

Abstract
Severe fever with thrombocytopenia syndrome (SFTS) is an emerging infectious disease with a high case fatality risk and is caused by the SFTS virus (SFTSV). A retrospective study conducted after the first identification of an SFTS patient in Japan revealed that SFTS is endemic to the region, and the virus exists indigenously in Japan. Since the nucleotide sequence of Japanese SFTSV strains contains considerable differences compared with that of Chinese strains, there is an urgent need to establish a sensitive and specific method capable of detecting the Chinese and Japanese strains of SFTSV. A conventional one-step reverse transcription-PCR (RT-PCR) (cvPCR) method and a quantitative one-step RT-PCR (qPCR) method were developed to detect the SFTSV genome. Both cvPCR and qPCR detected a Chinese SFTSV strain. Forty-one of 108 Japanese patients suspected of having SFTS showed a positive reaction by cvPCR. The results from the samples of 108 Japanese patients determined by the qPCR method were in almost complete agreement with those determined by cvPCR. The analyses of the viral copy number level in the patient blood samples at the acute phase determined by qPCR in association with the patient outcome confirmed that the SFTSV RNA load in the blood of the nonsurviving patients was significantly higher than that of the surviving patients. Therefore, the cvPCR and qPCR methods developed in this study can provide a powerful means for diagnosing SFTS. In addition, the detection of the SFTSV genome level by qPCR in the blood of the patients at the acute phase may serve as an indicator to predict the outcome of SFTS.
INTRODUCTION
Severe fever with thrombocytopenia syndrome (SFTS) is an emerging infectious disease with a high case mortality rate (approximately 12%) that is caused by the SFTS virus (SFTSV). The disease was first reported to be endemic to some parts of China in 2011 (1–3). The virus is classified within the Phlebovirus genus of the Bunyaviridae family.
In January 2013, a female patient who lived in western Japan died, and her death was confirmed to be due to SFTSV infection (4). Additionally, 10 SFTS patients were retrospectively identified (4). A phylogenetic study revealed that all eight strains isolated from Japanese patients were clustered in a lineage that was independent from the other lineages of the Chinese strains. Given that an isolate obtained from a South Korean patient with SFTS belongs to a Chinese lineage (5), Japanese SFTSV appears to be an indigenous virus.
For the rapid diagnosis of viral infectious diseases, a method of pathogen detection with both high sensitivity and high specificity is needed. Based on the nucleotide sequences of the Chinese SFTSV isolates, there have been some reports describing a reverse transcription-PCR (RT-PCR)-based method, a reverse transcription-loop-mediated isothermal amplification assay (RT-LAMP), reverse transcription-cross-priming amplification coupled (RT-CPA) with vertical flow (VF) visualization, and TaqMan-based quantitative real-time PCR for SFTSV detection (3, 6–8). However, since there are considerable sequence mismatches between the Chinese and Japanese lineages, especially in the reported TaqMan probe target regions, these primers/probes might not be suitable for use with the Japanese linage (4).
In this study, we developed a conventional one-step RT-PCR (cvPCR) and a quantitative one-step RT-PCR (qPCR) capable of detecting both the Chinese and Japanese SFTSV lineages. The efficacies of these methods were evaluated using clinical specimens collected from Japanese patients suspected of having SFTS. Furthermore, it was demonstrated that the SFTS viral RNA levels in the acute-phase peripheral blood samples were significantly higher from the patients who died than those from the patients who survived, as reported previously (9). On the other hand, there was a contradictory finding that the viral copy number in the serum during the initial 1 to 7 days after the onset was comparable between those from the patients who died and survived (10). We therefore also analyzed the association of the viral copy number in the peripheral blood specimens as determined by qPCR with the prognosis of the patients. The results support the report by Zhang et al. (9), indicating that the viral copy number in the acute-phase patient blood samples correlated with the outcome, with a higher level being associated with a poorer prognosis.
MATERIALS AND METHODS
Clinical specimens from patients suspected of having SFTS.
We asked medical personnel in Japan to inform us on a voluntary basis if they had seen any patients with symptoms similar to those of SFTS, as summarized by Takahashi et al. (4), from 30 January to 30 September 2013. Through the courtesy of prefectural and municipal public health institutes, 149 specimens consisting of serum, plasma, urine, and cerebrospinal fluid from 108 patients were collected and used in this study.
Viruses.
SFTSV strains YG1 and SPL005, previously isolated from serum samples from Japanese patients (4), and a Chinese strain of HB29 were utilized to develop the SFTSV genome amplification systems with RT-PCR. The supernatant of Vero cells infected with Rift Valley fever virus strain MP-12 (RVFV) and Forecariah virus (11, 12), which was a kind gift from Robert Tesh (University of Texas Medical Branch), was used in this study. The infectious dose of each of the SFTSV, RVFV, and Forecariah virus stock solutions was determined by calculating the 50% tissue culture infectious dose (TCID50) in Vero cells either through the visualization of infection by an indirect immunofluorescence assay, as described previously (4), or by observing the cytopathic effects (CPE). Briefly, cells infected with SFTSV were reacted with a rabbit anti-SFTSV NP antibody, followed by staining with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (H+L) (ZyMax Grade; Life Technologies, Carlsbad, CA).
Development of PCR references.
Positive-control plasmids for cvPCR and the RNA reference for qPCR that contained the target sequences of the cvPCR and qPCR primer-probe sets were prepared. To differentiate positive-control contamination of the samples during the cvPCR reaction, a pCR-SFTSV-posicon was constructed. Briefly, portions of two regions (positions 1044 to 1386 and 1267 to 1665) in the SFTSV YG1 small (S) segment were amplified with the addition of EcoRI site at each side (next to positions 1386 and 1267). These PCR products were ligated by a Rapid DNA ligation kit (Roche Applied Science, Penzberg, Germany) after EcoRI digestion. The ligated PCR product was then further ligated into pCR-Blunt II-TOPO using a Zero Blunt TOPO PCR cloning kit (Life Technologies), according to the manufacturer's protocol. The qPCR references for the primer-probe sets targeting the large (L), medium (M), or S segments were constructed with the insertion of the contamplicon sequence, which is, using a method described by Atkinson et al. (13), for the detection of RNA reference contamination by the contamplicon probe. Briefly, each of the virus genomes from positions 3710 to 3832 in the L segment, positions 236 to 357 in the M segment, and positions 1566 to 1702 in the S segment was cloned into pCR2.1-TOPO, which contains the T7 promoter sequence upstream of the cloning site. The complementary sequence of the contamplicon probe shown in Table 1 was replaced by positions 3732 to 3757 in the L segment, positions 291 to 316 in the M segment, or positions 1591 to 1616 in the S segment. The RNA references were produced from the plasmids containing partial L, M, or S segments using the MEGAscript T7 kit (Life Technologies) after XhoI digestion of the pUC plasmids. The copy numbers of the RNA references were determined based on the optical density (OD) values, and samples were diluted to an appropriate concentration in Tris-EDTA (TE) buffer containing 100 μg/ml of yeast tRNA (Life Technologies).
TABLE 1.
The primer-probe sets used for qPCR
| Primer set target (segment) | Primer sequence |
Probe sequencea | Target region (positions) | |
|---|---|---|---|---|
| Forward | Reverse | |||
| N (S) | TGTCAGAGTGGTCCAGGATT | ACCTGTCTCCTTCAGCTTCT | FAM-TGGAGTTTGGTGAGCAGCAGC-BHQ1 | 1566–1702 |
| GPC (M) | GGCAGCTACATGCAGACATA | CCTATCACCCCCAGAATCCA | TexasRed-GCCCTGTTTGGCAATGGGCT-BHQ2 | 236–357 |
| TexasRed-GCCTTGTTTGGCAATGGGCT-BHQ2 | ||||
| RdRp (L) | AACATCCTGGACCTTGCATC | CAATGTGGCCATCTTCTCCA | Cy5-TGGGAGCTCTACTCAGAAGTCCA-BHQ3 | 3710–3832 |
| Cy5-TGGGAGCTCTACTCAGAGGTCCA-BHQ3 | ||||
| Cy5-TGGGAGATCTACTCAGAAGTCCA-BHQ3 | ||||
| Cy5-TGGGAGCTTTACTCAGAAGTCCA-BHQ3 | ||||
| Contamplicon | JOE (HEX)-AGTAGCTTGCTCTTTCATCTGTTACG-BHQ1 | |||
The bases in bold type indicate the mismatch of the sequences between the probes. FAM, 6-carboxyfluorescein; BHQ1, black hole quencher 1; JOE, 2,7-dimethoxy-4,5-dichloro-6-carboxyfluorescein; HEX, hexacholoro-6-carboxyfluorescein.
The synthetic RNA sequences of the Heartland virus (used as a reference) (14) were made from an RNA solution extracted from the virus-infected culture supernatant, which was a kind gift from Hideki Ebihara (National Institute of Allergy and Infectious Diseases). Briefly, portions of three regions that contain potential target sequences for the cvPCR and qPCR (positions 2392 to 2947 in the L segment, 138 to 596 in the M segment, and 896 to 1749 in the S segment of Heartland virus patient 1) were amplified with the addition of a T7 promoter sequence. The synthesized RNA references were produced from the PCR products using the MEGAscript T7 kit (Life Technologies).
Total RNA extraction.
Total RNAs were extracted from 200 μl of the specimens using a High Pure viral RNA kit (Roche Applied Science) according to the manufacturer's protocol. The elution volume for RNA extraction was 50 μl.
Conventional one-step RT-PCR.
SFTSV genome-specific primer sets 1 and 2 (Table 2) were selected using the following procedures. First, based on a Chinese strain, HB29, several candidate primer sets were designed using the DNADynamo sequence analysis software program (BlueTractorSoftware, Ltd., United Kingdom). Next, the primer specificity for the SFTSV genome and cross-reactivity within SFTSV strains were checked by Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Primer sets 1 and 2, expected to be the most specific and broadly reactive primers to SFTSV strains, were selected. Moreover, to make sure they were compatible with both Chinese and Japanese SFTSV strains, a degenerate-base R (A or G) was substituted for a base in primer 1R. For the cvPCR assay, an aliquot of the extracted RNA solution was added to a reaction mixture of the SuperScript III one-step RT-PCR system with Platinum Taq DNA polymerase (Life Technologies), which contains 2× reaction mix, SuperScript III RT-Platinum Taq mix, H2O, and 0.2 μM either specific primer set 1 (expected product length, 458 bp) or 2 (expected product length, 461 bp). After being denatured at 95°C for 2 min, a reverse transcription step was performed at 55°C for 30 min, followed by 45 cycles of amplification under the following conditions: 94°C for 30 s, 52°C for 30 s, and 68°C for 30 s in a PCR machine (Eppendorf, Hamburg, Germany). The positive-control plasmid, which provides 584 bp or 587 bp of PCR products by primer sets 1 or 2, respectively, was subjected to RT-PCR simultaneously with the specimens. A total of 8 μl of the PCR products was digested by 10 U/1iter μl of EcoRI with 1 μl of 10× H buffer as necessary. After electrophoresis on 1% agarose gels, the PCR products with the expected sizes were visualized by staining with GelRed (Biotium, Hayward, CA). For a definite diagnosis of SFTSV infection, the PCR products were purified by a MonoFas DNA purification kit I (GL Sciences, Tokyo, Japan) and sequenced using the ABI Prism 3100 genetic analyzer (Life Technologies), according to the manufacturer's protocol.
TABLE 2.
Primer sets used for cvPCR
| Primer set | Forward | Reverse | Target region in the S segment (positions) |
|---|---|---|---|
| 1 | ATCGTCAAGGCATCAGGGAA | TTCAGCCACTTCACCCGRA | 1045–1502 |
| 2 | CATCATTGTCTTTGCCCTGA | AGAAGACAGAGTTCACAGCA | 1076–1536 |
Quantitative one-step RT-PCR.
Based on the nucleotide sequence of the Japanese strain, SFTSV YG1 (GenBank accession no. AB817995, AB817987, and AB817979), the specific quantitative one-step PCR (qPCR) primer and probe sets that were targeted to the RNA-dependent RNA polymerase (RdRp), glycoprotein (GPC), or nucleoprotein (NP) genes were designed using the DNADynamo sequence analysis software program (Table 1). The primer and probe specificities and cross-reactivities were checked by Primer-BLAST, as described above in the cvPCR primer selection procedure. For the qPCR assay, an aliquot of the extracted RNA solution was added to a reaction mixture for the QuantiTect probe RT-PCR kit (Qiagen, Hilden, Germany), which contains 2× QuantiTect probe RT-PCR master mix, QuantiTect RT mix, H2O, and 10× primer-probe mix, which contains 4 μM each specific primer, 2 μM TaqMan probe(s), and 2 μM contamplicon probe. After PCR activation at 95°C for 15 min, the reverse transcription reaction was carried out at 50°C for 30 min, followed by 45 cycles of amplification under the following conditions: 94°C for 15 s and 60°C for 60 s in a LightCycler Nano (Roche).
The qPCR method was performed according to a previous report (6), with the PCR products amplified from an SFTSV strain, HB29, for the standards and using the QuantiTect multiplex RT-PCR NoROX (NR) kit (Qiagen) instead of synthetic viral RNA transcripts of HB29 and AgPath-ID with the one-step RT-PCR kit (Life Technologies).
Statistical analysis.
One-way analysis of variance (ANOVA) with Bonferroni's multiple-comparison test was used to test the equality of the means of the viral RNA levels in the patient serum samples determined by either the reported qPCR method (6) or the qPCR method developed in this study, and to examine the relationship between the viral copy number level in the blood specimens as determined by qPCR and the prognoses of the patients by using the GraphPad Prism 5 software program (GraphPad software, La Jolla, CA). In the analysis, the copy number level was log10 transformed. If there were multiple blood specimens from a single patient, the specimen that contained the highest value was selected. A significant difference was considered to be present for any P value of <0.05.
Ethics statement.
The clinical specimens for this study were used after obtaining informed consent from the patients themselves (for those who survived) or their responsible family members (for those who died). All of the protocols and procedures were approved by the research and ethics committees of the National Institute of Infectious Diseases (no. 489).
RESULTS
Development of the cvPCR assay and application for the diagnosis of SFTS.
The cvPCR method, which adopted a one-step RT-PCR technique to facilitate the procedure, is targeted to the SFTSV NP gene in the S segment of the genome. As a positive control, a plasmid containing the target sequence, which was artificially modified by producing longer PCR products (the expected sizes for primer sets 1 and 2 are 584 bp and 587 bp, respectively) than those from the authentic viral genome (Fig. 1). In addition, the products were able to be digested by EcoRI (the expected sizes for primer set 1 are 347 bp and 237 bp, and those for primer set 2 are 316 bp and 271 bp) (Fig. 1). These characteristics of the method help to avoid false positives due to contamination with the positive control.
FIG 1.

Establishment of the cvPCR method and the limits of detection. (A) Authentic SFTSV strain HB29 viral RNA (HB29), the plasmid pCR-SFTSV-posicon (P.C.), which contains the artificial sequence, and the nontemplate control (H2O) were amplified by cvPCR using primer set 1 or 2. The PCR products were digested by EcoRI (+EcoRI). The sizes of the products were estimated by using a 2-log DNA ladder marker (NEB). (B and C) Isolated strains HB29, YG1, and SPL005, expanded in Vero cells, were diluted with serum from a healthy donor. The purified viral RNAs from the dilutions of virus-spiked serum were amplified. The resulting HB29, YG1, or SPL005 was amplified by primer set 1 (B) or 2 (C). The listed values are the dilutions of the viral strains. NTC, nontemplate control.
To determine the limit of detection of the cvPCR method, 10-fold serial dilutions of the SFTSV HB29, YG1, and SPL005 strains mixed with serum from a healthy donor were prepared. The purified viral RNA samples from each of the dilutions, which contained from 103 to 10−2 TCID50s of the virus genome per reaction, were loaded in the cvPCR reaction mixture. The viral RNA of each strain was detected until the lowest 100 TCID50/reaction with cvPCR using primer set 2, indicating that ≥125 TCID50 of SFTSV in a 1-ml blood specimen can be detected (Fig. 1B and C). Clinical specimens collected from a total of 108 suspected SFTS patients were tested for the SFTSV genome by the cvPCR method. If positive, the amplified PCR products were confirmed by the determination of the SFTSV nucleotide sequences. The isolation of SFTSV from some of the PCR-positive patient sera was attempted. With the exception of the sera expected to contain small amounts of the virus based on the qPCR results, SFTSV was not successfully isolated from other samples (data not shown). As a result, 41 (17 patients who died, 24 who survived) out of these suspected patients were confirmed to have SFTSV infection by positive amplification of the SFTSV genome using the cvPCR method.
Establishment of the qPCR method.
The amplification efficacy of qPCR targeted to the SFTSV NP, GPC, or RdRp gene was high, with PCR efficacy (EPCR) values of >95% and coefficients of determination (r2 values) of >0.997 (Fig. 2). These results indicated that the efficacies of all three qPCR strategies are close to the theoretical values (15).
FIG 2.

qPCR standard curves and amplification curves. The curves for NP (A and B), GPC (C and D), and RdRp (E and F) were derived from a dilution series of reference RNA. Values of the slope, correlation coefficient (r2), and PCR efficacy (EPCR) were calculated.
To determine the limits of detection of the qPCR methods, the SFTSV HB29, YG1, and SPL005 strains were used as described above in the cvPCR section. Based on the RNA copy number in the culture supernatant containing 1 TCID50 of SFTS, the theoretical values ranging from 103 to 10−2 TCID50 were plotted (Fig. 3A to C). The qPCR method using either of the primer-probe sets detected the viral RNA until the lowest 1 TCID50/reaction, which is equivalent to 17.8 (HB29), 20.0 (YG1), and 8.3 (SPL005) copies/reaction (average, 15.4 copies/reaction) at the theoretical value (Fig. 3A to C). The performance was equivalent to that of cvPCR (Fig. 1B and C). These results suggest that qPCR also possesses high efficacy for detecting Japanese and Chinese SFTSV strains.
FIG 3.

Limits of detection of the singleplex or multiplex qPCR to detect either the Chinese or the Japanese strain of SFTSV. Isolated strains HB29, YG1, and SPL005 were diluted with serum as described in Fig. 1, and purified RNA was amplified. The singleplex (A to C) or multiplex (D to F) qPCR copy numbers at each dilution of HB29 (A and D), YG1 (B and E), or SPL005 (C and F) that were detected by NP (square), GPC (triangle), or RdRp (diamond) are shown as dots and lines. The theoretical copy number of each strain estimated by undiluted viruses whose viral titers (TCID50/ml) were known are shown in orange.
Multiplex qPCR methods for NP and GPC with the contamplicon probe were also established. The limits of detection of multiplex qPCR (Fig. 3D to F), which was tested using the viral RNA samples, as described previously, were almost identical to those of the singleplex qPCR (Fig. 3A to C).
The cross-reactivities of the cvPCR and qPCR methods with the other phleboviruses were tested. Samples with 6 × 103 TCID50/reaction of RVFV, 1 × 105 TCID50/reaction of Forecariah virus, and 1 × 106 copies/reaction of synthetic RNA of the L, M, and S segments of Heartland virus were not amplified by either of the PCR methods (data not shown).
These results indicate that the application of the multiplex qPCR would be more convenient for the detection of viral genomes while maintaining its diagnostic efficacy.
Validation of qPCR using specimens from patients suspected to have SFTS.
The efficacies of cvPCR and qPCR for the detection of the SFTSV genome were evaluated using specimens from suspected SFTS patients. The results determined by cvPCR and qPCR were well correlated (Fig. 4). Only four, four, and two of the specimens measured by the primer-probe set targeting NP, GPC, and RdRp, respectively, showed results that conflicted with those of cvPCR (i.e., were cvPCR positive but qPCR negative, or vice versa). All of these specimens showing opposite results between cvPCR and qPCR were either cerebrospinal fluid or urine samples from patients with SFTSV genome amplification-positive serum samples, except for one specimen, which was a peripheral blood specimen collected from an SFTS patient 7 days after onset. The viral RNA was not detected from five out of seven urine specimens and one out of two cerebrospinal fluid samples, or if it was detected, the RNA level was low (i.e., <104 copies/ml), although around 105 to 108 copies/ml of viral RNA were circulating in the blood of these patients during the acute phase (Table 3). On the contrary, the SFTSV genome was always detected in the throat swab specimens taken from patients who were positive for SFTSV viremia. The cvPCR and qPCR results were 100% identical if the acute-phase blood specimens were used for validation.
FIG 4.
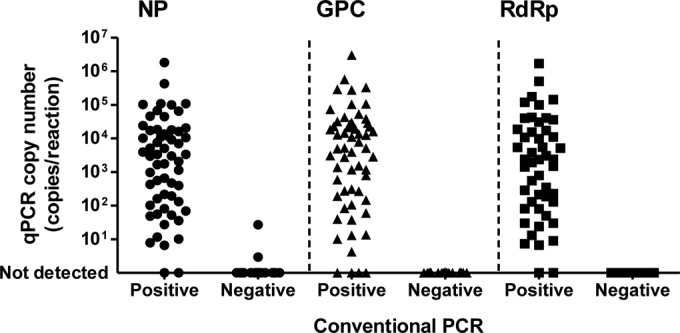
Agreement of cvPCR and qPCR results validated by clinical specimens from patients with suspected SFTS. The results (SFTSV positive or negative) determined by cvPCR (x axis) and the viral RNA copy number determined by qPCR (y axis) from each specimen are plotted as dots.
TABLE 3.
Results of various specimens from patients confirmed to have had SFTS
| Patient IDa | Outcome | Specimen source | Collection time (days after onset) | cvPCR resultc | The log10 viral RNA copy no. (in 1 ml of specimen) ofb: |
||
|---|---|---|---|---|---|---|---|
| NP | GPC | RdRp | |||||
| 010 | Died | Serum | 5 | + | 6.41 | 6.82 | 6.33 |
| Cerebrospinal fluid | 5 | + | − | − | 3.05 | ||
| 067 | Survived | Serum | 6 | + | 6.75 | 7.13 | 6.72 |
| Urine | 6 | − | − | − | − | ||
| 084 | Died | Serum | 5 | + | 8.35 | 8.57 | 8.32 |
| Throat swab | 8 | + | 7.13 | 7.54 | 7.09 | ||
| 087 | Survived | Serum | 5 | + | 7.10 | 6.19 | 7.34 |
| Cerebrospinal fluid | 11 | + | 2.99 | − | 2.92 | ||
| Throat swab | 5 | + | 3.84 | 3.70 | 4.00 | ||
| Urine | 5 | + | − | − | − | ||
| 089 | Died | Serum | 3 | + | 6.48 | 6.60 | 6.71 |
| Urine | 3 | − | 2.57 | − | − | ||
| 097 | Survived | Serum | 6 | + | 5.08 | 5.69 | 4.32 |
| Throat swab | 6 | + | 4.43 | 3.67 | 2.70 | ||
| Urine | 6 | − | − | − | − | ||
| Serum | 9 | + | 2.92 | 3.23 | − | ||
| Urine | 9 | − | − | − | − | ||
| 104 | Died | Serum | 3 | + | 5.69 | 5.79 | 6.01 |
| Throat swab | 3 | + | 3.16 | 3.21 | 3.29 | ||
| Urine | 3 | + | 3.65 | 2.73 | 3.07 | ||
| 108 | Died | Serum | 3 | + | 6.93 | 6.96 | 7.15 |
| Urine | 4 | + | 3.93 | − | − | ||
ID, identification.
NP, nucleoprotein; GPC, glycoprotein; RdRp, RNA-dependent RNA polymerase.
+, detected; −, not detected.
To compare the limits of detection of the cvPCR and qPCR methods developed in this study with that of the qPCR method reported previously (6), the viral copy numbers in some of the clinical specimens were measured by the method reported here (Table 4). All of the specimens determined to be negative by the developed cvPCR and qPCR methods were also determined to be negative by the previously reported qPCR method. However, the SFTSV genome was not amplified in the specimens (i.e., 064A and 097A) by using the S primer-probe set of the reported method. In addition, although the RNA copy numbers of HB29 determined by each of the methods as a control were not significantly different, those of 062A1, 067A, and 082A determined by the previously reported qPCR method were significantly lower than those determined by the new qPCR method. In particular, the viremia levels in 062A1 and 067A were >10 times higher than those determined by the previously reported qPCR method, especially for the detection of the Japanese strains.
TABLE 4.
Comparison of the PCR methods
| Specimen IDa | Reported qPCR method (log10 copies/ml of specimen) for segment: |
cvPCR result forb: |
qPCR result (log10 copies/ml of specimen): |
P valuec | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S | M | L | Mean | Set 1 | Set 2 | NP | GPC | RdRp | Mean | ||
| 057A | 5.02 | 5.75 | 5.60 | 5.46 | + | + | 6.23 | 6.57 | 5.24 | 6.01 | NSd |
| 062A1 | 4.61 | 4.87 | 4.81 | 4.76 | + | + | 5.62 | 6.14 | 5.58 | 5.78 | <0.01 |
| 064A | − | 3.53 | 3.62 | 3.57 | + | + | 4.01 | 4.54 | 3.57 | 4.04 | NS |
| 067A | 5.42 | 5.70 | 5.61 | 5.58 | + | + | 6.75 | 7.13 | 6.72 | 6.86 | <0.001 |
| 078A3 | 3.67 | 4.15 | 4.11 | 3.98 | + | + | 4.69 | 5.23 | 4.46 | 4.79 | NS |
| 082A | 4.76 | 4.89 | 4.89 | 4.85 | + | + | 5.81 | 5.99 | 5.47 | 5.76 | <0.05 |
| 090A | 5.28 | 5.57 | 5.51 | 5.45 | + | + | 6.06 | 6.46 | 6.14 | 6.22 | NS |
| 097A1 | − | 4.86 | 4.43 | 4.64 | + | + | 5.08 | 5.69 | 4.32 | 5.03 | NS |
| 076A | − | − | − | − | − | − | − | − | |||
| 079A | − | − | − | − | − | − | − | − | |||
| 081A | − | − | − | − | − | − | − | − | |||
| 092A | − | − | − | − | − | − | − | − | |||
| 093A | − | − | − | − | − | − | − | − | |||
| 094A1 | − | − | − | − | − | − | − | − | |||
| HB29 (2 × 105 TCID50/ml) | 6.01 | 6.13 | 6.02 | 6.05 | + | + | 6.46 | 6.48 | 6.38 | 6.44 | NS |
ID, identification.
+, detected; −, not detected.
P value was computed using a one-way ANOVA with Bonferroni's multiple-comparison test.
NS, not significant.
Viral copy number and patient outcome.
To examine if the viremia level in the peripheral blood of SFTS patients is relevant to their survival outcome, the viral copy number in the blood specimens as determined by qPCR and the prognoses of the patients were analyzed. There were statistically significant differences in the copy numbers between the patients with fatal and nonfatal outcomes (Fig. 5A). The mean viral copy numbers in the patients who died and survived (6.4 versus 5.2 for NP, 6.7 versus 5.4 for GPC, 6.4 versus 5.0 for RdRp, respectively) differed by >1.2 log10 copies/ml for all of the primer-probe sets.
FIG 5.
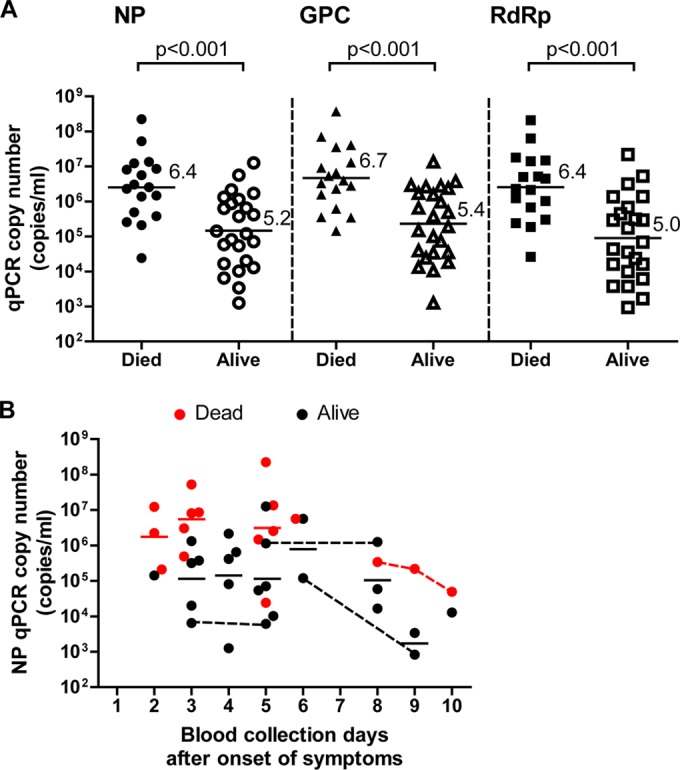
Relationship between the viral RNA level in the SFTS patient blood samples and the patient survival outcomes. (A) Viral RNA copy numbers in the blood samples from SFTS patients are plotted as dots and arranged based on the patients' survival outcomes. The mean of each group is indicated by a horizontal bar and the value. One-way ANOVA with Bonferroni's multiple-comparison test was used to determine the level of statistical significance. The calculated P values are shown above the groups that were compared. (B) Kinetics of the viral NP RNA copy number in the blood specimens after the onset of symptoms. The copy numbers in the blood specimens from SFTS patients who died (red) or survived (black) are plotted as dots for each of the collection days (days after the onset of symptoms). Horizontal solid lines for each day indicate the mean RNA copy number in the blood specimens collected from patients who died (red) or survived (black). The dots connected by dashed lines indicate specimens collected from the same patients.
We then analyzed the relationship of the viral NP gene copy number in the blood with the collection day (time after onset) (Fig. 5B). It was demonstrated that the average copy numbers on each blood collection day of the surviving patients were lower than those of the patients who died for ≥3 to 10 days after onset. Even when the data were analyzed using the GPC or RdRp gene copy numbers, the results indicated a similar tendency (data not shown).
DISCUSSION
This study suggests that both cvPCR and qPCR possess high efficacy for the diagnosis of SFTS caused by infection with either Chinese or Japanese SFTSV strains, because it was reported that the viremia level in the SFTS patients was >103 copies/ml (6). In addition, serum/plasma is the most suitable matrix for the detection of the SFTSV genome, since the serum/plasma specimens contained a higher viral RNA load than the other specimens. In addition, there was 100% agreement between the cvPCR and qPCR results in the serum specimens that were collected during the first 10 days after onset.
There were significant differences in the viral copy numbers in the peripheral blood between the patients who died and those who survived, even when the number of days elapsed after the onset of the disease was not considered for the statistical analysis (Fig. 5A). The reason for this is that the viral copy number was maintained at a low level in patients who survived SFTS, at least during the period beginning 3 days after onset and lasting until their recovery, but was maintained at a high level in the patients who died (Fig. 5B). Although there were previously conflicting findings about the relevance of the viral copy number in patient blood samples with regard to their survival outcome (9, 10), our study confirmed that the SFTS viral RNA level, even the Japanese lineage, in patient blood samples is associated with their prognosis. The small number of cases may be a reason for the results reported by Gai et al. (10) that did not find any significant relationship between these two factors. Since several other significant prognostic indicators (e.g., cytokine levels, chemokine levels, platelet counts, lactate dehydrogenase [LDH] levels, and aspartate transaminase [AST] levels) have already been reported (9, 10, 16, 17), the combined use of the viral RNA level with these indicators may provide more substantial information that can be used to select the therapeutic strategy and predict patient prognosis.
Interestingly, all of the throat swabs collected from patients who were confirmed to have viral RNA in their serum contained a detectable amount of the viral RNA (Table 3). This suggests that it may be possible to develop a rapid and simple diagnostic system for SFTS based on immunochromatography, such as those widely used for the identification of influenza (18).
On the other hand, it was very hard to detect the viral RNA from urine and cerebrospinal fluid samples (Table 3). It was previously reported that some patients exhibit neurological symptoms (1, 3, 4, 17). However, we tested cerebrospinal fluid samples from only two patients, so further studies are needed to determine whether the nervous tissue is a major site of virus multiplication. In addition, there was a possibility that blood contamination, especially in the cerebrospinal fluid samples, may have occurred during the collection.
The present study included only one Chinese strain (HB29) for the evaluation. However, our PCR systems detected a Japanese SFTSV strain that clustered in the Chinese genotype (data not shown). In addition, the PCR primer specificity for the SFTSV genome and cross-reactivities within SFTSV strains were checked using the Primer-BLAST Web-based program.
In summary, we established cvPCR and qPCR methods that were able to detect SFTSV lineages distributed in both China and Japan. Both the cvPCR and qPCR methods were validated as being comparable in terms of their sensitivity and specificity for the detection of SFTSV. In addition, it was confirmed that the qPCR method developed in this study has a lower limit of detection than the qPCR method reported in previous studies, especially for the detection of Japanese strains. The cvPCR likely would be useful for phylogenetic analysis following a definite diagnosis and sequencing. In addition, cvPCR is much less expensive than qPCR. The qPCR method has the advantage that it is possible to know the kinetics of the SFTSV circulating in patients and to determine their prognosis during the acute phase.
ACKNOWLEDGMENTS
The Chinese SFTSV strain HB29 was a kind gift from De-Xin Li and Mi-Fang Liang at the National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention.
We thank the Toyama Institute of Health for the collection of clinical specimens used in this study.
This work was supported by grants for Research on Emerging and Re-emerging Infectious Diseases from the Ministry of Health, Labor and Welfare of Japan (H25-Shinko-Ippan-004, H25-Shinko-Shitei-009, and H24-Shinko-Ippan-013) and by Grants-in-Aid for Challenging Exploratory Research (25670222) from the Japan Society for the Promotion of Science.
Footnotes
Published ahead of print 2 July 2014
REFERENCES
- 1.Yu XJ, Liang MF, Zhang SY, Liu Y, Li JD, Sun YL, Zhang L, Zhang QF, Popov VL, Li C, Qu J, Li Q, Zhang YP, Hai R, Wu W, Wang Q, Zhan FX, Wang XJ, Kan B, Wang SW, Wan KL, Jing HQ, Lu JX, Yin WW, Zhou H, Guan XH, Liu JF, Bi ZQ, Liu GH, Ren J, Wang H, Zhao Z, Song JD, He JR, Wan T, Zhang JS, Fu XP, Sun LN, Dong XP, Feng ZJ, Yang WZ, Hong T, Zhang Y, Walker DH, Wang Y, Li DX. 2011. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 364:1523–1532. 10.1056/NEJMoa1010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang YZ, Zhou DJ, Xiong Y, Chen XP, He YW, Sun Q, Yu B, Li J, Dai YA, Tian JH, Qin XC, Jin D, Cui Z, Luo XL, Li W, Lu S, Wang W, Peng JS, Guo WP, Li MH, Li ZJ, Zhang S, Chen C, Wang Y, de Jong MD, Xu J. 2011. Hemorrhagic fever caused by a novel tick-borne bunyavirus in Huaiyangshan, China. Zhonghua Liu Xing Bing Xue Za Zhi 32:209–220. [PubMed] [Google Scholar]
- 3.Xu B, Liu L, Huang X, Ma H, Zhang Y, Du Y, Wang P, Tang X, Wang H, Kang K, Zhang S, Zhao G, Wu W, Yang Y, Chen H, Mu F, Chen W. 2011. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: discovery of a new bunyavirus. PLoS Pathog. 7:e1002369. 10.1371/journal.ppat.1002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi T, Maeda K, Suzuki T, Ishido A, Shigeoka T, Tominaga T, Kamei T, Honda M, Ninomiya D, Sakai T, Senba T, Kaneyuki S, Sakaguchi S, Satoh A, Hosokawa T, Kawabe Y, Kurihara S, Izumikawa K, Kohno S, Azuma T, Suemori K, Yasukawa M, Mizutani T, Omatsu T, Katayama Y, Miyahara M, Ijuin M, Doi K, Okuda M, Umeki K, Saito T, Fukushima K, Nakajima K, Yoshikawa T, Tani H, Fukushi S, Fukuma A, Ogata M, Shimojima M, Nakajima N, Nagata N, Katano H, Fukumoto H, Sato Y, Hasegawa H, Yamagishi T, Oishi K, Kurane I, Morikawa S, Saijo M. 2014. The first identification and retrospective study of severe Fever with thrombocytopenia syndrome in Japan. J. Infect. Dis. 209:816-827. 10.1093/infdis/jit603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim KH, Yi J, Kim G, Choi SJ, Jun KI, Kim NH, Choe PG, Kim NJ, Lee JK, Oh MD. 2013. Severe fever with thrombocytopenia syndrome, South Korea, 2012. Emerg. Infect. Dis. 19:1892–1894. 10.3201/eid1911.130792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Y, Liang M, Qu J, Jin C, Zhang Q, Li J, Jiang X, Wang Q, Lu J, Gu W, Zhang S, Li C, Wang X, Zhan F, Yao W, Bi Z, Wang S, Li D. 2012. Early diagnosis of novel SFTS bunyavirus infection by quantitative real-time RT-PCR assay. J. Clin. Virol. 53:48–53. 10.1016/j.jcv.2011.09.031. [DOI] [PubMed] [Google Scholar]
- 7.Yang G, Li B, Liu L, Huang W, Zhang W, Liu Y. 2012. Development and evaluation of a reverse transcription loop-mediated isothermal amplification assay for rapid detection of a new SFTS bunyavirus. Arch. Virol. 157:1779–1783. 10.1007/s00705-012-1348-1. [DOI] [PubMed] [Google Scholar]
- 8.Cui L, Ge Y, Qi X, Xu G, Li H, Zhao K, Wu B, Shi Z, Guo X, Hu L, You Q, Zhang LH, Freiberg AN, Yu X, Wang H, Zhou M, Tang YW. 2012. Detection of severe fever with thrombocytopenia syndrome virus by reverse transcription-cross-priming amplification coupled with vertical flow visualization. J. Clin. Microbiol. 50:3881–3885. 10.1128/JCM.01931-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang YZ, He YW, Dai YA, Xiong Y, Zheng H, Zhou DJ, Li J, Sun Q, Luo XL, Cheng YL, Qin XC, Tian JH, Chen XP, Yu B, Jin D, Guo WP, Li W, Wang W, Peng JS, Zhang GB, Zhang S, Chen XM, Wang Y, Li MH, Li Z, Lu S, Ye C, de Jong MD, Xu J. 2012. Hemorrhagic fever caused by a novel bunyavirus in China: pathogenesis and correlates of fatal outcome. Clin. Infect. Dis. 54:527–533. 10.1093/cid/cir804. [DOI] [PubMed] [Google Scholar]
- 10.Gai ZT, Zhang Y, Liang MF, Jin C, Zhang S, Zhu CB, Li C, Li XY, Zhang QF, Bian PF, Zhang LH, Wang B, Zhou N, Liu JX, Song XG, Xu A, Bi ZQ, Chen SJ, Li DX. 2012. Clinical progress and risk factors for death in severe fever with thrombocytopenia syndrome patients. J. Infect. Dis. 206:1095–1102. 10.1093/infdis/jis472. [DOI] [PubMed] [Google Scholar]
- 11.Matsuno K, Weisend C, Travassos da Rosa AP, Anzick SL, Dahlstrom E, Porcella SF, Dorward DW, Yu XJ, Tesh RB, Ebihara H. 2013. Characterization of the Bhanja serogroup viruses (Bunyaviridae): a novel species of the genus Phlebovirus and its relationship with other emerging tick-borne phleboviruses. J. Virol. 87:3719–3728. 10.1128/JVI.02845-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hubálek Z. 2009. Biogeography of tick-borne Bhanja virus (Bunyaviridae) in Europe. Interdiscip. Perspect. Infect. Dis. 2009:372691. 10.1155/2009/372691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atkinson B, Chamberlain J, Logue CH, Cook N, Bruce C, Dowall SD, Hewson R. 2012. Development of a real-time RT-PCR assay for the detection of Crimean-Congo hemorrhagic fever virus. Vector Borne Zoonotic Dis. 12:786–793. 10.1089/vbz.2011.0770. [DOI] [PubMed] [Google Scholar]
- 14.McMullan LK, Folk SM, Kelly AJ, MacNeil A, Goldsmith CS, Metcalfe MG, Batten BC, Albariño CG, Zaki SR, Rollin PE, Nicholson WL, Nichol ST. 2012. A new phlebovirus associated with severe febrile illness in Missouri. N. Engl. J. Med. 367:834–841. 10.1056/NEJMoa1203378. [DOI] [PubMed] [Google Scholar]
- 15.Reynisson E, Josefsen MH, Krause M, Hoorfar J. 2006. Evaluation of probe chemistries and platforms to improve the detection limit of real-time PCR. J. Microbiol. Methods 66:206–216. 10.1016/j.mimet.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Cui N, Bao XL, Yang ZD, Lu QB, Hu CY, Wang LY, Wang BJ, Wang HY, Liu K, Yuan C, Fan XJ, Wang Z, Zhang L, Zhang XA, Hu LP, Liu W, Cao WC. 2014. Clinical progression and predictors of death in patients with severe fever with thrombocytopenia syndrome in China. J. Clin. Virol. 59:12–17. 10.1016/j.jcv.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 17.Weng Y, Chen N, Han Y, Xing Y, Li J. 2014. Clinical and laboratory characteristics of severe fever with thrombocytopenia syndrome in Chinese patients. Braz J. Infect. Dis. 18:88–91. 10.1016/j.bjid.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chartrand C, Leeflang MM, Minion J, Brewer T, Pai M. 2012. Accuracy of rapid influenza diagnostic tests: a meta-analysis. Ann. Intern. Med. 156:500–511. 10.7326/0003-4819-156-7-201204030-00403. [DOI] [PubMed] [Google Scholar]