inhibitor diphenylene iodonium preferentially reduced SS levels to those found in the salt-resistant control SS.13BN rats. Dinitrophenol, a mitochondrial uncoupler, eliminated the remaining O2·− production, indicating that Noxs and mitochondria accounted for nearly all of the tissue production in both strains. Inhibition of xanthine oxidase, NOS, and cyclooxygenase (COX) failed to lower tissue O2·− production, indicating that Noxs and the mitochondria are the major sources of ROS production in the outer medulla. Consistent with these observations, both total NAD(P)H oxidase activity and protein expression of the p22phox, p47phox, and p67phox Nox subunits are significantly higher in the outer medulla of SS compared with control SS.13BN rats (51,186).
Finally, yet other mechanisms that could contribute to enhanced ROS production in SS rats remain to be explored. For example, the COX-2 enzyme pathway has been found to be upregulated in response to high-salt intake apparently via an NF-κB pathway, resulting in increased synthesis of PGE2 (26, 69, 105, 122, 198, 200, 205, 207). Specific inhibition of COX-2 in the renal medulla acutely reduces sodium excretion and when inhibited chronically produces salt-sensitive hypertension in SD rats (104, 105, 122, 183, 199, 207). This is not the case with COX-1, which is constitutively expressed in the medullary collecting ducts and interstitial cells and is not altered by a salt diet (200). Studies which have utilized tissue homogenates of the renal medulla of SS rats found that neither COX-1 nor -2 could account for the excess ROS production compared with salt-insensitive SS.13BN rats. The COX inhibitor meclofenamate failed to lower ROS production in the renal medullary tissue of either SS or SS.13BN rats (186). There is at this time little evidence that elevated PGE2 contributes to the elevated levels of ROS in the medulla of SS rats.
Overall, the excess production of medullary O2·− in the outer medulla of SS rats appears to be explained by excess expression and activity of the Noxs until the final stages of the hypertension when NOS uncoupling contributes importantly to ROS production. It is the imbalance between both reduced NO and elevated O2·− (and H2O2) production in SS rats that appears to tilt the balance toward a state of oxidative stress even before a switch to a high-salt diet, which accelerates this process and leads to the rapid rise in arterial pressure and renal injury (32, 130, 133, 139).
Contribution of medullary O2·− and H2O2 to hypertension in the SS rat.
The contribution of excess ROS production in the renal medulla to hypertension has been investigated in a series of studies in which inhibitors of either Nox or H2O2 were infused chronically into the renal medulla of a single remaining kidney of SS rats. As summarized in Fig. 9, hypertension of SS rats was reduced by ∼50% by the infusion of apocynin into the medullary interstitium to inhibit Nox activity which was associated with a 60% reduction of medullary interstitial O2·− levels at the end of 5 days of a 4.0% NaCl diet. No effect on either blood pressure or interstitial O2·− was observed in the control SS.13BN rats (186).
Fig. 9.
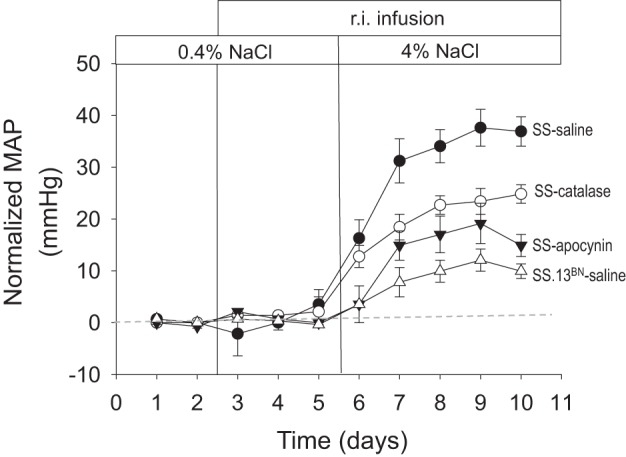
Summarized responses of MAP in to an increase in dietary salt. MAP has been normalized for each strain and treatment to the 0.4% salt control. All infusions were delivered directly into the renal interstitium (r.i.). Regraphed from results presented by Taylor et al. (185).
In addition to the evidence that H2O2 plays an important role in affecting MBF, Na+ excretion, and hypertension salt sensitivity (115, 186), there is also compelling evidence that chronic elevations of medullary H2O2 contribute importantly to the development of hypertension in SS rats. As also shown in Fig. 9, chronic infusion of catalase into the renal medulla of the SS rat significantly reduced salt sensitivity by ∼50% with medullary interstitial fibrosis also reduced (186). These results are consistent with observations that medullary H2O2 infusion in SD rats (115) produced a salt-sensitive form of hypertension with elevations of H2O2 concentrations in the medullary interstitium that were quite similar to those measured from the interstitium of SS rats fed a 4.0% NaCl diet (185, 186). Together, these studies indicate that both O2·− and H2O2 production contribute to the salt sensitivity of the SS rat either in an additive or possibly synergistic fashion.
The enhanced rate of Nox-related O2·− production together with reduced levels of catalase activity in the outer medulla probably account, in large measure, for the elevated interstitial levels of H2O2 observed in SS rats (185). However, studies indicate that fumarase insufficiency, an enzyme in the TCA cycle, also plays a functional role in the excess production of H2O2 and hypertension in SS rats (189). SS rats exhibit reduced levels of fumarase activity compared with SS.13BN rats. Medullary interstitial infusion of a fumarate precursor in the SS.13BN rats significantly increased medullary tissue H2O2 and exacerbated salt-induced hypertension. Consistent with these observations, a proteomic analysis of isolated mTAL mitochondria found nine proteins to be reduced in SS rats compared with SS.13BN rats (203). The downregulation of these proteins was predicted to reduce the efficiency of mitochondrial O2 utilization, which was confirmed by high-resolution respirometry of the mTAL cells and in mitochondria isolated from the outer medulla of SS rats. These studies revealed a largely unexplored role of mitochondrial and metabolic defects in the development of renal oxidative stress and hypertension in SS rats, which will require further investigation.
Which NAD(P)H oxidase isoforms likely contribute to excess renal medullary oxidative stress in SS rats?
As reviewed above, increased expression of Noxs appears to account for a large portion of the excess ROS production in the outer medulla of SS rats. The Nox family is composed of seven members, including Nox1-5 and the dual oxidases Duox1 and Duox2 (62). Nox1-4 are all localized in plasma membranes, and ongoing studies are assessing the presence and localization of the various family members within the kidneys of rats and mice. Only three of the seven known Noxs (Nox1, -2, and -4) are expressed in rodent kidneys (24, 76). Nox2 and Nox4 are abundantly expressed in the mTAL of the outer medulla, where Nox1 is either absent or in very low abundance (76).
p67phox.
p67phox is known to be essential for the activation of Nox2 (9, 111, 138). Immunohistochemical evidence from kidneys of SHR rats indicates that the Nox cytosolic subunit p67phox is expressed in the TAL, macula densa, distal convoluted tubule, cortical collecting duct, and perhaps the outer and inner medullary collecting ducts (24). Introgression of the salt-resistant p67phox allele from the Brown Norway rat into the SS rat (congenic SS.13BN26) (129) was found to significantly reduce salt-induced hypertension and renal injury (51). The congenic SS.13BN26 rats exhibited significantly lower levels of p67phox mRNA, protein, and NAD(P)H oxidase enzyme activity in the outer medullary tissue compared with SS rats (51). The overexpression of the p67phox found in the outer medullary tissue of SS rats was unique as expression of other subunits forming complex with Nox2 including p22phox, gp91phox, p47phox, or Rac1 (mRNA or proteins) were not different (51). The cloning and sequencing of p67phox yielded several genetic mutations of the SS allele of p67phox in the promoter region that contributed to higher promoter activity (51).
To directly assess the importance of p67phox in the salt-induced hypertension of the SS rat, site-directed mutagenesis by the application of zinc-finger nuclease technology (58, 59) was used to create SS rats containing a null mutation of the p67phox gene (51). As shown in Fig. 10, the functional importance of p67phox in the development of salt-sensitive hypertension was established as MAP was reduced in p67phox−/− rats by nearly 35% compared with their wild-type littermates. Renal oxidative stress was also reduced in these rats with levels of NAD(P)H oxidase, superoxide, and H2O2 all significantly decreased in homogenates of renal outer medullary tissue collected after 14 days of high salt (8% NaCl) compared with the wild-type littermates. There was also a significant attenuation of renal injury as evidenced by significant reductions of proteinuria and outer medullary interstitial fibrosis, reduced macrophage (ED-1 staining) and T cell (CD-43 staining) infiltration, and a sixfold reduction in tubular protein casts (51). Together, these data show that p67phox is an important contributor to renal oxidative stress, hypertension, and renal injury in SS rats. Although mTAL-specific p67phox knockout or transgenic rats have not been created yet, these approaches are now needed to determine the contribution of nonrenal tissues and organs such as blood vessels and the brain to the overall antihypertensive effects of the global mutation.
Fig. 10.
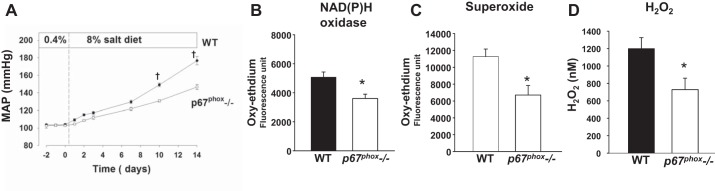
Salt-sensitive hypertension and renal oxidative stress are significantly attenuated in the p67phox−/− rats compared with their wild-type (WT) littermates. A: MAP of 5- to 6-wk-old rats on 0.4% salt and for 14 days after being fed an 8% salt diet. P < 0.001 significant difference between the 2 strains at that time point. B: outer medullary NAD(P)H oxidase activity on day 14 of 8% high salt. *P < 0.05 significant difference from WT. C: outer medullary superoxide on day 14 of 8% high salt. *P < 0.05 significant difference from WT. D: renal interstitial H2O2. *P < 0.05 significant difference from WT. Values are means ± SE. Regraphed from results by Feng et al. (51).
Nox4.
The biochemical features of Nox4 have been well characterized, although the roles of this isoform in kidney function remain to be determined. As with other Nox isoforms, it colocalizes with and directly binds to the integral membrane protein p22phox, which is necessary for its activity. Recent studies by Lyle and coworkers (110) have shown that Poldip2 [polymerase (DNA directed) delta-interacting protein 2] is a novel Nox4/p22phox-interacting protein which is a critical regulator of Nox4 activity. While these studies were focused on vascular smooth muscle cell migration and focal adhesion turnover, subsequent studies of RhoA/Rho kinase regulation upstream of the Poldip2 protein/Nox4 interaction in kidney myofibroblasts suggest a role in renal fibrosis (117). This transcription factor may be important in the local regulation of ROS production in the mTALs although further studies will be needed to clarify its specific role in renal ROS production. Uniquely, however, Nox4 does not require the binding of cytosolic proteins such as p67phox, p47phox, or Rac1. It is also remarkable that Nox4 largely produces H2O2 rather than O2·− (170), which is thought to be a consequence of a highly conserved histidine residue in the E-loop of Nox4 that promotes the rapid dismutation of O2·− within the protein itself (182). Beyond its presence in the plasma membrane, however, the localization of Nox4 in other intracellular locations is controversial as different studies have suggested expression in mitochondria, the endoplasmic reticulum, the nucleus, and focal adhesions (28). This lack of clarity may be attributed to the uncertainty of antibody selectivity, variations in staining approaches, splice variants (64), and possible transitioning between intracellular compartments (3, 53, 110, 128, 191).
Most studies related to the functional role of Nox4 have focused on the systemic and pulmonary vasculature where this isoform has been shown to be constitutively active, regulated at the gene level, and influenced by ANG II, shear stress/flow, hypoxia, and microRNAs (3, 53, 110, 128). However, Nox4 is expressed at particularly high levels in the kidney (61, 120), where it was first reported to be involved in cell growth (4, 63). It has been proposed that Nox4 mediates renal fibrogenesis (12) and is a major contributor to oxidative stress in diabetic nephropathy (4, 63, 83). Deletion of Nox4, but not Nox1 was found to provide renal protection from glomerular injury, attenuation of albuminuria, and preservation of the structure in ApoE−/− mice with streptozotocin-induced diabetes (83). To the contrary, in vivo studies using knockout mice or Nox4 inducible mice with three models of renal injury (streptozotocin diabetes, unilateral ureteral ligation, and ⅚ nephrectomy) concluded that Nox4 H2O2 production limited injury and disease progression (6). Other studies from the same group using mice reported that Nox4 protects the systemic vasculature during ischemic or inflammatory stress while Nox1 and Nox2 mediate endothelial dysfunction (168). [Renal Nox4 studies using rats are few. Mitochondria of rat renal NRK-52E cells (91) (proximal tubule-like) when stimulated with ANG II were reported to increase production of both O2·− and H2O2.] Using freshly isolated mTAL of SD rats, Nox4 siRNA was found to reduce O2·− responses to acute stimulation with either ANG II (120) or increased luminal flow (76). Given the various cell types (human and mouse) that have been studied in culture plus the application of various nonselective inhibitors, it is not surprising that results between laboratories have been inconsistent and difficult to extrapolate to the intact organism. Much remains to be learned about the role of Nox4 in both normal and pathological kidney function.
Excess O2·− and H2O2 Production Relative to NO in mTAL Leads to Cross Talk Between mTAL and Surrounding VR Pericytes in SS Rats
Blood flow through the VR of the renal outer medulla is uniquely controlled by numerous pericyte cell bodies that impart contractility, allowing control of vascular resistance of these vessels at the capillary level (145, 148, 156). As reviewed elsewhere, dysfunction of paracrine and autocrine signaling within the local outer medullary milieu may result in hypersensitivity of the medullary circulation to vasoconstrictor agents, medullary ischemia, and the development of hypertension (31). This was demonstrated by studies in SD rats in which the renal outer medulla was chronically infused with a nonhypertensive amount of the NOS inhibitor l-NAME, which hypersensitized the rat to the effects of intravenously administered ANG II (178), norepinephrine (180), or vasopressin (179). Low doses of these vasoconstrictor agents, which failed to produce hypertension when infused alone, resulted in sustained hypertension in SD rats when NO production in the renal medulla was specifically blunted by an infusion of l-NAME directly into this region. Importantly, the same hypersensitization to vasoconstrictors was found in SS rats, which naturally exhibit reduced production of medullary NO (181, 201).
Since mTAL produce large amounts of both ROS and NO, the possibility of paracrine-like communication between the mTAL and surrounding VR (e.g., tubulovascular cross talk) was explored. Cross talk of free radicals between cells and neighboring tissues has been the subject of much interest and controversy. Although it remains unclear how sufficient amounts of O2·−, H2O2, or NO could be released from mTAL epithelial cells and escape enzymatic degradation or interactions with other oxidizing molecules, there is compelling experimental evidence that this can occur under certain conditions (32, 90, 132, 140). Using freshly isolated thin tissue strips from the inner stripe of the outer medulla and time-resolved fluorescence videomicroscopy techniques, it was found that mTAL of SS rats exhibited greater rates of O2·− production in response to ANG II stimulation compared with SS.13BN rats (133). To determine whether cross talk from mTAL to surrounding VR pericytes occurred, nonperfused VR vessels were stimulated with ANG II either alone or in the presence of surrounding mTAL while maintaining their natural anatomic relationships while NO- or O2·−-sensitive fluorescent dye responses in the VR pericytes were recorded. These studies found that NO produced in mTAL of either SD or SS.13BN rats when stimulated by ANG II diffused to nearby VR pericytes (133). In contrast, however, cross talk of O2·− produced in the mTAL in response to ANG II was not observed in SD or salt-resistant SS.13BN rats, which was prevented by the scavenging effects of NO as shown by an absence of O2·− cross talk also in tissues pretreated with an NO scavenger (e.g., carboxy-PTIO) (133).
Consistent with these observations, O2·− cross talk from mTAL to the VR was clearly observed in the outer medullary tissue strips obtained from SS rats (133). Medullary NO levels have been found to be significantly reduced in SS rats, and O2·− cross talk is observed from the mTAL to the VR pericytes in a manner similar to the tissues from SS.13BN or SD rats when the tissue strips are pretreated with the NO scavenger (133). The SS rat strain is therefore a naturally occurring model in which O2·− signaling from mTAL to VR appears to occur, at least within the context of the freshly isolated tissue strip. It is this phenomenon, we propose, that produces the excess free radicals in the mTAL of the SS rat fed a high-salt diet and results in the constriction of VR and the reduction of MBF (32, 33).
Until recently, it was only assumed that if a vasoconstrictor released from the mTAL reached the contractile pericytes of the VR, there would be a resulting vasoconstriction and reduction of MBF. The first direct evidence was obtained using a unique in vitro model in which freshly isolated VR vessels were microperfused and stimulated with vasoactive agents either alone or in the presence of mTAL while maintaining their natural anatomic relationships. Dynamic 3D VR luminal diameter responses were determined together with changes in pericyte intracellular Ca2+ (fura 2) in response to the application of ANG II. Exchange of the vehicle superfusate with bath medium containing ANG II resulted in a rapid and sustained reduction of VR diameter in the absence of surrounding mTAL in both SS and SS.13BN control rats (Fig. 11). Compared with SS rats, only a small transient rise of pericyte intracellular [Ca2+] with an accompanying small transient reduction VR diameter was observed in VR isolated from SS.13BN when studied in the presence of surrounding mTAL (Fig. 11). The salt-insensitive SS.13BN rat has a mechanism(s) that buffers the VR vasoconstrictor effects of ANG II, and this buffering substance comes from the surrounding mTAL. As was also demonstrated in this study, the in vitro data predicted in vivo medullary flow responses to ANG II whereby ANG II reduced outer MBF in the SS but not SS.13BN as determined by laser-Doppler flowmetry (141).
Fig. 11.

Vasoconstrictor responses to angiotensin II (1 μM) in SS and SS.13BN outer medullary vasa recta (VR) and VR with mTAL. The X-axis is time after administration of angiotensin II to the bath media; y-axis, percentage of change in VR inner luminal diameter relative to mean of initial VR diameter measurements at 51 min before administration of angiotensin II. Values are means ± SE. *P < 0.05 for interaction between strain and response to angiotensin II. Reprinted with permission from O'Connor and Cowley (141).
These data indicate that tubular dysfunction in the SS rat contributes to enhanced medullary vascular sensitivity and may predispose these animals to the development of hypertension. However, it is not entirely clear which and how many different molecules may contribute to “tubulovascular cross talk.” In agreement with our previous findings, reduced NOS activity examined by l-NAME pretreatment reduced but did not abolish buffering responses seen in the SS.13BN rats. Pretreatment of medullary tissue strips with the P2 receptor antagonist suramin (albeit it is a relatively nonspecific antagonist) substantially blunted the cross talk from mTAL (141), indicating a role for P2Y or P2X receptors in the regulation of MBF (11). ATP and related nucleotides and PGE are known to be released in the renal outer medulla (8, 14, 23, 60, 102, 148, 172, 173). Although it has been found that exogenous ATP stimulates pericytes and constricts VR (36), the complex role played by ATP and adenosine in the in vivo regulation of MBF and in tubulovascular cross talk remains to be explored.
In brief, a number of studies have shown that both O2·− and NO (32, 33, 132, 133, 140) and perhaps ATP (141) produced in the mTAL can signal the pericyte cell bodies to constrict or dilate, responses which would modify renal MBF. The specific role of H2O2 remains to be explored, although given the nature of this molecule it is a likely candidate for cross talk in the outer medulla since a paracrine function of this molecule as a vascular mediator in the peripheral circulation has been described (3).
Other Intrarenal Sources of ROS Production in SS Rats
Summary.
The hypotheses based on the data presented in this review are summarized in Fig. 12. As shown, in a normal state (left, salt resistant) the data show that an increase in daily salt intake results in greater filtration of NaCl that would result in an increased delivery of Na+ and water to the mTAL. This stimulates increased production of NO and H2O2 via mechanical signaling and increased metabolism driven by greater Na+ transport. Illustrated also is the hypothesis that the resulting increased Na+ flux stimulates greater mitochondrial H2O2 production, which in some way signals greater activity of the membrane oxidases (Nox2 and -4). Under normal conditions, the O2·− produced in the mTAL does not successfully diffuse to the surrounding mTAL (e.g., cross-talk) due to the buffering actions of tissue NO. In contrast, when this redox balance is shifted toward greater production of ROS, in the face of reduced NO production (e.g., SS rats, right), medullary oxidative stress develops resulting in tubulovascular cross talk, reduction of MBF, Na+ retention, and hypertension. Although the many details of these events remain to be defined, there is little question that a shift in the redox balance and oxidative stress in the renal medulla results in a salt-sensitive form of hypertension.
Fig. 12.

Summary overview.
GRANTS
The work presented in this review was supported by National Heart, Lung, and Blood Institute Grants HL-29587 (A. W. Cowley, Jr.), HL-54998 (A. W. Cowley, Jr.), HL-082798 (A. W. Cowley, Jr.), and HL-116264 (A. W. Cowley, Jr.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.W.C.J. provided conception and design of research; A.W.C.J., M.A., T.M., P.M.O., and Y.O. interpreted results of experiments; A.W.C.J. drafted manuscript; A.W.C.J. edited and revised manuscript; A.W.C.J., M.A., T.M., P.M.O., Y.O., and N.N.Z. approved final version of manuscript; M.A., T.M., P.M.O., Y.O., and N.N.Z. performed experiments; M.A., T.M., P.M.O., Y.O., and N.N.Z. analyzed data; M.A., P.M.O., Y.O., and N.N.Z. prepared figures.
ACKNOWLEDGMENTS
The authors thank Dr. A. Staruschenko and M. Skelton for a critical review of the manuscript.
REFERENCES
- 1.Abe M, O'Connor P, Kaldunski M, Liang M, Roman RJ, Cowley AW Jr. Effect of sodium delivery on superoxide and nitric oxide in the medullary thick ascending limb. Am J Physiol Renal Physiol 291: F350–F357, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Adler S, Huang H. Impaired regulation of renal oxygen consumption in spontaneously hypertensive rats. J Am Soc Nephrol 13: 1788–1794, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Ardanaz N, Pagano PJ. Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp Biol Med (Maywood) 231: 237–251, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Asaba K, Tojo A, Onozato ML, Goto A, Quinn MT, Fujita T, Wilcox CS. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int 67: 1890–1888, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Aviv A, Hollenberg NK, Weder A. Urinary potassium excretion and sodium sensitivity in blacks. Hypertension 43: 707–713, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, Weissmann N, Anilkumar N, Shah AM, Schaefer L, Schroder K, Brandes RP. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 53: 842–853, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Barth E, Stammler G, Speiser B, Schaper J. Ultrastructural quantification of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 24: 669–681, 1992. [DOI] [PubMed] [Google Scholar]
- 8.Beach RE, Watts BA 3rd, Good DW, Benedict CR, DuBose TD Jr. Effects of graded oxygen tension on adenosine release by renal medullary and thick ascending limb suspensions. Kidney Int 39: 836–842, 1991. [DOI] [PubMed] [Google Scholar]
- 9.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Bienert GP, Moller Al Kristiansen KA, Schulz A, Moller IM, Schjoerring JK, Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem 282: 1183–1192, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Birch RE, Schwiebert EM, Peppiatt-Wildman CM, Wildman SS. Emerging key roles for P2X receptors in the kidney. Front Physiol 4: 262, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE, Barnes JL. NADPH oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol 21: 93–102, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonini MG, Rota C, Tomasi A, Mason RP. The oxidation of 2′,7′ -dichorofluorescein to reactive oxygen species: a self-fulfilling prophesy? Free Radic Biol Med 40: 969–975, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Bonvalet JP, Pradelles P, Farman N. Segmental synthesis and actions of prostaglandins along the nephron. Am J Physiol Renal Fluid Electrolyte Physiol 253: F377–F387, 1987. [DOI] [PubMed] [Google Scholar]
- 15.Boulden BM, Widder JD, Allen JC, Smith DA, Al-Baldawi RN, Harrison DB, Dikalov SI, Jo H, Dudley SC Jr. Early determinants of H2O2-induced endothelial dysfunction. Free Radic Biol Med 41: 810–817, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 134: 707–716, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Branco MR, Marinho HS, Cyrne L, Antunes F. Decrease of H2O2 plasma membrane permeability during adaptation to H2O2 in Saccharomyces cerevisiae. J Biol Chem 279: 6501–6506, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Brezis M, Rosen S, Silva P, Epstein FH. Renal ischemia: a new perspective. Kidney Int 26: 375–383, 1984. [DOI] [PubMed] [Google Scholar]
- 19.Brown KE, Dhaun N, Goddard J, Webb DJ. Potential therapeutic role of phosphodiesterase type 5 inhibition in hypertension and chronic kidney disease. Hypertension 63: 5–11, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Burg MR, Green N. Function of the thick ascending limb of Henle's loop. Am J Physiol 224: 659–668, 1973. [DOI] [PubMed] [Google Scholar]
- 21.Cabral PD, Garvin JL. Luminal flow regulates NO and O2− along the nephron. Am J Physiol Renal Physiol 300: F1047–F1053, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabral PD, Hong HJ, Garvin JL. Shear stress increases nitric oxide production in thick ascending limbs. Am J Physiol Renal Physiol 299: F1185–F1192, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao C, Edwards A, Sendeski M, Lee-Kwon W, Cui L, Cai CY, Patzak A, Pallone TL. Intrinsic nitric oxide and superoxide production regulates descending vasa recta contraction. Am J Physiol Renal Physiol 299: F1056–F1064, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 39: 269–274, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Chakraborti T, Ghosh SK, Michael JR, Batabyal SK, Chakraborti S. Targets of oxidative stress in cardiovascular system. Mol Cell Biochem 187: 1–10, 1998. [DOI] [PubMed] [Google Scholar]
- 26.Chandramohan G, Bai Y, Norris K, Rodriguez-Iturbe B, Vaziri ND. Effects of dietary salt on intrarenal angiotensin system, NAD(P)H oxidase, COX-2, MCP-1 and PAI-1 expressions and NF-kappaB activity in salt-sensitive and -resistant rat kidneys. Am J Nephrol 28: 158–167, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Chang MC, Pralle A, Isacoff EY, Chang CJ. A selective, cell-permeable optical probe for hydrogen peroxide in living cells. J Am Chem Soc 126: 15392–15393, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen F, Haigh S, Barman S, Fulton DJ. From form to function: the role of Nox4 in the cardiovascular system. Front Physiol 3: 412, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YF, Cowley AW Jr, Zou AP. Increased H2O2 counteracts the vasodilator and natriuretic effects of superoxide dismutation by tempol in renal medulla. Am J Physiol Regul Integr Comp Physiol 285: R827–R833, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Cowley AW., Jr Long term control of arterial blood pressure. Physiol Rev 72: 231–300, 1992a. [DOI] [PubMed] [Google Scholar]
- 31.Cowley AW., Jr The role of the renal medulla in volume and arterial pressure regulation. Am J Physiol Regul Integr Comp Physiol 273: R1–R15, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Cowley AW., Jr Renal medullary oxidative stress, pressure-natriuresis, and hypertension. Hypertension 52: 777–786, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cowley AW Jr, Mori T, Mattson D, Zou AP. Role of renal NO production in the regulation of medullary blood flow. Am J Physiol Regul Integr Comp Physiol 284: R1355–R1369, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Cowley AW Jr, Roman RJ, Kaldunski ML, Dumas P, Dickhout JG, Greene AS, Jacob HJ. Brown Norway chromosome 13 confers protection from high salt to consomic Dahl S rat. Hypertension 37: 456–461, 2001. [DOI] [PubMed] [Google Scholar]
- 35.Cowley AW Jr, Ryan RP, Kurth T, Skelton MM, Schock-Kusch D, Gretz N. Progression of GFR reduction determined in conscious Dahl S hypertensive rats. Hypertension, 62: 85–90, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crawford C, Wildman SS, Kelly MC, Kennedy-Lydon TM, Peppiatt-Wildman CM. Sympathetic nerve-derived ATP regulates renal medullary vasa recta diameter via pericyte cells: a role for regulating medullary blood flow? Front Physiol 4: 307, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeCoursey TE. Voltage-gated proton channels: molecular biology, physiology, and pathophysiology of H(V) family. Physiol Rev 93: 599–652, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 298: R1136–R1142, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Miguel C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and lead to hypertension and renal disease. Am J Physiol Renal Physiol 300: F734–F742, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Miguel C, Lund H, Mattson DL. High dietary protein exacerbates hypertension and renal damage in Dahl salt-sensitive (SS) rats by increasing infiltrating immune cells. Hypertension 57: 269–274, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc 130: 11561–11562, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7: 504–511, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dickinson BC, Peltier J, Stone D, Schaffer DV, Chang CJ. Nox2 redox signaling maintains essential cell populations in the brain. Nat Chem Biol 7: 106–112, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dikalov SI, Harrison DG. Methods for detection of mitochondrial and cellular reactive oxygen species. Antiox Redox Sign 20: 372–382, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Djouadi F, Bastin J, Gilber A, Rotig A, Rustin P, Merlet-Benichou C. Mitochondrial biogenesis and development of respiratory chain enzymes in kidney cells: role of glucocorticoids. Am J Physiol Cell Physiol 267: C245–C254, 1994. [DOI] [PubMed] [Google Scholar]
- 46.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 102: 488–496, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Downs CA, Kumar A, Kreiner LH, Johnson NM, Helms MN. H2O2 regulates lung epithelial sodium channel (ENaC) via ubiquitin-like protein Nedd8. J Biol Chem 288: 8135–8145, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drenjancevic-Peric I, Lombard JH. Reduced angiotensin II and oxidative stress contribute to impaired vasodilation in Dahl salt-sensitive rats on low-salt diet. Hypertension 45: 687–691, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Edwards A, Silldorff EP, Pallone TL. The renal medullary microcirculation. Front Biosci 5: E36–E52, 2000. [DOI] [PubMed] [Google Scholar]
- 50.Epstein FH, Balaban RS, Ross BD. Redox state of cytochrome aa3 in isolated perfused rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol 243: F356–F363, 1982. [DOI] [PubMed] [Google Scholar]
- 51.Feng D, Yang C, Geurts A, Kurth T, Liang M, Lazar J, Mattson D, O'Connor P, Cowley AW Jr. Increased expression of NAD(P)H oxidase subunit p67phox in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab 15: 201–208, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fenoy FJ, Ferrer P, Barbonell L, Garcia-Salom M. Role of nitric oxide on papillary blood flow and pressure natriuresis. Hypertension 25: 408–414, 1995. [DOI] [PubMed] [Google Scholar]
- 53.Fu Y, Zhang Y, Wang Z, Wang L, Wei X, Zhang B, Wen Z, Fang H, Pang Q, Yi F. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am J Nephrol 32: 581–589, 2010. [DOI] [PubMed] [Google Scholar]
- 54.Fujita M, Ando K, Nagae A, Fujita T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in salt-sensitive hypertension. Hypertension 50: 360–367, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Garcia-Estan J, Roman RJ. Role of renal interstitial hydrostatic pressure in the pressure diuresis response. Am J Physiol Renal Fluid Electrolyte Physiol 256: F63–F70, 1989. [DOI] [PubMed] [Google Scholar]
- 56.Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu Rev Physiol 73: 359–376, 2011. [DOI] [PubMed] [Google Scholar]
- 57.Garvin JL, Hong NJ. Cellular stretch increases superoxide production in the thick ascending limb. Hypertension 51: 488–493, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Ménoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325: 433, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, Menoret S, Jacob HJ, Anegon I, Buelow R. Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol Biol 597: 211–225, 2010. [DOI] [PubMed] [Google Scholar]
- 60.Geyti CS, Odgaard E, Overgaard MT, Jensen ME, Leipziger J, Praetorius HA. Slow spontaneous [Ca2+] I oscillations reflect nucleotide release from renal epithelia. Pflügers Arch 455: 1105–1117, 2008. [DOI] [PubMed] [Google Scholar]
- 61.Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxid Redox Signal 8: 1597–1607, 2006. [DOI] [PubMed] [Google Scholar]
- 62.Gorin Y, Block K. Nox4 and diabetic nephropathy: with a friend like this, who needs enemies? Free Radic Biol Med 23: 130–142, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 280: 39616–39626, 2005. [DOI] [PubMed] [Google Scholar]
- 64.Goyal P, Weissmann N, Rose F, Grimminger F, Schafers HJ, Seeger W, Hanze J. Identification of novel Nox4 splice variants with impact on ROS levels in A549 cells. Biochem Biophys Res Commun 329: 32–39, 2005. [DOI] [PubMed] [Google Scholar]
- 65.Granger JP, Alexander BT. Abnormal pressure-natriuresis in hypertension: role of nitric oxide. Acta Physiol Scand 168: 161–168, 2000. [DOI] [PubMed] [Google Scholar]
- 66.Greger R. Ion transport mechanisms in thick ascending limb of Henle's loop of mammalian nephron. Physiol Rev 65: 760–797, 1985. [DOI] [PubMed] [Google Scholar]
- 67.Guarasci DR, Kline RL. Pressure natriuresis following acute and chronic inhibition of nitric oxide synthase in rats. Am J Physiol Regul Integr Comp Physiol 270: R469–R478, 1996. [DOI] [PubMed] [Google Scholar]
- 68.Guyton AC. Dominant role of the kidneys and accessory role of whole-body autoregulation in the pathogenesis of hypertension. Am J Hypertens 2: 575–585, 1989. [DOI] [PubMed] [Google Scholar]
- 69.Harris RC, Breyer MD. Physiological regulation of cyclooxygenase-2 in the kidney. Am J Physiol Renal Physiol 281: F1–F11, 2001. [DOI] [PubMed] [Google Scholar]
- 70.Harrison DG. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest 100: 2153–2157, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He X, Liu Y, Usa K, Tian Z, Cowley AW Jr, Liang M. Ultrastructure of mitochondria and the endoplasmic reticulum in renal tubules of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 306: F1190–F1197, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hebert SC, Andreoli TE. Control of NaCl transport in the thick ascending limb. Am J Physiol Renal Fluid Electrolyte Physiol 246: F745–F756, 1984. [DOI] [PubMed] [Google Scholar]
- 73.Holstein-Rathlou NH, Marsh DJ. A dynamic model of the tubuloglomerular feedback mechanism. Am J Physiol Renal Fluid Electrolyte Physiol 258: F1448–F1459, 1990. [DOI] [PubMed] [Google Scholar]
- 74.Hong NJ, Garvin JL. Flow increases superoxide production by NADPH oxidase via activation of Na-K-2Cl cotransport and mechanical stress in thick ascending limbs. Am J Physiol Renal Physiol 292: F993–F998, 2007. [DOI] [PubMed] [Google Scholar]
- 75.Hong NJ, Garvin JL. Nitric oxide reduces flow-induced superoxide production via cGMP-dependent protein kinase in thick ascending limbs. Am J Physiol Renal Physiol 296: F1061–F1066, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hong NJ, Garvin JL. NADPH oxidase 4 mediates flow-induced superoxide production in thick ascending limbs. Am J Physiol Renal Physiol 303: F1151–F1156, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang A, Sun D, Kaley G, Koller A. Superoxide released to high intra-arteriolar pressure reduces nitric oxide-mediated shear stress- and agonist-induced dilations. Circ Res 83: 960–965, 1998. [DOI] [PubMed] [Google Scholar]
- 78.Huang BS, Leenen FH. Both brain angiotensin II and “ouabain” contribute to sympathoexcitation and hypertension in Dahl S rats on high salt intake. Hypertension 32: 1028–1033, 1998. [DOI] [PubMed] [Google Scholar]
- 79.Huie RE, Tadmaja S. The reaction of NO with superoxide. Free Radic Res Commun 18: 195–199, 1993. [DOI] [PubMed] [Google Scholar]
- 80.Ikenaga H, Suzuki H, Ishii N, Itoh H, Saruta T. Role of NO on pressure-natriuresis in Wistar-Kyoto and spontaneously hypertensive rats. Kidney Int 43: 205–211, 1993. [DOI] [PubMed] [Google Scholar]
- 81.Ilatovskaya DV, Pavlov TS, Levchenko V, Staruschenko A. ROS production as a common mechanism of ENaC regulation by EGF, insulin, and IGF-1. Am J Physiol Cell Physiol 304: C102–C111, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ito S, Hiratsuka M, Komatsu K, Tsukamoto K, Kanmatsuse K, Sved AF. Ventrolateral medulla AT1 receptors support arterial pressure in Dahl salt-sensitive rats. Hypertension 41: 744–750, 2003. [DOI] [PubMed] [Google Scholar]
- 83.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Manikoshi T, Thallas-Bonke VM, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. Genetic targeting or pharmacologic inhibition of NADPH oxidase Nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25: 1237–1254, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jin C, Hu C, Polichnowski A, Mori T, Skelton M, Ito S, Cowley AW Jr. Effects of renal perfusion pressure on renal medullary hydrogen peroxide and nitric oxide production. Hypertension 53: 1048–1053, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jin C, Sun J, Stilphen CA, Smith SM, Ocasio H, Bermingham B, Darji S, Guha A, Patel R, Geurts AM, Jacob HJ, Lambert NA, O'Connor PM. HV1 acts as a sodium sensor and promotes superoxide production in medullary thick ascending limb of Dahl salt-sensitive rats. Hypertension 64: 541–550, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin XH, Siragy HM, Carey RM. Renal interstitial cGMP mediates natriuresis by direct tubule mechanism. Hypertension 38: 309–316, 2001. [DOI] [PubMed] [Google Scholar]
- 87.Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, Shiraishi N, Adachi M, Zhang Z, Masilamani S, Tiomita K. Aberrant ENaC activation in Dahl salt-sensitive rats. J Hypertens 27: 1679–1689, 2009. [DOI] [PubMed] [Google Scholar]
- 88.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 52: 1–6, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Katusic ZS, Marshall JJ, Kontos HA, Vanhoutte PM. Similar responsiveness of smooth muscle of the canine basilar artery to EDRF and nitric oxide. Am J Physiol Heart Circ Physiol 257: H1235–H1239, 1989. [DOI] [PubMed] [Google Scholar]
- 90.Kennedy-Lydon TM, Crawford C, Wildman SS, Peppiatt-Wildman CM. Renal pericytes: regulators of medullary blood flow. Acta Physiol (Oxf) 207: 212–225, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim SM, Kim YG, Jeong KH, Lee SH, Lee TW, Ihm CG, Moon JY. Angiotensin II-induced mitochondrial Nox4 is a major endogenous sources of oxidative stress in kidney tubular cells. PLoS One 7: e39739, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kirchner KA. Greater loop chloride uptake contributes to blunted pressure natriuresis in Dahl salt sensitive rats. J Am Soc Nephrol 1: 180–186, 1990. [DOI] [PubMed] [Google Scholar]
- 93.Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol 14: 2775–2782, 2003. [DOI] [PubMed] [Google Scholar]
- 94.Kone BC, Madsen KM, Tisher CC. Ultrastructure of the thick ascending limb of Henle in the rat kidney. Am J Anat 171: 217–226, 1984. [DOI] [PubMed] [Google Scholar]
- 95.Kotchen TA, Cowley AW Jr, Frohlich ED. Salt in health and disease—a delicate balance. N Engl J Med 368: 1229–1237, 2013. [DOI] [PubMed] [Google Scholar]
- 96.Kotchen TA, Zhang HY, Covelli M, Blehschmidt N. Insulin resistance and blood pressure in Dahl rats and in one-kidney, one-clip hypertensive rats. Am J Physiol Endocrinol Metab 261: E692–E697, 1991. [DOI] [PubMed] [Google Scholar]
- 97.Krishna MC, Grahame DA, Samuni A, Mitchell JB, Russo A. Oxoammonium cation intermediate in the nitroxide-catalyzed dismutation of superoxide. Proc Natl Acad Sci USA 89: 5537–5541, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of O2− or as SOD mimics? J Biol Chem 271: 26026–26031, 1996. [DOI] [PubMed] [Google Scholar]
- 99.Kumar U, Shin Y, Wersinger C, Patel Y, Sidhu A. Diminished expression of constitutive nitric oxide synthases in the kidney of spontaneously hypertensive rat. Clin Exp Hypertens 25: 271–282, 2003. [DOI] [PubMed] [Google Scholar]
- 100.Lancaster JR., Jr Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proc Natl Acad Sci USA 91: 8137–8141, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lasseque B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lear S, Silva P, Kelley VE, Epstein FH. Prostaglandin E2 inhibits oxygen consumption in rabbit medullary thick ascending limb. Am J Physiol Renal Fluid Electrolyte Physiol 258: F1372–F1378, 1990. [DOI] [PubMed] [Google Scholar]
- 103.Lee DH, Riquier AD, Yang LE, Leong PK, Maunsbach AB, McDonough AA. Acute hypertension provokes acute trafficking of distal tubule Na-Cl cotransporter (NCC) to subapical cytoplasmic vesicles. Am J Physiol Renal Physiol 296: F810–F818, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li N, Chen L, Yi F, Zia M, Li PL. Salt-sensitive hypertension induced by decoy of transcription factor hypoxia-inducible factor-1 (alpha) in the renal medulla. Circ Res 102: 1101–1108, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li N, Yi F, dos Santos EA, Donley DK, Li Pl. Role of renal medullary heme oxygenase in the regulation of pressure natriuresis and arterial blood pressure. Hypertension 49: 148–154, 2007. [DOI] [PubMed] [Google Scholar]
- 106.Liang M, Knox FG. Production and functional roles of nitric oxide in the proximal tubule. Am J Physiol Regul Integr Comp Physiol 278: R1117–R1124, 2000. [DOI] [PubMed] [Google Scholar]
- 107.Liu R, Garvin JL, Ren Y, Pagano PJ, Carretaro OA. Depolarization of the macula densa induces superoxide production via NAD(P)H oxidase. Am J Physiol Renal Physiol 292: F1867–F1872, 2007. [DOI] [PubMed] [Google Scholar]
- 108.Lu S, Mattson DL, Cowley AW Jr. Renal medullary captopril delivery lowers blood pressure in spontaneously hypertensive rats. Hypertension 23: 337–345, 1994. [DOI] [PubMed] [Google Scholar]
- 109.Lusher TF, Raij L, Vanhoutte PM. Endothelium-dependent vascular responses in normotensive and hypertensive Dahl rats. Hypertension 9: 157–163, 1987. [DOI] [PubMed] [Google Scholar]
- 110.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Paunkova L, Du P, Papaharalambus C, Lasseque B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 105: 249–259, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maehara Y, Miyano K, Sumimoto H. Role for the first SH3 domain of p67phox in activation of superoxide-production NADPH oxidases. Biochem Biophys Res Commun 379: 589–593, 2009. [DOI] [PubMed] [Google Scholar]
- 112.Maghzal GJ, Krause KH, Stocker R, Jaquet V. Detection of reactive oxygen species derived from the family of NOX NADPH oxidases. Free Radic Biol Med 53: 1903–1918, 2012. [DOI] [PubMed] [Google Scholar]
- 113.Majid DS, Said KE, Omoro SA, Navar LG. Nitric oxide dependency of arterial pressure-induced changes in renal interstitial hydrostatic pressure in dogs. Circ Res 88: 347–351, 2001. [DOI] [PubMed] [Google Scholar]
- 114.Makino A, Skelton MM, Zou AP, Roman RJ, Cowley AW Jr. Increased renal medullary oxidative stress produces hypertension. Hypertension 39: 667–672, 2002. [DOI] [PubMed] [Google Scholar]
- 115.Makino A, Skelton MM, Zou AP, Cowley AW Jr. Increased renal medullary H2O2 leads to hypertension. Hypertension 42: 25–30, 2003. [DOI] [PubMed] [Google Scholar]
- 116.Makino N, Sasaki K, Hashida K, Sakakura Y. A metabolic model describing the H2O2 elimination by mammalian cells including H2O2 permeation through cytoplasmic and peroxisomal membranes: comparison with experimental data. Biochim Biophys Acta 1673: 149–159, 2004. [DOI] [PubMed] [Google Scholar]
- 117.Manickam N, Patel M, Greindling KK, Gorin Y, Barnes JL. RhoA/Pho kinase mediated TFG-β1-induced myofibroblast activation through Poldip2/Nox4-derived reactive oxygen species. Am J Physiol Renal Physiol 307: F159–F171, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Manning RD Jr, Meng S, Tian N. Renal and vascular oxidative stress and salt-sensitivity of arterial pressure. Acta Physiol Scand 179: 243–250, 2003. [DOI] [PubMed] [Google Scholar]
- 119.Manning RD Jr, Tian N, Meng S. Oxidative stress and antioxidant treatment in hypertension and the associated renal damage. Am J Nephrol 25: 311–317, 2005. [DOI] [PubMed] [Google Scholar]
- 120.Massey KJ, Hong NJ, Garvin JL. Angiotensin II stimulates superoxide production in the thick ascending limb by activating NOX4. Am J Physiol Cell Physiol 303: C781–C789, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension, and renal injury. Am J Physiol Renal Physiol 307: F499–F508, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mattson DL, Higgins DJ. Influence of dietary sodium intake on renal medullary nitric oxide synthase. Hypertension 27: 688–692, 1996. [DOI] [PubMed] [Google Scholar]
- 123.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 48: 149–156, 2006. [DOI] [PubMed] [Google Scholar]
- 124.McDonough AA. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am J Physiol Regul Integr Comp Physiol 298: R851–R861, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Miller EW, Dickinson BC, Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci USA 107: 15681–15686, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miyata N, Cowley AW Jr. Renal intramedullary infusion of l-arginine prevents reduction of medullary blood flow in Dahl salt-sensitive rats. Hypertension 33: 445–450, 1999. [DOI] [PubMed] [Google Scholar]
- 127.Miyata N, Zou AP, Mattson DL, Cowley AW Jr. Renal medullary interstitial infusion of l-arginine prevents hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 275: R1667–R1673, 1998. [DOI] [PubMed] [Google Scholar]
- 128.Montezano AC, Burger D, Ceravolo GS, Yusug H, Montero M, Touyz RM. Novel Nox homologues in the vasculature: focusing on Nox4 and Nox5. Clin Sci (Lond) 120: 131–141, 2011. [DOI] [PubMed] [Google Scholar]
- 129.Moreno C, Kaldunski ML, Want T, Roman RJ, Greene AS, Lazar J, Jacob HJ, Cowley AW Jr. Multiple blood pressure loci on rat chromosome 13 attenuate development of hypertension in the Dahl S hypertensive rat. Physiol Genomics 31: 228–235, 2007. [DOI] [PubMed] [Google Scholar]
- 130.Mori T, Cowley AW Jr. Angiotensin II-NAD(P)H oxidase-stimulated superoxide modifies tubulovascular nitric oxide cross-talk in renal outer medulla. Hypertension 42: 588–593, 2003. [DOI] [PubMed] [Google Scholar]
- 131.Mori T, Cowley AW Jr. High perfusion pressure accelerates renal injury in salt-sensitive hypertension. J Am Soc Nephrol 19: 1472–1482, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mori T, Cowley AW Jr, Ito S. Molecular mechanisms and therapeutic strategies of chronic renal injury: physiological role of angiotensin II-induced oxidative stress in renal medulla. J Pharmacol Sci 100: 2–8, 2006. [DOI] [PubMed] [Google Scholar]
- 133.Mori T, O'Connor PM, Abe M, Cowley AW Jr. Enhanced superoxide production in renal outer medulla of Dahl salt-sensitive rats reduces nitric oxide tubular-vascular cross-talk. Hypertension 49: 1336–1341, 2007. [DOI] [PubMed] [Google Scholar]
- 134.Mozaffari MS, Jirakulsomchok S, Shao ZH, Wyss JM. High-NaCl diets increase natriuretic and diuretic responses in salt-resistant but not salt-sensitive SHR. Am J Physiol Renal Fluid Electrolyte Physiol 260: F890–F897, 1991. [DOI] [PubMed] [Google Scholar]
- 135.Munter P, Newsome B, Kramer H, Peralta CA, Kim Y, Jacobs DR Jr, Kiefe CI, Lewis CE. Racial differences in the incidence of chronic kidney disease. Clin J Am Soc Nephrol 7: 101–107, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension 57: 355–362, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nistala R, Whaley-Connell A, Sowers JR. Redox control of renal function and hypertension. Antioxid Redox Signal 10: 2047–2089, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Noack D, Rae J, Cross AR, Munoz J, Salmen S, Mendoza JA, Rossi N, Curnutte JR, Heyworth PG. Autosomal recessive chronic granulomatous disease caused by novel mutations in NC-2, the gene encoding the p67-phox component of phagocyte NADPH oxidase. Hum Genet 105: 460–467, 1999. [DOI] [PubMed] [Google Scholar]
- 139.O'Connor PM, Lu L, Schreck C, Cowley AW Jr. Enhanced amiloride-sensitive superoxide production in renal medullary thick ascending limb of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 295: F726–F733, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.O'Connor PM, Cowley AW Jr. Modulation of pressure-natriuresis by renal medullary reactive oxygen species and nitric oxide. Curr Hypertens Rep 12: 86–92, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.O'Connor PM, Cowley AW Jr. Medullary thick ascending limb buffer vasoconstriction of renal outer-medullary vasa recta in salt-resistant but not salt-sensitive rats. Hypertension 60: 965–972, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ohsaki Y, O'Connor PM, Mori T, Ryan RP, Dickinson BC, Chang CJ, Lu Y, Ito S, Cowley AW Jr. Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am J Physiol Renal Physiol 302: F95–F102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ortiz PA, Garvin JL. Interaction of O2− and NO in the thick ascending limb. Hypertension 39: 591–596, 2002. [DOI] [PubMed] [Google Scholar]
- 144.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. Am J Physiol Renal Physiol 287: F274–F280, 2004. [DOI] [PubMed] [Google Scholar]
- 145.Pallone TL. Transport of sodium chloride and water in rat ascending vasa recta. Am J Physiol Renal Fluid Electrolyte Physiol 261: F519–F525, 1991. [DOI] [PubMed] [Google Scholar]
- 146.Pallone TL, Edwards A, Mattson DL. Renal medullary circulation. Compr Physiol 2: 97–140, 2012. [DOI] [PubMed] [Google Scholar]
- 147.Pallone TL, Robertson CR, Jamison RL. Renal medullary microcirculation. Physiol Rev 70: 885–920, 1990. [DOI] [PubMed] [Google Scholar]
- 148.Pallone TL, Silldorff EP. Pericyte regulation of renal medullary blood flow. Exp Nephrol 9: 165–170, 2001. [DOI] [PubMed] [Google Scholar]
- 149.Pallone TL, Silldorff EP, Turner MR. Intrarenal blood flow: microvascular anatomy and the regulation of medullary perfusion. Clin Exp Pharmacol Physiol 25: 383–392, 1998. [DOI] [PubMed] [Google Scholar]
- 150.Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Ryan RP, Cowley AW Jr, Staruschenko A. Real-time electrochemical detection of ATP and H2O2 release in freshly isolated kidneys. Am J Physiol Renal Physiol 305: F134–F141, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care 31: S170–S180, 2008. [DOI] [PubMed] [Google Scholar]
- 152.Pavlov TS, Levchenko V, O'Connor PM, Ilatovskaya DV, Palygin O, Mori T, Mattson DL, Sorokin A, Lombard JH, Cowley AW Jr, Staruschenko A. Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol 24: 1053–1062, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Plata C, Meade P, Vazquez N, Hebert SC, Gamba G. Functional properties of the apical Na+-K+-2Cl− cotransporter isoforms. J Biol Chem 277: 11004–11012, 2002. [DOI] [PubMed] [Google Scholar]
- 154.Pollock CA, Lawrence JR, Field MJ. Tubular sodium handling and tubuloglomerular feedback in experimental diabetes mellitus. Am J Physiol Renal Fluid Electrolyte Physiol 260: F946–F952, 1991. [DOI] [PubMed] [Google Scholar]
- 155.Quiroz Y, Pons H, Gordon KL, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Larfo R, Egido J, Johnson RJ, Rodriquez-Iturbe B. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from nitric oxide synthase inhibition. Am J Physiol Renal Physiol 281: F38–F47, 2001. [DOI] [PubMed] [Google Scholar]
- 156.Rhinehart K, Handelsman CA, Silldorff EP, Pallone TL. ANG II AT2 receptor modulates AT1 receptor-mediated descending vasa recta endothelial Ca2+ signaling. Am J Physiol Heart Circ Physiol 284: H779–H789, 2003. [DOI] [PubMed] [Google Scholar]
- 157.Rodriquez-Iturbe B, Vaziri ND, Herrera Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells and salt-sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol 286: F606–F616, 2004. [DOI] [PubMed] [Google Scholar]
- 158.Rodriquez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001. [DOI] [PubMed] [Google Scholar]
- 159.Roman RJ. Abnormal renal hemodynamics and pressure-natriuresis relationship in Dahl salt-sensitive rats. Am J Physiol Renal Fluid Electrolyte Physiol 251: F57–F65, 1986. [DOI] [PubMed] [Google Scholar]
- 160.Roman RJ, Alonso-Galicia M, Wilson TW. Renal P450 metabolites of arachidonic acid and the development of hypertension in Dahl salt-sensitive rats. Am J Hypertens 10: 63S–67S, 1997. [PubMed] [Google Scholar]
- 161.Roman RJ, Kaldunski ML. Enhanced chloride reabsorption in the loop of Henle in Dahl salt-sensitive rats. Hypertension 17: 1018–1024, 1991. [DOI] [PubMed] [Google Scholar]
- 162.Roman RJ, Kaldunski M. Pressure natriuresis and cortical and papillary blood flow in inbred Dahl rats. Am J Physiol Regul Integr Comp Physiol 261: R595–R602, 1991. [DOI] [PubMed] [Google Scholar]
- 163.Sanada H, Jones JE, Jose PA. Genetics of salt-sensitive hypertension. Curr Hypertens Rep 13: 55–66, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schlatter E, Greger R. cAMP increases the basolateral Cl− conductance in the isolated perfused medullary thick ascending limb of Henle's loop of the mouse. Pflügers Arch 405: 367–376, 1985. [DOI] [PubMed] [Google Scholar]
- 165.Schock-Kusch D, Sadick M, Henninger N, Kraenzlin B, Claus G, Kloetzer HM, Weiss C, Pill J, Gretz N. Transcutaneous measurement of glomerular filtration rate using FITC-sinistrin in rats. Nephrol Dial Transplant 24: 2997–3001, 2009. [DOI] [PubMed] [Google Scholar]
- 166.Schock-Kusch D, Shulhevich Y, Zie Q, Hesser J, Stsepankou D, Neudecker S, Friedemann J, Koenig S, Heinrich R, Hoecklin F, Pill J, Gretz N. On-line feedback-controlled renal constant infusion clearances in rats. Kidney Int 82: 314–320, 2012. [DOI] [PubMed] [Google Scholar]
- 167.Schrieiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, Koenig S, Heinrich R, Hoecklin F, Pill J, Friedemann J, Schweda F, Gretz N, Schock-Kusch D. Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol 303: F783–F788, 2012. [DOI] [PubMed] [Google Scholar]
- 168.Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Leudike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 110: 1217–1225, 2012. [DOI] [PubMed] [Google Scholar]
- 169.Seaver LC, Imlay JA. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol 183: 7182–7189, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406: 105–114, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 91: 406–413, 2002. [DOI] [PubMed] [Google Scholar]
- 172.Silldorff EP, Kreisberg MS, Pallone TL. Adenosine modulates vasomotor tone in outer medullary descending vasa recta of the rat. J Clin Invest 98: 18–23, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Silva GB, Garvin JL. TRPV4 mediates hypotonicity-induced ATP release by the thick ascending limb. Am J Physiol Renal Physiol 295: F1090–F1095, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Silva GB, Ortiz PA, Hong NJ, Garvin JL. Superoxide stimulates NaCl absorption in the thick ascending limb via activation of protein kinase C. Hypertension 48: 467–472, 2006. [DOI] [PubMed] [Google Scholar]
- 175.Sousa-Lopes A, Antunes F, Cyrne L, Marinho HS. Decreased cellular permeability to H2O2 protects Saccharomyces cerevisiae cells in stationary phase against oxidative stress. FEBS Lett 578: 152–156, 2004. [DOI] [PubMed] [Google Scholar]
- 176.Stoner LC, Trimble ME. Effects of MK-196 and furosemide on rat medullary thick ascending limbs of Henle in vitro. J Pharmacol Exp Ther 221: 715–720, 1982. [PubMed] [Google Scholar]
- 177.Sullivan JM, Prewitt RL, Ratts TE. Sodium sensitivity in normotensive and borderline hypertensive humans. Am J Med Sci 295: 370–377, 1988. [DOI] [PubMed] [Google Scholar]
- 178.Szentivanyi M Jr, Maeda CY, Cowley AW Jr. Local renal medullary l-NAME infusion enhances the effect of long-term angiotensin II treatment. Hypertension 33: 440–445, 1999. [DOI] [PubMed] [Google Scholar]
- 179.Szentivanyi M Jr, Park F, Maeda CY, Cowley AW Jr. Nitric oxide in the renal medulla protects from vasopressin-induced hypertension. Hypertension 35: 740–745, 2000. [DOI] [PubMed] [Google Scholar]
- 180.Szentivanyi M Jr, Zou AP, Maeda CY, Mattson DL, Cowley AW Jr. Increase in renal medullary nitric oxide synthase activity protects from norepinephrine-induced hypertension. Hypertension 35: 418–423, 2000. [DOI] [PubMed] [Google Scholar]
- 181.Szentivanyi M Jr, Zou AP, Mattson DL, Soares P, Moreno C, Roman RJ, Cowley AW Jr. Renal medullary nitric oxide deficit of Dahl S rats enhances hypertensive actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 283: R266–R272, 2002. [DOI] [PubMed] [Google Scholar]
- 182.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 286: 13304–13313, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tan DY, Meng S, Cason GW, Manning RD Jr. Mechanisms of salt-sensitive hypertension: role of inducible nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol 279: R2297–R2303, 2000. [DOI] [PubMed] [Google Scholar]
- 184.Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol 286: R431–R444, 2004. [DOI] [PubMed] [Google Scholar]
- 185.Taylor NE, Cowley AW Jr. Effect of renal medullary H2O2 on salt-induced hypertension and renal injury. Am J Physiol Regul Integr Comp Physiol 289: R1573–R1579, 2005. [DOI] [PubMed] [Google Scholar]
- 186.Taylor NE, Glocka P, Liang M, Cowley AW Jr. NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats. Hypertension 47: 692–698, 2006. [DOI] [PubMed] [Google Scholar]
- 187.Taylor NE, Maier KG, Roman RJ, Cowley AW Jr. NO synthase uncoupling in the kidney of Dahl S rats: role of dihydrobiopterin. Hypertension 48: 1066–1071, 2006. [DOI] [PubMed] [Google Scholar]
- 188.Tian N, Moore RS, Phillips WE, Lin L, Braddy S, Pryor JS, Stoctsill RL, Hughson MD, Manning RD Jr. NADPH oxidase contributes to renal damage and dysfunction in Dahl salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol 295: R1858–R1865, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tian Z, Liu Y, Usa K, Mladinov D, Fang Y, Ding X, Greene AW, Cowley AW Jr, Liang M. Novel role of fumarate metabolism in Dahl salt-sensitive hypertension. Hypertension 54: 255–260, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Trac D, Liu B, Pao AC, Thomas SV, Park M, Downs CA, Ma HP, Helms MN. Fulvene-5 inhibition of NADPH oxidases attenuates activation of epithelial sodium channels in A6 distal nephron cells. Am J Physiol Renal Physiol 305: F995–F1005, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Von Lohneysen K, Noack D, Wood MR, Friedman JS, Knaus UG. Structural insights into Nox4 and Nox2, motifs involved in function and cellular localization. Mol Cell Biol 30: 961–975, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med 43: 995–1022, 2007. [DOI] [PubMed] [Google Scholar]
- 193.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension 27: 481–490, 1996. [DOI] [PubMed] [Google Scholar]
- 194.Wilcox CS. Reactive oxygen species: roles in blood pressure and kidney function. Curr Hypertens Rep 4: 160–166, 2002. [DOI] [PubMed] [Google Scholar]
- 195.Wilcox CS, Welch WJ. Oxidative stress: cause or consequence of hypertension. Exp Biol Med (Maywood) 226: 619–620, 2001. [DOI] [PubMed] [Google Scholar]
- 196.Williams JM, Sarkis A, Lopez B, Ryan RP, Flasch AK, Roman RJ. Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension 49: 687–694, 2007. [DOI] [PubMed] [Google Scholar]
- 197.Yang C, Stingo FC, Ahn KW, Liu P, Vannucci M, Laud PW, Skelton M, O'Connor P, Kurth T, Ryan RP, Moreno C, Tsaih SW, Patone G, Hummel O, Jacob HJ, Liang M, Cowley AW Jr. Increased proliferative cells in the medullary thick ascending limb of the loop of Henle in the Dahl salt-sensitive rat. Hypertension 61: 208–215, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yang T, Singh I, Pham H, Sun D, Smart A, Schnermann JB, Briggs JP. Regulation of cyclooxygenase expression in the kidney by dietary salt intake. Am J Physiol Renal Physiol 274: F481–F489, 1998. [DOI] [PubMed] [Google Scholar]
- 199.Yao B, Harris RC, Zhang MZ. Interactions between 11β-hydroxysteroid dehydrogenase and XOC-1 in kidney. Am J Physiol Regul Integr Comp Physiol 288: R1767–R1773, 2005. [DOI] [PubMed] [Google Scholar]
- 200.Ye W, Zhang H, Hillas E, Kohan DE, Miller RL, Nelson RD, Honeggar M, Yang T. Expression and function of COX isoforms in renal medulla: evidence for regulation of salt-sensitivity and blood pressure. Am J Physiol Renal Physiol 290: F542–F549, 2006. [DOI] [PubMed] [Google Scholar]
- 201.Yuan B, Cowley AW Jr. Evidence that reduced renal medullary nitric oxide synthase activity of Dahl S rats enables small elevations of arginine vasopressin to produce sustained hypertension. Hypertension 37: 524–528, 2001. [DOI] [PubMed] [Google Scholar]
- 202.Zhang YB, Magyar CE, Holstein-Rathlou NH, McDonough AA. The cytochrome P-450 inhibitor cobalt chloride prevents inhibition of renal Na, K-ATPase and redistribution of apical NHE-3 during acute hypertension. J Am Soc Nephrol 9: 531–537, 1998. [DOI] [PubMed] [Google Scholar]
- 203.Zheleznova NN, Yang C, Ryan RP, Halligan BD, Liang M, Greene AS, Cowley AW Jr. Mitochondrial proteomic analysis reveals deficiencies in oxygen utilization in medullary thick ascending limb of Henle in the Dahl salt-sensitive rat. Physiol Genomics 44: 829–842, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhou XJ, Vaziri ND, Zhang J, Wang HW, Wang XQ. Association of renal injury with nitric oxide deficiency in aged SHR: prevention by hypertension control with AT1 blockade. Kidney Int 62: 914–921, 2002. [DOI] [PubMed] [Google Scholar]
- 205.Zou AP, Cowley AW Jr. Nitric oxide in renal cortex and medulla: an in vivo microdialysis study. Hypertension 29: 194–198, 1997. [DOI] [PubMed] [Google Scholar]
- 206.Zou AP, Li N, Cowley AW Jr. Production and actions of superoxide in the renal medulla. Hypertension 37: 547–553, 2001. [DOI] [PubMed] [Google Scholar]
- 207.Zwede T, Mattson DL. Inhibition of cyclooxygenase-2 in the rat renal medulla leads to sodium-sensitive hypertension. Hypertension 44: 424–428, 2004. [DOI] [PubMed] [Google Scholar]