

Abstract
Interaction of polycystin-1 (PC1) and Gα12 is important for development of kidney cysts in autosomal dominant polycystic kidney disease (ADPKD). The integrity of cell polarity and cell-cell adhesions (mainly E-cadherin-mediated adherens junction) is altered in the renal epithelial cells of ADPKD. However, the key signaling pathway for this alteration is not fully understood. Madin-Darby canine kidney (MDCK) cells maintain the normal integrity of epithelial cell polarity and adherens junctions. Here, we found that deletion of Pkd1 increased activation of Gα12, which then promoted the cystogenesis of MDCK cells. The morphology of these cells was altered after the activation of Gα12. By using liquid chromatography-mass spectrometry, we found several proteins that could be related this change in the extracellular milieu. E-cadherin was one of the most abundant peptides after active Gα12 was induced. Gα12 activation or Pkd1 deletion increased the shedding of E-cadherin, which was mediated via increased ADAM10 activity. The increased shedding of E-cadherin was blocked by knockdown of ADAM10 or specific ADAM10 inhibitor GI254023X. Pkd1 deletion or Gα12 activation also changed the distribution of E-cadherin in kidney epithelial cells and caused β-catenin to shift from cell membrane to nucleus. Finally, ADAM10 inhibitor, GI254023X, blocked the cystogenesis induced by PC1 knockdown or Gα12 activation in renal epithelial cells. Our results demonstrate that the E-cadherin/β-catenin signaling pathway is regulated by PC1 and Gα12 via ADAM10. Specific inhibition of this pathway, especially ADAM10 activity, could be a novel therapeutic regimen for ADPKD.
Keywords: G proteins, polycystin-1, E-cadherin, ADAM10, ADPKD
heterotrimeric g proteins contain a Gα and Gβγ subunit and are classically coupled to seven plasma membrane-bound transmembrane receptors. However, they are also associated with atypical receptors or modulators and transduce signaling via nontraditional mechanisms. G proteins are composed of four major families, Gs, Gi/o, Gq, and G12. The G12 family includes Gα12 and Gα13. They share some common downstream signaling molecules, but their functions are not completely redundant during embryonic development. Gα13-deficient mice are embryonic lethal at around embryonic day 10, whereas Gα12-deficient mice are apparently normal (42). Gα12 is ubiquitously expressed and signals through seven transmembrane receptors and atypical receptors on the cell membrane. Activation of G12 plays a role in cell growth and proliferation, cytoskeleton rearrangement, cell polarity, paracellular permeability, cell-cell adhesion, and migration and invasion, particularly in pathological situations (54, 61). In tumor cells, Gα12 is very important for regulating cell migration, invasion, and probably tumor metastasis. Activation of Gα12 triggers downstream signaling, such as Rho activation, cofilin and myosin light chain-2 phosphorylation, and stress fiber formation (13, 14). There are several signaling molecules related to Gα12 such as monomeric GTPases, mitogen-activated protein kinases, and nonreceptor tyrosine kinases (non-RTKs). Their effectors include cadherins, radixin of the ezrin/radixin/moesin protein family, non-RTKs, protein phosphatases, A-kinase anchoring proteins, the tight junction protein zonula occludens-1, Hsp90, and regulators of G protein signaling RGS1, RGS16, and axin, etc. (15, 19).
We have previously shown that Gα12 activation affects renal epithelial cell-matrix adhesions and cystogenesis (20, 21). The activation of Gα12 regulates apoptosis and tight junctions in renal epithelial cells (67), and knockout of Gα12 protects kidney epithelial cells from ischemic injury (65). Gα12 is directly associated with the cytoplasmic tail of polycystin-1 (PC1, encoded by PKD1) (68). Taken together, these findings indicate that Gα12 could be downstream signaling molecule for PC1 and very important for development of kidney cysts induced by PKD1 mutation in autosomal dominant polycystic kidney disease (ADPKD).
ADPKD is one of the most common life-threatening genetic diseases and is characterized by early formation and gradual enlargement of multiple kidney cysts, which eventually results in end-stage renal disease. Mutation in PKD1 or PKD2 (polycystin-2, PC2) accounts for 85 and 15% of this disease, respectively (10, 50, 58, 70). PC1 acts as a G protein-coupled receptor and activates all four families of heterotrimeric G proteins (39, 68). PC1 also regulates mTOR (mammalian target of rapamycin) (16), planar cell polarity and Wnt signaling (15, 36), and focal adhesions (17, 60). Renal cysts initiate from a focal area within a tubule (usually within the distal tubule or collecting duct) and lead to tubular widening. In ADPKD, cyst formation begins in utero (59). As a cyst expands in size, it fills with fluid derived from unreabsorbed glomerular filtrate and fluid secreted from surrounding cells. Once cysts expand to ∼2 mm in diameter, they detach from their parental tubules and become isolated fluid-filled sacs lined by an epithelial cell layer. These isolated cysts continue to expand in size at a relatively constant rate after birth (59). Over time, the kidneys become enlarged to four to eight times their normal size. By the fifth to seventh decade of life, there is significant loss of renal function. Approximately one-half of ADPKD patients progress to chronic renal failure by age 60 yr and require dialysis or transplantation (43, 59). Regardless of the initial pathogenic mechanism, kidney cysts are accompanied by partial differentiation of the epithelial cells, dysregulation of epithelial cell proliferation and apoptosis, and disruption of cell polarity, cell-matrix and cell-cell contacts. There is also chronic focal ischemia, inflammation, and fibrosis (6, 31).
E-cadherin is a cell membrane protein with a single transmembrane domain. It forms adherens junction between cell-cell contact via homophilic interaction of its ectodomain. This special structure between epithelial cells is important for maintaining the integrity of planar polarity and cell-cell adhesions (9, 23). Following the ectodomain interactions, the cytoplasmic domain of E-cadherin forms a complex with p120-, α-, β-, and γ-catenin (plakoglobin), which connects to the actin cytoskeleton and promotes the maturation of adherens junctions in epithelia (37, 38). E-cadherin-mediated adhesion is also involved in Wnt signaling cascades that regulate gene expression and cytoskeleton remodeling (8). The link between the cytoplasmic tail of E-cadherin and the actin cytoskeleton is very important for regulating morphology, junction stabilization, cell-cell adhesion, cell migration, and tissue remodeling (8, 53).
On the cytoplasmic side, E-cadherin function is regulated by altering the composition of the cadherin-catenin complex, the presence of growth factors, tyrosine phosphorylation of the cadherin-catenin complex, p120 binding, and the activity of small GTPases (5, 55). In addition, E-cadherin can be removed from the cell surface by proteolytic cleavage, resulting in an 80 kDa fragment as a soluble E-cadherin (sE-cad). sE-cad was initially found in breast cancer cells (57), but it has also been reported in patients with viral and bacterial infections, organ failure, and other benign diseases. There are several proteases that cleave E-cadherin, such as the A disintegrin and metalloprotease (ADAM) family, bacterial proteases (gingipains and BFT/fragilysin), cathepsins (B, L, S), the family of matrix metalloproteases (MMPs) (MMP-2, 3, 7, 9, and 14), KLK7, and plasmin (2, 18, 27, 29, 35, 62).
PC1 forms multiple protein complexes in cell membrane, which including E-cadherin and the catenins (15). In human renal cystic epithelial cells, PC1 deletion is associated with the concomitant loss of surface E-cadherin (44, 49). However, it is still unclear how E-cadherin is regulated in renal cystic epithelial cells after Pkd1 inactivation. We demonstrate here that Pkd1 deletion increases the activation of Gα12, which then promotes the maturation of ADAM10 that is subsequently responsible for cleaving the ectodomain of E-cadherin in kidney epithelial cells.
METHODS
Cell culture and materials.
Tet-off inducible Gα12 and Gα12QL (constitutively active) Madin-Darby canine kidney (MDCK) cell lines were cultured and maintained as previously described (20, 32). Plastic ware and tissue culture supplies were from BD Bioscience (San Jose, CA). Chemicals were from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. Gα12 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Collagen type I (rat tail) and Matrigel were from BD Biosciences. E-cadherin antibody was from Cell Signaling (cat. #3195, clone: 24E10), ADAM10 antibody was from Abcam (ab1997), and β-catenin antibody from Cell Signaling (cat. #9582, clone: 6B3). The secondary antibodies were from Life Technologies (Grand Island, NY).
Animals and kidney specimens.
Inducible Pkd1 knockout mice (Mx1Cre+Pkd1flox/flox) were described elsewhere (51). In brief, exon 2 through exon 6 of Pkd1 were flanked by two LoxP sites. These mice were crossed with the mice expressing transgenic Cre recombinase under the control of the INF-inducible Mx1 promoter (Mx1Cre mice). All genotyping was done via PCR on tail DNA. The primers were as follows: Gα12WT, 5′-GTG CTC ATC CTT CTT GGT TTC C-3′; 5′-CGG GTC GCC CTT GAA ATC TGG-3′. Pkd1flox/flox, 5′-TTG CTG CCA GCT CTG TGT AT-3′; 5′-CAC AGC GGT AGG AAG AGG AG-3′. Mx1Cre, 5′-TCC CAA CCT CAG TAC CAA GCC AAG-3′; 5′-ACG ACC GGC AAA CGG ACA GAA GCA-3′. In all experiments, littermates without Cre recombinase or Mx1Cre+Pkd1flox/flox mice without inducible deletion of Pkd1 were used as controls. Cre recombinase was induced to conditionally knockout Pkd1 by intraperitoneal injection of 62.5 μg, 250 μg of IFN inducer pI:pC (Sigma) at consecutive 5 days at 1 or 5 wk of age. Most of these mice were kept to 9 wk of age. Animal protocols were approved by the Standing Committee on Animals of Harvard Medical School.
Kidney specimens (including cystic fluid and paraffin-embedded tissue) from control patients and ADPKD patients were obtained from Drs. J. Zhou and B. Denker (Harvard Polycystic Kidney Disease Center). The specimens had been taken from routine diagnostic or therapeutic samples and patient data had been made anonymous. The sample collection was approved by the ethical committee and the institutional review board of Harvard Medical School, and each patient gave written informed consent.
Liquid chromatography-tandem mass spectrometry.
MDCK cells were grown to 90% confluence in 100 mm plates in the presence or absence of doxycycline (Dox) without serum for 48 h. Then the culture media were collected and centrifuged at 2,000 g at 4°C for 5 min. The supernatants were collected and filtered through Amicon spin tubes (10 kDa, Centricon, Millipore) from 5 ml to ∼500 μl. Concentrated supernatants were resolved by SDS-PAGE using reducing condition. The gel was stained with Coomassie blue (Bio-Rad, Hercules, CA). All of the stained bands > 10 kDa were excised and sent to the Taplin Biological Mass Spectrometry Facility (Harvard Medical School). Excised SDS-polyacrylamide gel bands were cut into small pieces and digested in gels with trypsin. Then the samples were run in a mass spectrometer. All of the criteria and data were acquired and analyzed as reported previously (63).
Immunoblotting.
Monolayers were scraped in lysis buffer (150 mM NaCl, 2.5 mM EDTA, 25 mM HEPES, pH 7.5, 1 mM PMSF, 1% Triton X-100, and protease inhibitors; Roche, Indianapolis, IN) and phosphatase inhibitor cocktail (Sigma-Aldrich) plus 1 mM NaVO4 and 25 mM NaF. Cells were lysed at 4°C for 2 h and centrifuged. Lysates were used for GST-fused tetratricopeptide repeat (TPR) pull-down or Western blots as detailed previously (20, 67). Mouse primary kidney epithelial cells and collecting duct epithelial cells were separated and purified as previously described (36). The density of Western blot bands was quantified by with NIH ImageJ (Version 1.48) after subtracting background.
Immunohistochemistry.
Histopathological analysis was performed after hematoxylin and eosin staining on formalin-fixed, paraffin-embedded tissues. Immunofluorescence of cells was performed as described previously (20). In brief, cells grown on slide chambers were washed with PBS and fixed (3.7% paraformaldehyde, 15 min). After blocking (1 h in PBS containing 1% BSA and 2% goat serum at 25°C), primary antibody (1:50–100 dilution with 1% BSA in PBS) was added for 1 h at room temperature. After three washes, secondary antibodies conjugated with Alexa Fluor-430 (green), and Alexa Fuor-532 (red) were added for 1 h at 25°C (Invitrogen). After three washes, the slides were air-dried and mounted in solution containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA).
Selected frozen mouse kidney tissue was sectioned in 4 μm thickness and air dried at room temperature for 10 min. Sections were washed for 10 min in PBS and fixed in 95% alcohol for 10 min followed by two PBS washes of 5 min each. The sections were incubated in 1% BSA/3% normal donkey serum/PBS-Tween for 40 min to block nonspecific protein-protein interaction. Primary rabbit polyclonal Adam10 antibody (Abcam, ab1997) was applied on the sections at 1:400 dilution. E-cadherin (Cell Signaling, cat. #3195, clone: 24E10) and β-catenin (Cell Signaling, cat. #9582, clone: 6B3) were applied on the sections at 1:50 dilution. The sections were incubated overnight at 4°C for 20 h followed by two PBS washes of 5 min each. Then the sections were incubated for 30 min at room temperature with Alexa Fluor 568 donkey anti-rabbit IgG (Life Technologies, A10042) at 1:250 dilution with DAPI, 1 μg/ml, and Alexa Fluor 488 donkey anti-rabbit IgG (Life Technologies A21206) at 1:250 dilution, with DAPI, 1 μg/ml, and DBA-1:500 incubated 30 min. The sections were then washed in PBS twice for 5 min each and mounted with Fluorescent Mounting Medium (Vector Laboratories). Fluorescence images were acquired by using confocal microscopy (Zeiss LSM 510).
Three-dimensional cell culture.
MDCK Tet-off, inducible Gα12WT [wild type (WT)] and Gα12QL cell lines were characterized and cultured as before (20). For three-dimensional (3D) culture, MDCK cells were grown to 60–80% confluence on 100 mm dishes, trypsinized, and resuspended at a concentration of 4 × 104 cells/ml in the matrix with 40% collagen-I and 60% Matrigel mixture, 10× DMEM, and HEPES (at 8:1:1) on ice. The single-cell suspension was plated on to six-well plates for 30 min at 37°C until solid. We added 2 ml of media with 10% FBS into each chamber (three wells for each cell line) together with or without HGF (20 ng/ml, Sigma) and with or without Dox (40 ng/ml). ADAM10 inhibitor GI254023X (Sigma-Aldrich) was added into the culture medium to a final concentration of 5 μM. DMSO was used as vehicle control. This experiment was repeated at least three times per cell line.
Statistical analyses.
Statistics were done in GraphPad Prism (San Diego, CA). Significance was determined by ANOVA. P < 0.05 was considered statistically significant.
RESULTS
Activation of Gα12 blocks the tubulogenesis in PC1 overexpressing MDCK cells.
In a 3D culture system, MDCK cells grew in tubular patterns (3). We have reported that activation of Gα12 promoted cystic growth of these cells (20). To explore the biological links between Gα12 and PC1, we used MDCK cells in this 3D culture system. Both vector control and PC1-overexpressing cells formed tubular growth patterns (Fig. 1A). PC1-silenced MDCK cells spontaneously formed cysts, similar to the effect caused by thrombin-stimulated Gα12 activation. As previously reported, thrombin stimulation also changed the tubular growth to cystic growth in PC1-overexpressing cells (Fig. 1A). Gα12 was associated with PC1 in kidney epithelial cells (67). Therefore, we hypothesize that PC1 regulates Gα12 activity and then results in subsequent biological changes in kidney epithelial cells.
Fig. 1.

Deletion of Pkd1 increased the activation of Gα12 and promoted cystogenesis of renal epithelial cells. A: vector control and overexpressing polycystin-1 (PC1) Madin-Darby canine kidney MDCK cells formed tubular growth. The stimulation with thrombin (to activate Gα12) induced cystic growth. Knockdown of PC1 promoted cystogenesis, which was independent of stimulation with thrombin. B: the level of Gα12 expression in mouse kidney tissue. WT, wild type; Pkd1−/−, Pkd1+/−, conditional deletion of Pkd1 in 1 wk old mice, homozygously and heterozygously, respectively. C: expression levels of Gα12 (mRNA and protein) in kidney tissue from autosomal dominant polycystic kidney disease (ADPKD) patients. D: active form of Gα12 was pulled down with GST-TPR beads in the epithelial primary cells from mouse kidney tissue. E: the mean ratio of Gα12/GAPDH. Ctrl, control; IB, immunoblotting; Th, thrombin.
The levels of Gα12 mRNA and protein were significantly increased in the kidney tissue after conditional Pkd1 deletion in mice (Fig. 1B). In normal kidney tissue, the level of Gα12 mRNA was very low. Its level was elevated in the kidney tissue of ADPKD mice. The level of Gα12 protein was barely detected. In kidney tissue from ADPKD patients, mRNA and protein of Gα12 were dramatically increased (Fig. 1C). The active form of Gα12 was barely detected in normal kidney epithelial cells. To confirm that active Gα12 was also elevated after Pkd1 deletion, we used GST-TPR (GST-fused TPR domain of protein phosphatase 5: only binding to active form of Gα12) to precipitate active Gα12 in mouse kidney tissue. Equal amount of lysates were used to pull down active Gα12. In kidney epithelial cells from Pkd1−/− mice, active Gα12 was significantly higher than in control (WT). However, after stimulation with thrombin, there was no significant difference of active Gα12 between Pkd1−/− and WT mice (Fig. 1, D and E).
Activation of Gα12 results in morphological changes in MDCK cells.
The integrity of cell-cell contacts and polarity is disrupted in the kidney epithelial cells of ADPKD patients (44, 49). We noticed a dramatic change in the morphology of MDCK cells after we induced Gα12QL by removing Dox from the culture medium (Fig. 2A). Cell morphology is mostly determined by cell-cell contacts, polarity, and cellular stress fibers. To identify the proteins in the extracellular environment that could be related to these changes, we collected the conditioned medium from MDCK cells after Gα12QL was induced. We used mass spectrometry to identify these proteins (Table 1). Compared with the control (without inducible expression of Gα12QL), one of the most specific peptides was from E-cadherin (Table 1). In addition, two cell membrane metalloproteinases, ADAM10 and ADAM17 were found in the media, which could be also important for the shedding of these cell membrane proteins in kidney epithelial cells.
Fig. 2.
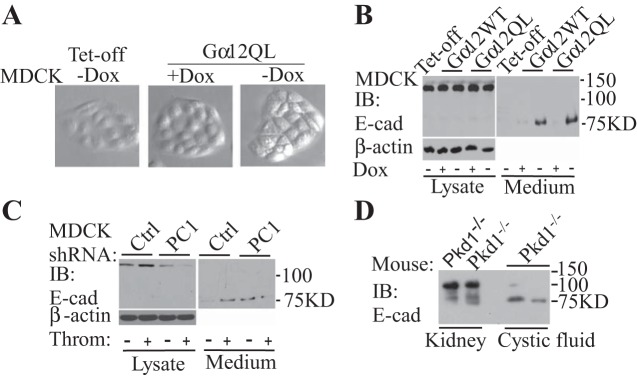
Activation of Gα12 led to cell morphology change and shedding of E-cadherin (E-cad). A: Gα12WT and Gα12QL (constitutively active) were induced by removal of doxycycline (Dox) for 48 h in MDCK cells. Typical phase contrast images are shown here. B: the expression of Gα12 increased the shedding of E-cadherin. C: knockdown of PC1 increased the shedding of E-cadherin. D: in renal cystic fluid of ADPKD mice, the shed fragment of E-cadherin was increased.
Table 1.
Peptides in conditioned media after Gα12 activation
| Gα12QL+Dox | Gα12QL−Dox |
|---|---|
| Calsyntenin 1 isoform 2 (4) | Calsyntenin 1 isoform 2 (4) |
| Syndecan-4 (3) | Syndecan-4 (6) |
| JAM-A (2) | JAM-A (1) |
| Lactadherin (1) | Lactadherin (2) |
| APP (1) | APP (8) |
| E-cadherin (1) | E-cadherin (5) |
| L1CAM (1) | Cadherin-6 (2) |
| ADAM10 (1) | ADAM17 isoform 2 (1) |
| Integrin beta-4 (2) | |
| CD44 (1) | |
| Clusterin (23) | Clusterin (21) |
| Osteopontin (10) | Osteopontin (10) |
| IGF-binding protein7 (6) | IGF-binding protein7 (4) |
| Stanniocalcin-1 (5) | Stanniocalcin-1 (5) |
| Semaphorin 3C (3) | Semaphorin 3C (3) |
| Collagen alpha 2(V) (13) | Collagen alpha 2(V) (12) |
| Galectin-3 binding protein (10) | Galectin-3 binding protein (10) |
| Thombospondin 1 (3) | Thombospondin 1 (4) |
| Fibulin-1 (1) | Fibulin-1 (4) |
| Fibronectin 1 isoform 1 (5) | |
| Urokinase plasminogen activator (1) | |
| Galectin-3 (5) | |
| Agrin (1) | |
| Podocan isoform 1 (1) | |
| Fibronectin (2) |
Peptides were identified by liquid chromatography-tandem mass spectrometry in conditioned media. Only membrane and extracellular proteins are listed. Peptide numbers are shown in parentheses.
To confirm the result from mass spectrometry, we used kidney cell lines and mouse kidney tissue. In MDCK cells, the cleaved ectodomain of E-cadherin was barely detected in culture medium. However, inducible expression of Gα12 increased the shedding of E-cadherin (Fig. 2B). After endogenous Gα12 was activated by the addition of thrombin, the shed fragment of E-cadherin was also increased. In PC1 knockdown cells, the shed amount of E-cadherin was elevated, which was independent of thrombin stimulation (Fig. 2C). In the renal cysts induced by inducible inactivation of Pkd1 in mice, the cleaved fragments of E-cadherin were also present (Fig. 3D).
Fig. 3.

The expression of Gα12 increased the active form of A disintegrin and metalloprotease (ADAM)10 and the ectodomain shedding of E-cadherin. A: inducible expression of Gα12WT, Gα12QL, or knockdown of PC1 promoted the active (mature) form (M). P, premature. The efficiency of ADAM10 siRNA in MDCK cells (B). Knockdown of ADAM10 reduced (siRNA oligo 3) or blocked (siRNA oligo 2) the shedding of E-cadherin induced by Gα12QL (C). The ADAM10-specific inhibitor GI254023X also blocked the ectodomain shedding of E-cadherin promoted by Gα12 expression or knockdown of PC1 (D).
Activation of Gα12 promotes the shedding of E-cadherin, which is mediated via ADAM10.
There are several proteolytic enzymes that cleave E-cadherin (29, 40). According to our mass spectrometry, ADAM17 and ADAM10 were present in the conditioned medium (Table 1). It has been reported that both of these metalloproteinases can cleave E-cadherin. In MDCK cells, there are two forms of ADAM10: the premature form and the mature form. The inducible expression of Gα12WT or Gα12QL increased the mature form. In addition, knockdown of PC1 also promoted the mature form of ADAM10 (Fig. 3A). In these cells, we did not observe any changes in the expression level of ADAM17 after Gα12 was induced (data not shown). To confirm that ADAM10 is the major sheddase for E-cadherin, we first used siRNA to knock down the expression of ADAM10 in these cells. The siRNA oligos 2 and 3 were more efficient in inhibiting ADAM10 expression (Fig. 3B). Therefore, we transfected these two ADAM10 siRNA oligos into MDCK Gα12QL cells. After the expression of Gα12QL was induced, the shed fragment of E-cadherin was dramatically reduced with siRNA oligo 3, and no E-cadherin fragment in the conditioned medium was detected with the siRNA oligo 2 (Fig. 3C). After the ADAM10-specific inhibitor GI254023 was added into MDCK cells with the expression of Gα12QL, or knockdown of PC1, there was no cleaved fragment of E-cadherin detected in the conditioned media as well (Fig. 3D).
Gα12 activation disrupts the distribution of E-cadherin and β-catenin in kidney epithelial cells.
In MDCK cells, the pattern and intensity of E-cadherin were altered after Gα12 activation. In vector control (Tet-off) or noninducible Gα12QL-expressing cells (+Dox), E-cadherin was homogeneously located on the cell membrane. However, after Gα12QL was induced, this distribution pattern of E-cadherin disappeared. Instead, some E-cadherin accumulated on certain regions of the cell membrane (Fig. 4A). In kidney tissue obtained from WT Pkd1+/+ mice, E-cadherin was localized on the cell membrane between tubular epithelial cells (Fig. 4B). However, in the renal epithelial cells of the cystic walls from Pkd1−/− null mice, E-cadherin staining was much weaker and accumulated only on certain spots (Fig. 4B). β-Catenin is associated with the cytoplasmic tail of E-cadherin and is important for maintaining the integrity of adherens junctions in epithelial cells (15). In kidney tissue of WT Pkd1+/+ mice, β-catenin was located on cell membrane (Fig. 4C). Pkd1 deletion promoted the transition of β-catenin from the cell membrane to the nucleus (Fig. 4C), where it regulates gene expression and causes cytoskeletal change (8, 38).
Fig. 4.

Distribution of E-cadherin and β-catenin in kidney epithelial cells. A: active Gα12 was induced for 48 h in MDCK cells. Green, E-cadherin; blue (DAPI), nucleus. Immunostaining of E-cadherin (B, white arrowheads) and β-catenin (C, white arrowheads) in the kidney tissue from WT and Pkd1−/− (null) mice. White bar: 10 μm.
In the 3D culture system, MDCK cells grow in tubular patterns. We have shown that both active Gα12 and PC1 knockdown resulted in cystic growth of MDCK cells (Fig. 5). Addition of a specific ADAM10 inhibitor, GI254023X, completely blocked the cystic growth of all the cells, and caused these cells to grow in tubular patterns (Fig. 5).
Fig. 5.
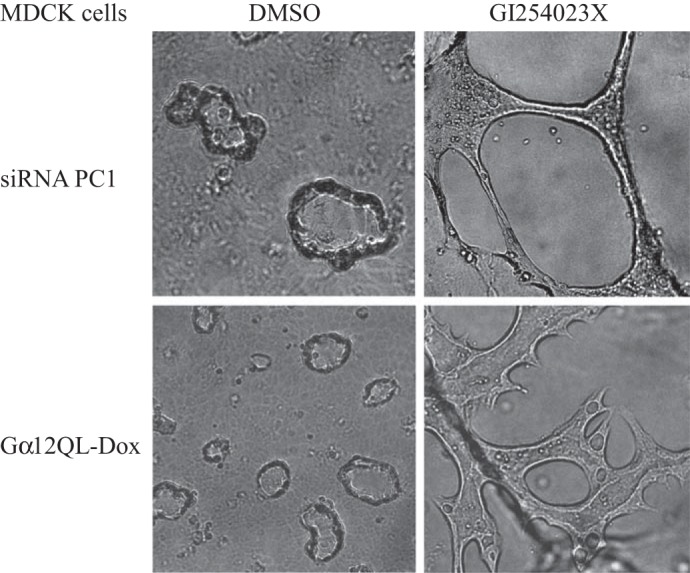
The ADAM10 inhibitor GI254023X completely blocked the cystic growth of MDCK induced by knockdown of PC1 or expression of Gα12QL.
DISCUSSION
MDCK cells (12) are widely used for studying the biological function of epithelia since they maintain the integrity of apico-basolateral polarity and cell junctions (tight and adherens). They polarize in 2D and 3D cell culture and are suitable for confocal imaging (3). In our laboratory, we have used MDCK Tet-off cells to investigate the functional roles of Gα12 in kidney diseases for over 10 yr. Activation of Gα12 leads to cystic growth of these kidney epithelial cells in a 3D culture (20). The cytoplasmic tail of PC1 is directly associated with Gα12 (66, 67). We used this system with the initial goal to understanding the relationship between Gα12 and PC1 and especially to determine if Gα12 is a downstream signaling molecule of PC1. Our results demonstrate that activation of Gα12 blocked tubulogenesis in the MDCK cells expressing ectopic PC1, which indicates that Gα12 could be a signaling mediator of PC1. We recently reported that the loss of PC1 led to hyperactive Gα12 signaling (66, 67), which was further supported by the GST-TPR immunoprecipitation of activated Gα12 (67) (Fig. 1D). The expression level of Gα12 was elevated in kidney tissue from Pkd−/− null mice and ADPKD patients (Fig. 1, B and C). Although PC1 is physically associated with Gα12 (66, 67), a few possibilities may be responsible for loss of PC1 resulted in activation of Gα12. Deletion of PC1 increased the expression of Gα12 (Fig. 1, B and C), which may increase the basal level of the activation of Gα12. PC1 may affects the stability of Gα12 protein. Direct binding of PC1 to Gα12 may prevent the exchange of G12 with GTP. Further study is needed to address the underlying mechanism.
We noticed that activation of Gα12 resulted in the morphology change of MDCK cells, which could result from the disruption of PC1, Gα12, ADAM10, and E-cadherin/β-catenin signaling. The signaling complex could be critical for renal cystogenesis in ADPKD.
In ADPKD, cell-cell adhesion, cell-matrix adhesion, and polarity are altered (43). We have reported that cell-matrix adhesion was disrupted after Gα12 activation, which was through changing the affinity of integrins and phosphorylation of focal adhesion molecules (20). The normal integrity of adherens junctions is dependent on intact E-cadherin. E-cadherin forms complexes with a few intracellular proteins and is involved in Wnt/β-catenin signaling to regulate cell polarity and adherens junctions. PC1 is colocalized with E-cadherin and can be coimmunoprecipitated with E-cadherin and the catenins (15). Gα12 is directly associated with the cytoplasmic tail of PC1 (67). Gα12 is associated with E-cadherin/catenins (30). Therefore, PC1/Gα12/E-cadherin/catenin is a unique complex in kidney epithelial cells and is important for regulating the polarity and adherens injunction.
The ectodomain shedding of E-cadherin is a unique way to control the biological function of E-cadherin (44, 49). Metalloproteinases are the major sheddase of E-cadherin. In MDCK cells, ADAM10 and ADAM17 are the two major proteinases. Both ADAM10 and Gα12 are involved in migration and invasion of tumor cells (19, 40). ADAM10 is responsible for shedding dozens of substrates that are related to cancer progression, inflammatory disease, and Alzheimer's disease, such as Notch, E-cadherin, EGF, ErbB2, amyloid precursor protein (APP), and inflammatory cytokines (29, 40). APP is related to cell proliferation and Alzheimer's disease. We also observed that syndecan-4 and cadherin-6 (K-cadherin) were present among the shed proteins (Table 1). However, we did not find any differences in the expression level of K-cadherin after active Gα12 was induced (data not shown). Syndecan-4 is a cell surface protein that affects cellular proliferation, migration, mechanotransduction, and endocytosis via various signaling pathways such as growth factor receptors, mTOR, AKT1, and the Rho family of GTPases. Associated with extracellular matrix and cytoskeletal signaling proteins, syndecan-4 is also involved in regulating integrin turnover and cell-matrix adhesions (7). We have shown that Gα12 regulates the cytoskeleton and integrin and focal adhesion (20, 21). Further studies are needed to elucidate any functional linkage between Gα12 and syndecan-4 in kidney epithelial cells. In addition, there were dozens of extracellular proteins, which seems insignificant after active Gα12 is induced (Table 1).
In MDCK cells, the majority of ADAM10 is the premature form. After removal of the prodomain from its premature form, ADAM10 becomes the mature form: enzymatically active. Our result showed that expression of Gα12WT or Gα12QL increased the mature form of ADAM10 to a similar level (Fig. 3A). Theoretically, expression of Gα12WT only elevates the amount of Gα12 but not the active form of Gα12. However, in the kidney tissue from WT mice, there was a decent amount of active Gα12 (Fig. 1D). In MDCK cells, we did not see dramatic difference of the ectodomain shedding of E-cadherin between Gα12WT and Gα12QL (Fig. 2B). In addition, there is no difference in the adhesion of MDCK cells on collagen-1 between the expression of Gα12WT and Gα12QL (20). So, we assume that certain amount of active Gα12 would be enough to activate ADAM10 or that another mechanism is involved, increasing stress fibers and disrupting integrins (20, 21). To confirm that the increased shedding of E-cadherin by active Gα12 or knockdown of PC is dependent on ADAM10, we used ADAM10-specific inhibitor GI254023X, which completely blocked its effects on the cleavage of E-cadherin (Fig. 3D). This result demonstrates that ADAM10 is the key proteolytic enzyme, which is regulated by PC1 and Gα12 and is responsible for cleaving the ectodomain of E-cadherin in kidney epithelial cells.
The disruption of adherens junctions results in the dissociation of β-catenin from the cytoplasmic tail of E-cadherin and its translocation into the nucleus to regulate the expression of certain genes. Subsequently, this disruption causes the changes in morphology, polarity, and cytoskeleton (8, 38). We observed that activation of Gα12 altered the distribution of E-cadherin and β-catenin in MDCK cells (Fig. 4). In transgenic mice, the Wnt/β-catenin signaling pathway is activated in kidney epithelial cells of ADPKD. Targeted expression of a mutant β-catenin results in development of renal cysts similar to that seen in ADPKD (45). PC1 multiprotein complexes also comprise E-cadherin and β-catenin that are located at the plasma membrane and cell-cell contacts (15, 43). In addition, recent evidence also indicates that the proteolytic cleaved COOH-terminal tail (CTT) of PC1 binds to active β-catenin. Then this complex moves to the nucleus together (22). It is unclear if the cleavage is mediated via ADAM10 in a similar way to the shedding of E-cadherin.
The disrupted signaling pathways in ADPKD are mostly involved in Ca2+, cAMP, and mammalian target of rapamycin complex (mTORC). mTORC1 controls cell growth, proliferation, and autophagy (47, 49, 62). Gα12 is reportedly involved in mTORC2 signaling that regulates the cytoskeleton and resistance to apoptosis (11, 25, 26). Several medications that reduce cAMP levels inhibit mTOR, inhibit cell proliferation, and reduce fluid secretion have been through clinical trials for ADPKD (4, 52). Based on changes in total kidney volume (TKV), total cyst volume, renal function, and adverse effects, a recent meta-analysis of all major clinic trials for ADPKD shows that TORC1 inhibitors and eicosapentaenoic acid have no therapeutic effects; somatostatin analogs decrease TKV by 9% only; the vasopressin receptor antagonist slows TKV increase to 3%/yr and attenuates kidney function decline. All of their adverse events are significant in all trials compared with placebo (16, 34). At present, there are no safe and effective clinical therapeutic regimens for ADPKD. Further understanding and identification of the signaling pathways and cellular mechanisms associated with ADPKD are essential to develop new therapeutic targets that block or slow down renal cystogenesis. Inhibition of ADAM10 activity could be useful for blocking the proliferation, differentiation, adhesion, and migration of cancer cells (33). ADPKD epithelial cells also demonstrate abnormalities in proliferation, differentiation, adhesion, and migration (10, 50, 58). Triptolide, a bioactive small molecule isolated from a Chinese herb, shows both antitumor activity (1) and reduction of renal cystogenesis in ADPKD mice (24). Triptolide has been reported to inhibit ADAM10 activity and affect Wnt/β-catenin signaling pathway (46, 48).
In summary, Pkd1 mutation or deletion leads to the activation of Gα12, which promotes the maturation of ADAM10 that increases the shedding of E-cadherin. Subsequently, the cleavage of the ectodomain fragment of E-cadherin causes the transition of β-catenin from cell membrane into the nucleus. Eventually cell polarity and cell-cell contacts could be disrupted, favoring the cystogenesis of renal epithelial cells. Blocking this signaling pathway, especially inhibiting ADAM10 activity, could be a novel target for ADPKD.
GRANTS
These studies were supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants K01 DK-080179, 3K01DK-080179, and DK-096160 (T. Kong). These studies were also supported by the Baltimore Polycystic Kidney Disease Research and Clinical Core Center, P30DK-090868.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.X.X., T.-S.L., S.L., Y.W., L.D., and T.K. performed experiments; J.X.X., T.-S.L., B.M.D., J.V.B., and T.K. analyzed data; J.X.X., T.-S.L., B.M.D., J.V.B., and T.K. interpreted results of experiments; J.X.X. and T.K. prepared figures; J.X.X., T.-S.L., S.L., Y.W., L.D., B.M.D., J.V.B., and T.K. approved final version of manuscript; T.-S.L. and T.K. edited and revised manuscript; T.K. conception and design of research; T.K. drafted manuscript.
ACKNOWLEDGMENTS
We thank Dr. G. Germino for providing PC1-overexpressing MDCK cells, Dr. G. Gusella for providing PC1-silenced MDCK cells, and Dr. J. Zhou for Mx1Cre+Pkd1flox/flox mice.
REFERENCES
- 1.Alsaied OA, Sangwan V, Banerjee S, Krosch TC, Chugh R, Saluja A, Vickers SM, Jensen EH. Sorafenib and triptolide as combination therapy for hepatocellular carcinoma. Surgery 156: 270–279, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Biswas MH, Du C, Zhang C, Straubhaar J, Languino LR, Balaji KC. Protein kinase D1 inhibits cell proliferation through matrix metalloproteinase-2 and matrix metalloproteinase-9 secretion in prostate cancer. Cancer Res 70: 2095–2104, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boletta A, Qian F, Onuchic LF, Bhunia AK, Phakdeekitcharoen B, Hanaoka K, Guggino W, Monaco L, Germino GG. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol Cell 6: 1267–1273, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Chang MY, Ong AC. Mechanism-based therapeutics for autosomal dominant polycystic kidney disease: recent progress and future prospects. Nephron Clin Pract 120: c25–c35, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Citi S, Spadaro D, Schneider Y, Stutz J, Pulimeno P. Regulation of small GTPases at epithelial cell-cell junctions. Mol Membr Biol 28: 427–444, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Cowley BD., Jr Recent advances in understanding the pathogenesis of polycystic kidney disease: therapeutic implications. Drugs 64: 1285–1294, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Elfenbein A, Simons M. Syndecan-4 signaling at a glance. J Cell Sci 126: 3799–3804, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fagotto F. Looking beyond the Wnt pathway for the deep nature of β-catenin. EMBO Rep 14: 422–433, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gall TM, Frampton AE. Gene of the month: E-cadherin (CDH1). J Clin Pathol 66: 928–932, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic disease. Adv Chronic Kidney Dis 17: 118–130, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gan X, Wang J, Wang C, Sommer E, Kozasa T, Srinivasula S, Alessi D, Offermanns S, Simon MI, Wu D. PRR5L degradation promotes mTORC2-mediated PKC-δ phosphorylation and cell migration downstream of Gα12. Nat Cell Biol 14: 686–696, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaush CR, Hard WL, Smith TF. Characterization of an established line of canine kidney cells (MDCK). Proc Soc Exp Biol Med 1966 122: 931–935, 1966. [DOI] [PubMed] [Google Scholar]
- 13.Goulimari P, Kitzing TM, Knieling H, Brandt DT, Offermanns S, Grosse R. Gα12/13 is essential for directed cell migration and localized Rho-Dia1 function. J Biol Chem 280: 42242–42251, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Gu JL, Müller S, Mancino V, Offermanns S, Simon MI. Interaction of G alpha(12) with G alpha(13) and G alpha(q) signaling pathways. Proc Natl Acad Sci USA 99: 9352–9357, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huan Y, van Adelsberg J. Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J Clin Invest 104: 1459–1468, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibraghimov-Beskrovnaya O, Natoli TA. mTOR signaling in polycystic kidney disease. Trends Mol Med 17: 625–633, 2011. [DOI] [PubMed] [Google Scholar]
- 17.Israeli S, Amsler K, Zheleznova N, Wilson PD. Abnormalities in focal adhesion complex formation, regulation, and function in human autosomal recessive polycystic kidney disease epithelial cells. Am J Physiol Cell Physiol 298: C831–C846, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson SK, Ramani VC, Hennings L, Haun RS. Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer 109: 1811–1820, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Juneja J, Casey PJ. Role of G12 proteins in oncogenesis and metastasis. Br J Pharmacol 158: 32–40, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong T, Xu D, Yu W, Takakura A, Boucher I, Tran M, Kreidberg JA, Shah J, Zhou J, Denker BM. G alpha 12 inhibits alpha2 beta1 integrin-mediated Madin-Darby canine kidney cell attachment and migration on collagen-I and blocks tubulogenesis. Mol Biol Cell 20: 4596–4610, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong T, Xu D, Tran M, Denker BM. Regulation of integrin expression by Gα12: an additional potential mechanism modulating cell attachment. Cell Adh Migr 4: 372–376, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lal M, Song X, Pluznick JL, Di Giovanni V, Merrick DM, Rosenblum ND, Chauvet V, Gottardi CJ, Pei Y, Caplan MJ. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet 17: 3105–3117, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leckband DE, de Rooij J. Cadherin adhesion and mechanotransduction. Annu Rev Cell Dev Biol 30: 291–315, 2014. [DOI] [PubMed] [Google Scholar]
- 24.Leuenroth SJ, Bencivenga N, Chahboune H, Hyder F, Crews CM. Triptolide reduces cyst formation in a neonatal to adult transition Pkd1 model of ADPKD. Nephrol Dial Transplant 25: 2187–2194, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lieberthal W, Levine JS. Mammalian target of rapamycin and the kidney part I: the signaling pathway. Am J Physiol Renal Physiol 303: F301–F310, 2012. [DOI] [PubMed] [Google Scholar]
- 26.Lieberthal W, Levine JS. Mammalian target of rapamycin and the kidney part II: pathophysiology and therapeutic implications. Am J Physiol Renal Physiol 303: F180–F391, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol 139: 1861–1872, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luyten A, Su X, Gondela S, Chen Y, Rompani S, Takakura A, Zhou J. Aberrant regulation of planar cell polarity in polycystic kidney disease. J Am Soc Nephrol 21: 1521–1532, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maretzky T, Scholz F, Koten B, Proksch E, Saftig P, Reiss K. ADAM10-mediated e-cadherin release is regulated by proinflammatory cytokines and modulates keratinocyte cohesion in eczematous dermatitis. J Invest Dermatol 128: 1737–1746, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Meigs TE, Fields TA, McKee DD, Casey PJ. Interaction of Galpha 12 and Galpha 13 with the cytoplasmic domain of cadherin provides a mechanism for beta-catenin release. Proc Natl Acad Sci USA 98: 519–524, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menon V, Rudym D, Chandra P, Miskulin D, Perrone R, Sarnak M. Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clin J Am Soc Nephrol 6: 7–13, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer TN, Schwesinger C, Denker BM. Zonula occludens-1 is a scaffolding protein for signaling molecules. Gα12 directly binds to the Src homology 3 domain and regulates paracellular permeability in epithelial cells. J Biol Chem 277: 24855–24858, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Moss ML, Stoeck A, Yan W, Dempsey PJ. ADAM10 as a target for anti-cancer therapy. Curr Pharm Biotechnol 9: 2–8, 2008. [DOI] [PubMed] [Google Scholar]
- 34.Myint TM, Rangan GK, Webster AC. Treatments to slow progression of autosomal dominant polycystic kidney disease: systematic review and meta-analysis of randomized trials. Nephrology 19: 217–226, 2014. [DOI] [PubMed] [Google Scholar]
- 35.Najy AJ, Day KC, Day ML. The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J Biol Chem 283: 18393–18401, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, Ingber DE, Loghman-Adham M, Zhou J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol 17: 1015–1025, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J 8: 1711–1717, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozawa M, Kemler R. Altered cell adhesion activity by pervanadate due to the dissociation of alpha-catenin from the E-cadherin-catenin complex. J Biol Chem 273: 6166–6170, 1998. [DOI] [PubMed] [Google Scholar]
- 39.Parnell SC, Magenheimer BS, Maser RL, Rankin CA, Smine A, Okamoto T, Calvet JP. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem Biophys Res Commun 251: 625–631, 1998. [DOI] [PubMed] [Google Scholar]
- 40.Pruessmeyer J, Ludwig A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin Cell Dev Biol 20: 164–174, 2009. [DOI] [PubMed] [Google Scholar]
- 41.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 16: 179–183, 1997. [DOI] [PubMed] [Google Scholar]
- 42.Robishaw JD, Berlot CH. Translating G protein subunit diversity into functional specificity. Curr Opin Cell Biol 16: 206–209, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Roitbak T, Ward CJ, Harris PC, Bacallao R, Ness SA, Wandinger-Ness A. A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Mol Biol Cell 15: 1334–1346, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russo RJ, Husson H, Joly D, Bukanov NO, Patey N, Knebelmann B, Ibraghimov-Beskrovnaya O. Impaired formation of desmosomal junctions in ADPKD epithelia. Histochem Cell Biol 124: 487–497, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Saadi-Kheddouci S, Berrebi D, Romagnolo B, Cluzeaud F, Peuchmaur M, Kahn A, Vandewalle A, Perret C. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene 20: 5972–5981, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Shao H, Ma J, Guo T, Hu R. Triptolide induces apoptosis of breast cancer cells via a mechanism associated with the Wnt/β-catenin signaling pathway. Exp Ther Med 8: 505–508, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 103: 5466–5471, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soundararajan R, Sayat R, Robertson GS, Marignani PA. Triptolide: An inhibitor of a disintegrin and metalloproteinase 10 (ADAM10) in cancer cells. Cancer Biol Ther: 2054–2062, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Streets AJ, Newby LJ, O'Hare MJ, Bukanov NO, Ibraghimov-Beskrovnaya O, Ong AC. Functional analysis of PKD1 transgenic lines reveals a direct role for polycystin-1 in mediating cell-cell adhesion. J Am Soc Nephrol 14: 1804–1815, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Sutters M, Germino GG. Autosomal dominant polycystic kidney disease: molecular genetics and pathophysiology. J Lab Clin Med 141: 91–100, 2003. [DOI] [PubMed] [Google Scholar]
- 51.Takakura A, Contrino L, Beck AW, Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol 9: 2351–2363, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int 76: 149–168, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer 14: 121–134, 2014. [DOI] [PubMed] [Google Scholar]
- 54.Voyno-Yasenetskaya TA, Faure MP, Ahn NG, Bourne HR, Voyno-Yasenetskaya TA, Faure MP, Ahn NG, Bourne HR. Galpha12 and Galpha13 regulate extracellular signal-regulated kinase and c-Jun kinase pathways by different mechanisms in COS-7 cells. J Biol Chem 271: 21081–21087, 1996. [DOI] [PubMed] [Google Scholar]
- 55.Watanabe T, Sato K, Kaibuchi K. Cadherin-mediated intercellular adhesion and signaling cascades involving small GTPases. Cold Spring Harb Perspect Biol 1: 3009, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Watnick T, Germino GG. mTOR inhibitors in polycystic kidney disease. N Engl J Med 363: 879–881, 2010. [DOI] [PubMed] [Google Scholar]
- 57.Wheelock MJ, Buck CA, Bechtol KB, Damsky CH. Soluble 80-kd fragment of cell-CAM 120/80 disrupts cell-cell adhesion. J Cell Biochem 34: 187–202, 1987. [DOI] [PubMed] [Google Scholar]
- 58.Wilson PD. Polycystic kidney disease. N Engl J Med 350: 151–164, 2004. [DOI] [PubMed] [Google Scholar]
- 59.Wilson PD. Mouse models of polycystic kidney disease. Curr Top Dev Biol 84: 311–350, 2008. [DOI] [PubMed] [Google Scholar]
- 60.Wilson PD, Geng L, Li X, Burrow CR. The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest 79: 1311–1323, 1999. [PubMed] [Google Scholar]
- 61.Worzfeld T, Wettschureck N, Offermanns S. G(12)/G(13)-mediated signalling in mammalian physiology and disease. Trends Pharmacol Sci 29: 582–589, 2008. [DOI] [PubMed] [Google Scholar]
- 62.Wu SG, Lim KC, Huan J, Saidi RF, Sears C. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc Natl Acad Sci USA 95: 14979–14984, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu D, Hemler ME. Metabolic activation-related CD147-CD98 complex. Mol Cell Proteomics 4: 1061–1071, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamaguchi Y, Katoh H, Mori K, Negishi M. Galpha(12) and Galpha(13) interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr Biol 12: 1353–1358, 2002. [DOI] [PubMed] [Google Scholar]
- 65.Yu W, Beaudry S, Negoro H, Boucher I, Tran M, Kong T, Denker BM. H2O2 activates G protein, α12 to disrupt the junctional complex and enhance ischemia reperfusion injury. Proc Natl Acad Sci USA 109: 6680–6685, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu W, Kong T, Beaudry S, Tran M, Negoro H, Yanamadala V, Denker BM. Polycystin-1 protein level determines activity of the Galpha12/JNK apoptosis pathway. J Biol Chem 285: 10243–10251, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu W, Ritchie BJ, Su X, Zhou J, Meigs TE, Denker BM. Identification of polycystin-1 and Gα12 binding regions necessary for regulation of apoptosis. Cell Signal 23: 213–221, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yuasa T, Takakura A, Denker BM, Venugopal B, Zhou J. Polycystin-1L2 is a novel G-protein-binding protein. Genomics 84: 126–138, 2004. [DOI] [PubMed] [Google Scholar]
- 69.Zafar I, Belibi FA, He Z, Edelstein CL. Long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease (PKD). Nephrol Dial Transplant 24: 2349–2353, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou J, Pei Y. Autosomal dominant polycystic kidney disease. In: The Molecular and Genetic Basis of Kidney Disease: A Companion to Brenner and Rector's The Kidney, edited by Mount DB, Pollak MR. Philadelphia, PA: Saunders, 2007, p. 85–171. [Google Scholar]