

Abstract
While all verotoxin-producing Escherichia coli O157:H7 bacteria are considered potential pathogens, their genetic subtypes appear to differ in their levels of virulence. The aim of this study was to compare the distribution of subtypes of E. coli O157:H7 in the cattle reservoir and in human cases with and without severe complications in order to gain clues about the relationship between subtype and relative virulence. A lineage-specific polymorphism assay (LSPA-6), multilocus variable-number tandem-repeat analysis (MLVA), and a novel real-time PCR assay to identify clade 8 were applied to a large and representative set of isolates from cattle from 1996 to 2009 (n = 381) and human cases from 2008 to 2011 (n = 197) in Sweden. Draft genome sequences were produced for four selected isolates. The E. coli O157:H7 isolates in Swedish cattle generally belonged to four groups with the LSPA-6 profiles 211111 (clade 8/non-clade 8), 213111, and 223323. The subtype composition of the cattle isolates changed dramatically during the study period with the introduction and rapid spread of the low-virulence 223323 subtype. The human cases presumed to have been infected within the country predominantly carried isolates with the profiles 211111 (clade 8) and 213111. Cases progressing to hemolytic-uremic syndrome (HUS) were mostly caused by clade 8, with MLVA profiles consistent with Swedish cattle as the source. In contrast, infections contracted abroad were caused by diverse subtypes, some of which were associated with a particular region. The work presented here confirms the high risk posed by the clade 8 variant of E. coli O157:H7. It also highlights the dynamic nature of the E. coli O157:H7 subtype composition in animal reservoirs and the importance of this composition for the human burden of disease.
INTRODUCTION
Historically, the Escherichia coli serotype O157:H7 has been the most common cause of enterohemorrhagic E. coli (EHEC) infections in Sweden, causing several major outbreaks (1, 2). In recent years, other serotypes, in particular E. coli O26 and O103, have been increasingly common causes of gastrointestinal disease (3); however, there is a strong association between E. coli O157:H7 and rare but severe complications, such as hemolytic-uremic syndrome (HUS), which in particular make these bacteria a public health concern (4). Prevalence studies in animal reservoirs of this zoonotic pathogen have shown E. coli O157:H7 to be present in 3.3% of the fecal samples from Swedish cattle (5) and 1.8% from Swedish sheep (6) at slaughter. In the human population, 21 to 61 domestic cases of E. coli O157:H7 per year were reported in Sweden from 2006 to 2012, corresponding to an incidence of 0.2 to 0.7 cases per 100,000 population (7). The overall incidence of verotoxin-producing E. coli (VTEC) in Sweden is high, by European standards (8).
Previous studies have shown that subclones of E. coli O157:H7, referred to as lineages and clades, seem to differ in their pathogenicity or tendency to infect humans (9–11). Thus, the prevalence of high-virulence subgroups might be more relevant than the overall E. coli O157:H7 prevalence, partially explaining the marked differences between countries and regions regarding E. coli O157:H7 morbidity. We previously showed a strong connection between one such group, clade 8, and Swedish farms linked to cases of infection in humans versus E. coli O157:H7-positive dairy farms in general (12). The representatives of this clade caused the biggest domestic EHEC outbreak in Sweden, with 135 cases in 2005 being associated with the consumption of lettuce (1).
Several molecular methods are used for typing E. coli O157:H7. Multilocus variable-number tandem-repeat analysis (MLVA) is fast and cost-effective and has been applied to E. coli O157 (12, 13), as well as many other bacteria. This method can be used, e.g., to trace spread of the bacteria between animals and from animals to humans. While MLVA profiles are expected to change over time, a system including six variable-number tandem repeats (VNTRs) and insertions/deletions forming a lineage-specific polymorphism assay (LSPA-6) uses more stable genetic variation to divide E. coli O157:H7 into lineages I, II, and I/II (14, 15). Based on a set of informative single nucleotide polymorphisms, Manning et al. (9) defined a number of clades, among which the previously mentioned clade 8 is of particular interest for the Swedish situation and can be identified using a single nucleotide polymorphism (SNP) in a gene encoding a hypothetical protein (ECs2357) (12, 16). Whole-genome sequencing-based SNP typing has largely confirmed the division of E. coli O157:H7 into lineages and clades and identified further markers for these subgroups (17–20). In contrast with traditional methods, such as phage typing and pulsed-field gel electrophoresis, which are still commonly used, the new-generation typing methods can easily be implemented in standardized ways, allowing a direct comparison of the data between institutes and countries.
In the present study, we used a combination of MLVA, LSPA-6, SNP data, and verotoxin profiles to phylogenetically subdivide E. coli O157:H7 isolated from cattle in prevalence studies in 1996 to 2009, as well as all human case isolates of E. coli O157:H7 submitted to the Public Health Agency of Sweden from 2008 to 2011. We compared the relative prevalences of subclones in the cattle reservoir over time and used over- and underrepresentation in the human cases infected within the country compared to cattle prevalence as an indicator of relative virulence. We also investigated the phylogenetic characteristics of the isolates known to have caused HUS. Finally, we used whole-genome sequencing to compare the representatives of the major groups found, both to each other and to available E. coli O157:H7 reference sequences from other countries.
MATERIALS AND METHODS
The isolates of E. coli O157:H7 from slaughterhouse prevalence studies performed between 1996 and 2009 (n = 381) (5) were held in storage at −70°C at the National Veterinary Institute, Uppsala, Sweden. The human case isolates were sent to The Public Health Agency of Sweden by the clinical laboratories in Sweden for confirmation and typing (2008 to 2011, n = 197). There were no significant outbreaks during the study period, but around 20 of the included case isolates had known or suspected family associations to other cases in the set, forming familial outbreak groups of 2 to 4 cases each.
Clade 8 status was determined using an SNP in the ECs2357 gene (16), targeted with a novel TaqMan assay using the primers ECs2357f1 (5′-CAGGCGTCGCAATAATGGTTT-3′) and ECs2357r1 (5′-ATGGCTGGGTGCGAATCTG-3′), as well as two competing probes: ECs2357_G (5′-6-FAM-CCAGTGGCTCGGAAT-BHQplus-3′) (FAM, 6-carboxyfluorescein; BHQ, black hole quencher) and ECs2357_T (5′-CAL Fluor orange-TCCAGTGGCTCGTAA-BHQplus-3′) (Biosearch Technologies, Novato, CA, USA) run in a single 15-μl reaction with quantitative PCR (qPCR) Fastmix, Low ROX (Quanta BioSciences, Gaithersburg, MD, USA), 0.33 μM each primer, and 0.13 μM each probe. An ABI7500 Fast real-time PCR instrument (Life Technologies, Carlsbad, CA, USA) was used with the program of 95°C for 3 min, followed by 35 cycles of 95°C for 3 s and 66°C for 30 s. The clade 8 isolates produced a threshold cycle (CT) of <30 for the CAL Fluor orange probe and no CT for the FAM probe, while the non-clade 8 isolates produced a FAM CT of <30 and no CAL Fluor orange CT.
MLVA analysis according to the method developed by Hyytiä-Trees et al. (13) was performed as described previously (12) but using a new forward primer for VNTR 34 (5′-AACAAGGTTCTGGCGTGTTACCAACGCG-3′), due to previous issues with false negatives. A panel of eight isolates selected to reasonably cover the observed range of copy number variation was sequenced using previously described methods (21) for all eight loci and used for size correction to remove systematic errors from the capillary gel electrophoresis. The profiles are reported as the number of repeats at each locus, and this approach results in EDL933 being assigned the profile 9-10-11-5-6-6-8-7. Clustering of the MLVA profiles was performed using the minimum spanning tree algorithm implemented in BioNumerics 6.6 (Applied Maths, Sint-Martens-Latem, Belgium), using each VNTR as a categorical variable. The descriptive statistics for the MLVA analysis are presented in Table 1.
TABLE 1.
Descriptive statistics for MLVA performed on Swedish O157:H7 isolates
| Statistics | MLVA | VNTR 3 | VNTR 34 | VNTR 9 | VNTR 25 | VNTR 17 | VNTR 19 | VNTR 36 | VNTR 37 |
|---|---|---|---|---|---|---|---|---|---|
| Total no. of alleles | 379 | 22 | 6 | 6 | 18 | 15 | 10 | 13 | 16 |
| PCR negatives (%) | 17 | 0.1 | 3.3 | 0.5 | 1.4 | 1.0 | |||
| SID, cattlea | 0.99 | 0.90 | 0.35 | 0.91 | 0.66 | 0.72 | 0.70 | 0.81 | 0.74 |
| SID, human cases infected in Sweden | 0.98 | 0.90 | 0.38 | 0.91 | 0.73 | 0.69 | 0.60 | 0.82 | 0.70 |
| SID, human cases infected abroad | 0.98 | 0.94 | 0.61 | 0.91 | 0.67 | 0.87 | 0.80 | 0.77 | 0.90 |
SID, Simpson's index of diversity (1 − D).
LSPA-6 was performed according to the original protocol (14) but with all forward primers labeled with FAM and analyzed using capillary gel electrophoresis in the same way as for the MLVA assay. EDL933 was used for size correction (profile 111111). All previously undescribed alleles were sequenced as described above for a representative of each profile in which they occurred; the sequence variation in these alleles is described in Table 2. For the cattle isolates, due to the low diversity and large number of isolates, a total of 138 representative isolates were selected and typed by the LSPA-6 so that all MLVA profiles either contained a typed isolate or were within one locus variation of a typed isolate. The LSPA-6 profiles of the remaining isolates were inferred based on this. The same approach was applied to the human clade 8 isolates, while all human non-clade 8 isolates were typed. No conflicts in the data were observed using this approach.
TABLE 2.
Atypical LSPA-6 alleles confirmed by sequencing
| Target | Sequenced in isolate: | Allele no. | Size of EDL933 (bp) | Size change (bp) | Sequence difference | Full isolate profile |
|---|---|---|---|---|---|---|
| Z5935 | T234/00 | 6 | 133 | +27 | +2× TACCTGGAA +1× CACCTGGAA | 262123 |
| Z5935 | E256/10 | 7 | 133 | −9 | −1× TACCTGGAA | 273111 |
| Z5935 | E14/11 | 4 | 133 | +18 | +2× CACCTGGAA | 242123 |
| yhcG | T458/00 | 3 | 394 | −2 | Deletion CACC(AT)TGCC | 223323 |
| yhcG | T171/00 | 3 | 394 | −2 | Deletion CACC(AT)TGCC | 213111 |
| yhcG | E280/11 | 4 | 394 | −1 | Deletion CACCA(T)TGCC | 224311 |
| yhcG | E322/11 | 4 | 394 | −1 | Deletion CACCA(T)TGCC | 214311 |
| rtcB | 08-BKT055439 | 3 | 270 | +18 | +2× TGGGAACGC | 222133 |
Four representative isolates underwent whole-genome sequencing using 454 technology on a Genome Sequencer FLX system (Roche, Basel, Switzerland) with single-end reads, producing ∼8 to 12× average coverage with 98.3 to 99.1% high-quality assembly positions (with a quality score of ≥Q40). The read data were assembled using Newbler 2.5.3. The SNP states for the known variable positions for clade typing (9) and testing association with lineage II (17) were extracted from contigs using local BLAST+ databases. The SNP contexts that could not be extracted from one or more of the sequenced genomes were excluded from the final analysis. Phylogenetic analysis of the SNP data was performed using the NeighborNet algorithm in SplitsTree4 (22), with the default settings.
Nucleotide sequence accession numbers.
Raw sequence data are available from the NCBI Sequence Read Archive (SRA) under the project PRJNA215274, and the assembled contigs have been deposited in the whole-genome shotgun (WGS) database under accession no. JJOJ00000000, JJOK00000000, JJOL00000000, and JJOM00000000 (Table 3).
TABLE 3.
Genetic characteristics of representatives from each of the four major subclones of E. coli O157:H7 in the Swedish cattle reservoir
| Isolate | Accession no. | Clade | LSPA-6 | Gene (position) |
Verotoxin(s) | % LII SNP statea | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| tir (255) | fimH (468) | agaF (271) | ECs2357(539) | wrbA | cdtV | ||||||
| 09-BKT048303 | JJOM00000000 | 7 | 223323 | A + 16-bp insert | C | G | C | Intact | − | 2 | 49 |
| 08-BKT061141 | JJOL00000000 | 7 | 213111 | T | C | G | C | Intact | − | 1 + 2 | 21 |
| T1543/06 | JJOK00000000 | 8 | 211111 | T | C | A | T | Intact | − | 2 | 0 |
| 09-BKT002497 | JJOJ00000000 | 5/6 | 211111 | T | A | G | C | Intact | + | 2 | 1 |
LII, lineage II.
RESULTS AND DISCUSSION
Four subtypes of E. coli O157:H7 dominate in Swedish cattle, two of which are also prevalent among isolates from human cases.
LSPA-6 typing and ECs2357 SNP typing generally agreed with the verotoxin gene profiling results in the division of cattle isolates into four major subtypes. The isolates with the LSPA-6 profile 213111 were very common in the isolates studied, regardless of the source (Fig. 1). The majority of the cattle isolates (98%) and human case isolates (95%) with this profile were vtx1 positive. In both cases, virtually all of these isolates also carried vtx2 genes. The majority of the isolates with any LSPA-6 profile other than 213111 carried only the vtx2 gene; 3% of the human isolates and only 1% of the cattle isolates were vtx1 positive in the non-213111 profiles. Clustering analysis of the MLVA data from the 213111 isolates revealed a high degree of variability within this group, but close links between the cattle and human case isolates were seen. However, only a single perfect MLVA profile match between the groups was observed. The isolates with this LSPA-6 profile were frequently negative for VNTR 3 (53% in cattle isolates and 43% in patient isolates). This characteristic was not seen in conjunction with any other profile except a single occurrence in 273111, likely representing a derivate of 213111 caused by the loss of a repeat unit in Z5935 (Table 2). The LSPA-6 profile 223323 is prevalent among Swedish cattle isolates but was found in only three isolates from humans, all infected within Sweden. Two of these isolates had identical MLVA profiles (20-7-12-3-4-5-8-7), a profile that was also observed in a cattle isolate. The lineage I/II non-clade 8 isolates from cattle form a tight complex of MLVA profiles. This is to some degree due to a very low variability in VNTR 3, which is one of the most variable markers for other strains. It is conceivable that the low copy number contributes to the low mutation rate in this VNTR. We have previously shown that this subtype generally belongs to phage type 14 and that it frequently carries vtx2c genes and cytolethal distending toxin (12, 21). The few cattle isolates included in this study that do not cluster with the main group of 211111 isolates have been shown to belong to other phage types (23) that appear to be present only transiently or at very low frequencies. The isolates in this project with the characteristics consistent with each of the four major groups found in cattle were previously isolated from Swedish sheep at slaughter (6).
FIG 1.
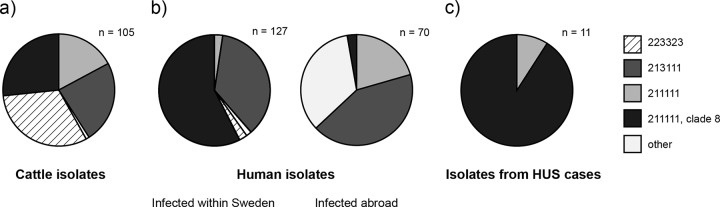
(a) Subtype composition of E. coli O157:H7 strains isolated from Swedish cattle at slaughter in 2008 to 2009. Comparison with isolates from Swedish human cases (2008 to 2011) with and without a recent history of travel abroad before developing symptoms (b) and the subset of the same human case isolates that caused hemolytic-uremic syndrome (c).
Subtype composition of E. coli O157:H7 in the Swedish cattle reservoir has changed dramatically in the last decade by emergence and expansion of a lineage II clone.
A comparison of the LSPA-6 and SNP data from isolates collected from 1996 to 2009 reveals a marked shift in the composition of E. coli O157:H7 subtypes in the cattle reservoir over time (Fig. 2). Strikingly, the most common subtype in the latest prevalence study was not observed before the year 2000 and was still very rare before 2005. All of these isolates had the same lineage II LSPA-6 profile (223323), and typing based on whole-genome sequencing of a representative isolate revealed a high degree of lineage II-related SNPs, as well as a tir allele that is characteristic for this lineage. Lineage II (LII) strains in the United States are common in cattle but rare in human cases (11), suggesting that they are less virulent and/or less likely to be transmitted to humans. These strains generally carry vtx2c genes alone and produce very little verotoxin (24). The same appears to be true for the Swedish LII clone, which is virtually absent among human patients (Fig. 1). Whole-genome analysis of LII isolates has demonstrated traits consistent with improved adaptation to the bovine host environment (17), and it is conceivable that such genetic advantages explain the rapid expansion of the Swedish LII between 2000 and 2009. It is also noteworthy that 213111 isolates have become far less prevalent in the same time frame. The overall prevalence from the different studies performed during this time period have increased from 1.2% positive fecal samples at slaughter in 1996 to 1997 to 3.3% in 2008 to 2009 (5); however, an improved isolation methodology makes a comparison of the results from the different studies problematic. In the absence of a dramatic increase in overall prevalence, the expansion of low-pathogenicity subtypes in animal reservoirs at the expense of more pathogenic ones is likely to lower the burden of disease for the human population.
FIG 2.
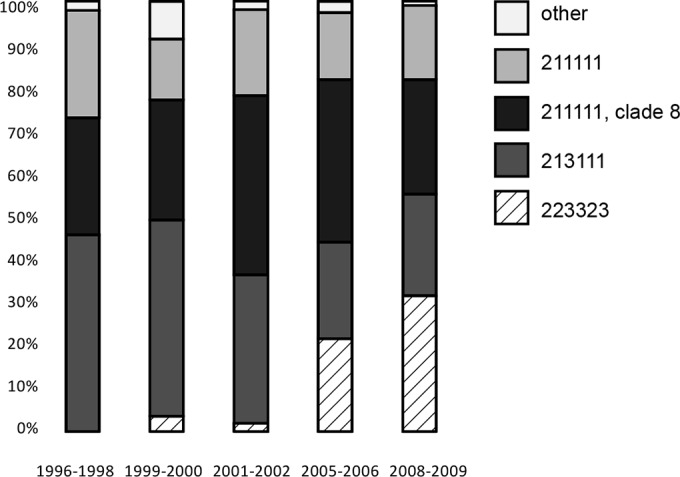
Subtype composition of O157:H7 strains isolated from cattle at slaughter in Sweden, from 1996 to 2009. The division into subtypes is by LSPA-6 and clade 8 typing based on real-time PCR determination of a SNP state in ECs2357.
Clade 8 is overrepresented among human cases compared to cattle and causes HUS more often than do other types of E. coli O157:H7.
Human cases of E. coli O157:H7 infection in the study were 16 times more likely to progress to HUS if they were infected by clade 8 than by non-clade 8 strains (Fig. 1c). This association was statistically significant (Fisher's exact test, P < 0.001), although it was based on a total of only 11 observations of HUS. There was also a clear overrepresentation of clade 8 in the isolates from human cases infected within the country compared to the prevalence of this subclone in the cattle reservoir (Fig. 1). We previously observed an overrepresentation of clade 8 isolates among Swedish farms connected to human cases of disease compared to farms in general (12). The majority of the Swedish clade 8 isolates from humans and cattle cluster together in terms of MLVA profiles (Fig. 3). SNP typing based on a large number of markers revealed a high similarity between a Swedish clade 8 representative and clade 8 isolates from the United States. Clade 8 has caused outbreaks in the United States with high HUS and hospitalization rates compared to outbreaks caused by less virulent subtypes (9). Argentina has a high clade 8 prevalence among E. coli O157:H7 in cattle (∼50%) and even higher among human cases (∼80%) (25), together with an extremely high HUS incidence (26) compared to countries with little or no clade 8 strains. A few studies on clade 8 have been performed in Europe; in a set of Norwegian isolates investigated by Haugum et al. (27), clade 8 occurred more commonly in human than in nonhuman sources but was not significantly associated with HUS (27). A recent study comparing cattle and human isolates of E. coli O157:H7 from the Netherlands found no specific association of clade 8 with strains of human clinical origin (28). However, this study used rhsA typing to define clade 8, a definition that included strains with a variety of LSPA-6 profiles, while clade 8 isolates from Sweden and the United States always have been typed with LSPA-6 as 211111. We have applied the SNP-based typing method from the current study to a set of E. coli O157:H7 isolates from the Netherlands (provided by E. Franz, RIVM, the Netherlands) to resolve this, with the results indicating an overrepresentation of clade 8 among human isolates (57%) compared to that in animal isolates (17%), using the SNP-based definition (our unpublished data). The results of the present study support the notion that clade 8 strains pose a greater threat to humans than do other subtypes of E. coli O157:H7.
FIG 3.

Minimum spanning tree clustering of MLVA profiles from Swedish E. coli O157:H7 isolates from cattle (gray) and human cases (black), with clade 8 isolates highlighted with gray circles. The short lines correspond to single-locus differences and longer lines to two. The disks representing the profiles are scaled according to the number of observations.
The other major clone causing infections in humans in Sweden is characterized by the LSPA-6 profile 213111, which generally carries Vtx1 often in combination with Vtx2 and belongs to phage type 8 or “reacts but does not conform” (RDNC). This variant causes almost half of the infections contracted within the country but rarely or never HUS. This is in agreement with data from a clonal outbreak of E. coli O157:H7 in the United Kingdom in 2010 to 2011 caused by a phage type 8 (PT 8) vt1+ vt2+ strain with ≥250 reported cases, only four of which progressed to HUS (http://www.hpa.org.uk/NewsCentre/NationalPressReleases/2011PressReleases/110930Ecolioutbreakassocwithsoilonveg/). Swedish isolates with this LSPA-6 profile form a diffuse cluster by MLVA typing, some branches of which appear to be more frequently involved with human infections. The isolates belonging to this group have also been found to be variable in terms of genome content and tccP type (21). Thus, further investigation of the diversity of this group is warranted.
E. coli O157:H7 strains isolated from people who recently traveled abroad are diverse and reflect travel destination.
A highly diverse set of isolates was found in human cases presumed to have been infected while abroad. There was a tendency for profiles to be geographically associated, with repeated isolations from infected people returning from particular countries or regions with profiles found in no other isolates in the study (Table 4). In contrast, lineage I/II strains and the lineage I/II-like 213111 strains appeared to have a wide distribution, with the lineage I/II-like 213111 profile in particular being prevalent in all sets of isolates, regardless of the source. Eighty percent of the Swedish cases infected with 211111 isolates have been infected abroad and by strains with highly variable MLVA profiles. This suggests that while 211111 isolates are diverse on a global scale, they are represented by a single low-virulence clone in Swedish cattle, explaining the discrepancy evident in Fig. 1b. However, a single HUS case was caused by a domestic 211111 isolate, which should be investigated further.
TABLE 4.
Isolates of E. coli O157:H7 from cases in humans with a recent history of travel, classified by LSPA-6 profile and destination region
| Travel destination | No. by profile: |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 111111 | 211111 | 211111 (clade 8) | 213111 | 212111 | 211113 | 212123 | 223121 | 242123 | 262123 | 221123 | 214311 | 223123 | 224311 | 221133 | 2N2113 | 242133 | 244311 | |
| Northern Europe | 1 | 1 | 4 | 1 | ||||||||||||||
| Southern Europe | 3 | 10 | 1 | |||||||||||||||
| West Asia | 1 | 11 | 1 | 4 | 4 | 1 | 1 | |||||||||||
| East Asia | 2 | 1 | 1 | |||||||||||||||
| Southeast Asia | 1 | 2 | ||||||||||||||||
| North Africa | 6 | 1 | 6 | 1 | 2 | 1 | ||||||||||||
| East Africa | 2 | |||||||||||||||||
| Total | 1 | 12 | 2 | 31 | 1 | 1 | 1 | 1 | 4 | 1 | 2 | 1 | 4 | 4 | 1 | 1 | 1 | 1 |
Lineage I (LSPA-6 111111) has caused several outbreaks in Asia and the United States, including a major outbreak in 1999, which caused 177 deaths in Xuzhou, China (29). In the present study, a single LI isolate was found from a human case. To date, this lineage has not been found in Swedish livestock.
While 38% of the diagnosed cases of E. coli O157:H7 infection in Sweden during the study period were probably acquired abroad, all clade 8 infections except two occurred within the country. Thus, severe infection appears to mostly be domestically contracted and by isolates that are genotypically consistent with Swedish cattle as the source. All HUS cases caused by clade 8, except one in the present study, were believed to have been infected within the country. It should be pointed out that in all likelihood, some isolates have been misclassified as contracted abroad and vice versa; however, the large difference between the genotype distributions (Fig. 1b) indicates that the classification is mostly sound. A comprehensive study of E. coli O157:H7 carriage in asymptomatic people in Sweden would be very interesting for comparison but has not been performed to date.
Conclusions.
This study highlights the difference in virulence between the subgroups of E. coli O157:H7, as well as the importance of the prevalence of specific subtypes compared to the importance of the overall prevalence. E. coli O157:H7 HUS cases in Sweden are generally domestically acquired and caused by clade 8, which is genotypically consistent with Swedish ruminants being the source. Using the single-reaction real-time PCR protocol presented here, clade 8 isolates can be identified quickly and with minimal effort. LSPA-6 can be used to further distinguish between non-clade 8 groups that cause disease frequently but with comparatively mild symptoms and groups that cause mild or rare cases of human disease. This will allow a risk assessment of the isolates of E. coli O157:H7 found in a farm environment or foodstuffs and can be used to predict disease progression in infected patients. The same methods can also to some extent be used to distinguish infections contracted abroad from those with Swedish ruminants as the ultimate source. By comparing isolates from different slaughterhouse prevalence studies going back to 1996, we also show the dynamic nature of the E. coli O157:H7 population in the cattle reservoir, in particular the rapid spread of a low-pathogenicity LII clone during the last decade.
ACKNOWLEDGMENTS
This work was supported by grants from the Elsa and Ivar Sandberg Foundation, The Swedish Board of Agriculture, and the Swedish Civil Contingencies Agency. The contribution of E.B.-R. has been partially supported by COST-BMBS action BM1006 “Next generation sequencing data analysis network,” SeqAhead, the AllBio project “Broadening the bioinformatics infrastructure to unicellular, animal, and plant science,” a EU FP7 (theme KBBE.2011.3.6-02), grant agreement 289452.
We thank the staff at the SVA EHEC laboratory for technical assistance, members of the Public Health Agency of Sweden core sequencing facility and the Bo Segerman group, SVA, for assistance with high-throughput sequencing.
Footnotes
Published ahead of print 20 August 2014
REFERENCES
- 1.Söderström A, Osterberg P, Lindqvist A, Jönsson B, Lindberg A, Blide Ulander S, Welinder-Olsson C, Löfdahl S, Kaijser B, De Jong B, Kühlmann-Berenzon S, Boqvist S, Eriksson E, Szanto E, Andersson S, Allestam G, Hedenström I, Ledet Muller L, Andersson Y. 2008. A large Escherichia coli O157 outbreak in Sweden associated with locally produced lettuce. Foodborne Pathog. Dis. 5:339–349. 10.1089/fpd.2007.0065. [DOI] [PubMed] [Google Scholar]
- 2.Sartz L, De Jong B, Hjertqvist M, Plym-Forshell L, Alsterlund R, Löfdahl S, Osterman B, Ståhl A, Eriksson E, Hansson HB, Karpman D. 2008. An outbreak of Escherichia coli O157:H7 infection in southern Sweden associated with consumption of fermented sausage; aspects of sausage production that increase the risk of contamination. Epidemiol. Infect. 136:370–380. 10.1017/S0950268807008473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.SVA. 2013. Surveillance of infectious diseases in animals and humans in Sweden 2012. National Veterinary Institute (SVA), Uppsala, Sweden: http://www.sva.se/upload/Redesign2011/Pdf/Om_SVA/publikationer/Surveillance2012.pdf. [Google Scholar]
- 4.Salvadori M, Bertoni E. 2013. Update on hemolytic uremic syndrome: diagnostic and therapeutic recommendations. World J. Nephrol. 2:56–76. 10.5527/wjn.v2.i3.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eriksson E. 2010. Verotoxinogenic Escherichia coli O157:H7 in Swedish cattle and pigs. Doctoral thesis Acta Universitatis Agriculturae Sueciae, Uppsala, Sweden. [Google Scholar]
- 6.Söderlund R, Hedenström I, Nilsson A, Eriksson E, Aspán A. 2012. Genetically similar strains of Escherichia coli O157:H7 isolated from sheep, cattle and human patients. BMC Vet. Res. 8:200. 10.1186/1746-6148-8-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smittskyddsinstitutet. 2012. Epidemiologisk årsrapport. Smittskyddsinstitutet, Solna, Sweden: http://www.folkhalsomyndigheten.se/documents/statistik-uppfoljning/smittsamma-sjukdomar/2012/norovirus-epidemiologisk-arsrapport-2012-2013-101-8.pdf. [Google Scholar]
- 8.ECDC. 2013. Annual epidemiological report 2013. European Centre for Disease Prevention and Control, Stockholm Sweden. [Google Scholar]
- 9.Manning SD, Motiwala AS, Springman AC, Qi W, Lacher DW, Ouellette LM, Mladonicky JM, Somsel P, Rudrik JT, Dietrich SE, Zhang W, Swaminathan B, Alland D, Whittam TS. 2008. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. U. S. A. 105:4868–4873. 10.1073/pnas.0710834105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laing CR, Buchanan C, Taboada EN, Zhang Y, Karmali MA, Thomas JE, Gannon VPJ. 2009. In silico genomic analyses reveal three distinct lineages of Escherichia coli O157:H7, one of which is associated with hyper-virulence. BMC Genomics 10:287. 10.1186/1471-2164-10-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Nietfeldt J, Benson AK. 1999. Octamer-based genome scanning distinguishes a unique subpopulation of Escherichia coli O157:H7 strains in cattle. Proc. Natl. Acad. Sci. U. S. A. 96:13288–13293. 10.1073/pnas.96.23.13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eriksson E, Söderlund R, Boqvist S, Aspan A. 2011. Genotypic characterization to identify markers associated with putative hypervirulence in Swedish Escherichia coli O157:H7 cattle strains. J. Appl. Microbiol. 110:323–332. 10.1111/j.1365-2672.2010.04887.x. [DOI] [PubMed] [Google Scholar]
- 13.Hyytiä-Trees E, Smole SC, Fields PA, Swaminathan B, Ribot EM. 2006. Second generation subtyping: a proposed PulseNet protocol for multiple-locus variable-number tandem repeat analysis of Shiga toxin-producing Escherichia coli O157 (STEC O157). Foodborne Pathog. Dis. 3:118–131. 10.1089/fpd.2006.3.118. [DOI] [PubMed] [Google Scholar]
- 14.Yang Z, Kovar J, Kim J, Nietfeldt J, Smith DR, Moxley RA, Olson ME, Fey PD, Benson AK. 2004. Identification of common subpopulations of non-sorbitol-fermenting, beta-glucuronidase-negative Escherichia coli O157:H7 from bovine production environments and human clinical samples. Appl. Environ. Microbiol. 70:6846–6854. 10.1128/AEM.70.11.6846-6854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Laing C, Steele M, Ziebell K, Johnson R, Benson AK, Taboada E, Gannon VPJ. 2007. Genome evolution in major Escherichia coli O157:H7 lineages. BMC Genomics 8:121. 10.1186/1471-2164-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riordan JT, Viswanath SB, Manning SD, Whittam TS. 2008. Genetic differentiation of Escherichia coli O157:H7 clades associated with human disease by real-time PCR. J. Clin. Microbiol. 46:2070–2073. 10.1128/JCM.00203-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eppinger M, Mammel MK, LeClerc JE, Ravel J, Cebula TA. 2011. Genome signatures of Escherichia coli O157:H7 isolates from the bovine host reservoir. Appl. Environ. Microbiol. 77:2916–2925. 10.1128/AEM.02554-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leopold SR, Shaikh N, Tarr PI. 2010. Further evidence of constrained radiation in the evolution of pathogenic Escherichia coli O157:H7. Infect. Genet. Evol. 10:1282–1285. 10.1016/j.meegid.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eppinger M, Mammel MK, Leclerc JE, Ravel J, Cebula TA. 2011. Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc. Natl. Acad. Sci. U. S. A. 108:20142–20147. 10.1073/pnas.1107176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leopold SR, Magrini V, Holt NJ, Shaikh N, Mardis ER, Cagno J, Ogura Y, Iguchi A, Hayashi T, Mellmann A, Karch H, Besser TE, Sawyer SA, Whittam TS, Tarr PI. 2009. A precise reconstruction of the emergence and constrained radiations of Escherichia coli O157 portrayed by backbone concatenomic analysis. Proc. Natl. Acad. Sci. U. S. A. 106:8713–8718. 10.1073/pnas.0812949106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Söderlund R, Aspán A, Ragione RM, Eriksson E, Boqvist S. 2010. Microarray-based detection of virulence genes in verotoxigenic Escherichia coli O157:H7 strains from Swedish cattle. Epidemiol. Infect. 139:1088–1096. 10.1017/S095026881000213X. [DOI] [PubMed] [Google Scholar]
- 22.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267. 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 23.Aspán A, Eriksson E. 2010. Verotoxigenic Escherichia coli O157:H7 from Swedish cattle; isolates from prevalence studies versus strains linked to human infections–a retrospective study. BMC Vet. Res. 6:7. 10.1186/1746-6148-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Laing C, Zhang Z, Hallewell J, You C, Ziebell K, Johnson RP, Kropinski AM, Thomas JE, Karmali M, Gannon VP. Lineage and host source are both correlated with levels of Shiga toxin 2 production by Escherichia coli O157:H7 strains. Appl. Environ. Microbiol. 76:474–482. 10.1128/AEM.01288-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellor GE, Sim EM, Barlow RS, D'Astek BA, Galli L, Chinen I, Rivas M, Gobius KS. 2012. Phylogenetically related Argentinean and Australian Escherichia coli O157 isolates are distinguished by virulence clades and alternative Shiga toxin 1 and 2 prophages. Appl. Environ. Microbiol. 78:4724–4731. 10.1128/AEM.00365-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leotta GA, Miliwebsky ES, Chinen I, Espinosa EM, Azzopardi K, Tennant SM, Robins-Browne RM, Rivas M. 2008. Characterisation of Shiga toxin-producing Escherichia coli O157 strains isolated from humans in Argentina, Australia and New Zealand. BMC Microbiol. 8:46. 10.1186/1471-2180-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haugum K, Brandal LT, Løbersli I, Kapperud G, Lindstedt BA. 2011. Detection of virulent Escherichia coli O157 strains using multiplex PCR and single base sequencing for SNP characterization. J. Appl. Microbiol. 110:1592–1600. 10.1111/j.1365-2672.2011.05015.x. [DOI] [PubMed] [Google Scholar]
- 28.Franz E, van Hoek AHAM, van der Wal FJ, de Boer A, Zwartkruis-Nahuis A, van der Zwaluw K, Aarts HJM, Heuvelink AE. 2012. Genetic features differentiating bovine, food, and human isolates of Shiga toxin-producing Escherichia coli O157 in the Netherlands. J. Clin. Microbiol. 50:772–780. 10.1128/JCM.05964-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong Y, Wang P, Lan R, Ye C, Wang H, Ren J, Jing H, Wang Y, Zhou Z, Bai X, Cui Z, Luo X, Zhao A, Wang Y, Zhang S, Sun H, Wang L, Xu J. 2012. A novel Escherichia coli O157:H7 clone causing a major hemolytic uremic syndrome outbreak in China. PLoS One 7:e36144. 10.1371/journal.pone.0036144. [DOI] [PMC free article] [PubMed] [Google Scholar]