

Abstract
In critically ill patients, the development of pneumonia results in significant morbidity and mortality and additional health care costs. The accurate and rapid identification of the microbial pathogens in patients with pulmonary infections might lead to targeted antimicrobial therapy with potentially fewer adverse effects and lower costs. Major advances in next-generation sequencing (NGS) allow culture-independent identification of pathogens. The present study used NGS of essentially full-length PCR-amplified 16S ribosomal DNA from the bronchial aspirates of intubated patients with suspected pneumonia. The results from 61 patients demonstrated that sufficient DNA was obtained from 72% of samples, 44% of which (27 samples) yielded PCR amplimers suitable for NGS. Out of the 27 sequenced samples, only 20 had bacterial culture growth, while the microbiological and NGS identification of bacteria coincided in 17 (85%) of these samples. Despite the lack of bacterial growth in 7 samples that yielded amplimers and were sequenced, the NGS identified a number of bacterial species in these samples. Overall, a significant diversity of bacterial species was identified from the same genus as the predominant cultured pathogens. The numbers of NGS-identifiable bacterial genera were consistently higher than identified by standard microbiological methods. As technical advances reduce the processing and sequencing times, NGS-based methods will ultimately be able to provide clinicians with rapid, precise, culture-independent identification of bacterial, fungal, and viral pathogens and their antimicrobial sensitivity profiles.
INTRODUCTION
The development of ventilator-associated pneumonia (VAP) in intubated patients in intensive care units (ICU) remains a major nationwide clinical challenge and economic burden, despite advances in antibiotic therapy (1). Patients with VAP have estimated crude mortality rates of 20 to 70% and attributable mortality rates of 10 to 40% (2–4). Because appropriate antimicrobial therapy has been shown to reduce crude mortality rates in patients if administered within the first 48 h of VAP diagnosis (5), patients suspected of VAP are generally started on broad-spectrum antibiotics to ensure that the most common pathogens are targeted. However, establishing the precise microbiologic cause of VAP allows clinicians to replace this preemptive broad-spectrum antibiotic therapy with targeted antibiotics against an identified pathogen or group of bacteria, thereby reducing the risk of antibiotic-associated side effects, decreasing the potential for antibiotic resistance, and reducing the overall health care costs (6).
Establishing an accurate microbial cause of the pneumonia in ventilated patients can be challenging. Lower tract aspirate cultures are recommended in patients suspected of having hospital-acquired pneumonia or VAP (7) and have been shown to help guide appropriate antimicrobial therapy (8, 9). Culture-based microbiological diagnosis remains complicated because the presence of just a few colonizing bacteria in the respiratory tract can result in significant microbial growth on agar cultures and thus can be erroneously interpreted as being indicative of infection. The diagnostic process is further complicated by false-negative results from pathogens that cannot be cultured using standard laboratory procedures, possibly due to prior antibiotic administration or lack of anaerobic transport conditions (10). Combined, these false positives and false negatives significantly reduce the clinical value of deep tracheal aspirate cultures (11–13). Microbiologic yields of 33 to 60% have been reported when only conventional diagnostic methods were used (14–16). Furthermore, whereas a primary pathogen might be identified using standard culture techniques, the bacterial community of other abundant, but not necessarily pathogenic bacteria, may be an important factor modifying the virulence of the predominant pathogen through mechanisms such as quorum sensing. Therefore, analysis of the pulmonary bacterial community, also called the lung microbiome (17), may be an important factor in understanding the pathogenesis of bacteria in VAP and in managing the infection.
The rapid development of DNA sequencing technology has led to an increasing potential for culture-independent methods of identifying microbiological pathogens in various clinical settings (18). Advances in DNA sequencing now allow increasingly rapid and massive parallel sequencing of thousands to millions of DNA strands simultaneously, thereby allowing a snapshot of all bacteria present in a given sample. Thus, we sought to determine whether the microbiome of human endobronchial aspirates might be effectively characterized by next-generation sequencing (NGS). Prior approaches to bacterial identification by DNA sequencing have included fluorescent Sanger sequencing of hundreds of small variable regions of 16S ribosomal amplimer clones (19, 20), length heterogeneity (LH) PCR for 16S ribosomal variable regions (21), restriction fragment polymorphisms in 16S (19, 21), arrays with bacterium-specific probes, such as PhyloChip (22, 23), and early applications of NGS (24, 25). These methods have been applied to various clinical situations, such as the microbiome of suboptimally controlled asthma (22), cystic fibrosis (19, 21), chronic obstructive pulmonary disease (COPD) (24, 26, 27), bronchiectasis (28), pleurisy (20), forensic assessment of drowning (25), and intubated patients (23). These studies have revealed unexpectedly large numbers of bacterial genera in patients with clinical illnesses and in healthy individuals (29, 30). In the present study, single-molecule NGS of thousands of full-length 16S small subunit ribosomal DNA amplimers was used to characterize the repertoire of pulmonary bacteria in intubated patients being clinically assessed for possible VAP.
MATERIALS AND METHODS
Design and participants.
The protocol for this study was approved by the George Washington University (GWU) institutional review board and included the provisions for collection of discarded deep tracheal aspirate samples for bacterial sequencing and deidentified clinical and microbiological data. The indication for endotracheal culture was solely based on the clinical evaluation of the patient's attending physician, as was any decision regarding initiation of antibiotic therapy. Intubated patients in the intensive care unit (ICU) who had a deep tracheal aspirate sputum sample sent for standard bacterial culture were identified. When available, a portion of the residual aspirate was salvaged and stored for genomic analysis.
Clinical data.
Demographic information, including age, sex, pertinent medical conditions, and the presence of any preexisting respiratory conditions was obtained from the charts of the study subjects (Table 1). Clinical measures, including the length of the hospital stay at the time of aspirate sampling, current antibiotic usage, results of blood cultures obtained within 24 h of aspirate sampling, and results of routine aspirate cultures were recorded. The clinical pulmonary infection score (CPIS) was calculated for each of the subjects based on body temperature, white blood cell count, aspirate quality, the partial arterial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) ratio, and chest radiography at the time that the deep endotracheal suctioning was performed.
TABLE 1.
Demographic and clinical parameters of the patients
| Criterion | Results for patients with CPIS that was: |
P | |
|---|---|---|---|
| High | Low | ||
| Total no. of patients | 16 | 28 | |
| Age (mean [SD]) (yr) | 56.7 (4.3) | 55.3 (2.4) | NSa |
| Gender (% male) | 69 | 79 | NS |
| Intubation (mean [SD]) (days) | 6.2 (2.2) | 12.1 (4.8) | NS |
| PaO2 (mean [SD]) (mm Hg) | 151 (22.5) | 128 (12.4) | NS |
| FiO2 (mean [SD]) (%) | 58 (5.5) | 45 (2.4) | 0.03 |
| Temp (mean [SD]) (°C) | 37.2 (0.33) | 37.4 (0.12) | NS |
| WBCb (mean [SD]) (103/μl) | 13.3 (1.7) | 12.5 (1.1) | NS |
| Bands (mean [SD]) (%) | 9.4 (4.9) | 5.8 (2.0) | NS |
| gDNA positive (%) | 88 | 89 | NS |
| PCR positive (%) | 63 | 61 | NS |
| NGS positive (%) | 63 | 61 | NS |
NS, not significant.
WBC, white blood cells.
Sampling procedures.
The deep endotracheal aspirate samples were collected by advancing a suction catheter through the subjects' endotracheal tubes and infusing 2.5 ml of sterile saline, which was then suctioned into a sterile collection container. The aspirate was submitted to the GWU Hospital microbiology laboratory for routine Gram staining and microbial culture. In short, the most purulent or blood-tinged portions were used for a Gram stain and bacterial cultures on sheep blood, chocolate, and MacConkey agars. The cultures on sheep blood and chocolate agars were incubated in 5% CO2 at 35°C for at least 48 h, while the cultures on MacConkey agar were incubated in a non-CO2 atmosphere at 35°C for at least 24 h. Significant growth was defined as moderate to heavy growth of an isolate in the second, third, or fourth quadrants of each plate. Organisms were identified and susceptibility was determined using Vitek 2 identification (ID) and antibiotic susceptibility testing (AST) cards (bioMérieux, Marcy l'Etoile, France) following the standard operating procedures utilized by the GWU Hospital microbiology laboratory. The residual aspirate samples were frozen at −80°C until processing for DNA extraction. After thawing, the samples were transferred into a 50-ml conical tube and processed by minor modifications to the method optimized for Mycobacterium tuberculosis isolation from human aspirate (31). To help dissolve the mucus, N-acetylcysteine (NAC-50; Remel Microbiology Products, USA) mucolytic agent (5 mg/ml, pH 6.8) was added 1:1 and vortexed. To pellet the bacteria, the samples were spun at 2,500 × g for 30 min in a swinging bucket centrifuge, and the pellet was then resuspended in 1 ml of 70% isopropanol and stored at −80°C until DNA isolation.
DNA isolation.
Before the isolation of lung aspirate genomic DNA (gDNA) from clinical samples, several commercially available bacterial DNA extraction kits were tested. Standard bacterial samples (Staphylococcus aureus, ATCC 25923; Enterococcus faecalis, ATCC 51299; Pseudomonas aeruginosa, ATCC 27853; Escherichia coli, ATCC 25922; and Streptococcus pneumoniae, ATCC 6303, and E. coli dH5α NEB C2987) were used to optimize DNA isolation from both Gram-positive (G+) and Gram-negative (G−) samples. Bacteria were grown in LB broth for 16 h, and 1 ml was used for DNA isolation by different methods. The following kits were used for optimization of the DNA extraction protocol: TRIzol (Invitrogen), QuickExtract (Epicentre), DNAZol (MP Biologicals), the Wizard SV genomic DNA purification system (Promega), and the GenElute bacterial DNA kit (Sigma). After the yield of gDNA was analyzed, a combination of G+/G− lysis buffer based on the Sigma GenElute kit was used for patients' gDNA isolation (bacterial G+ lysis solution supplemented with lysozyme [2.2 × 106 units/ml], lysostaphin [200 units/ml], and mutanolysin [5,000 units/ml] combined in equal volumes with lysis solution T for G− bacteria). Aspirate samples (1 ml) were spun for 5 min at 1,500 × g, and the pellet was resuspended in 200 μl of the modified lysis solution described above. The concentration of gDNA was measured using both a NanoDrop spectrophotometer (ND-1000; Thermo Scientific, USA) and a Qubit 2.0 fluorometer (Thermo Scientific, USA). Typical gDNA yields were 500 ng to 20 μg per 1 ml of aspirate.
16S ribosomal DNA PCR.
The overall workflow for amplification of bacterial 16S rDNA, library preparation, and long-read single-molecule sequencing is shown schematically in Fig. 1. Prokaryotic genes coding for 16S small subunit rRNA were amplified using “universal” primers annealing to the constant regions (B27F, 5′-AGAGTTTGATCCTGGCTCAG-3′; and U1492R, 5′-GGTTACCTTGTTACGACTT-3′) with E. coli 16S for numbering, yielding a predicted product of 1,466 bp. These primers were originally described by Weisburg et al. (32) and more recently characterized for effectiveness in a broad range of Gram-positive and Gram-negative bacteria (33).
FIG 1.
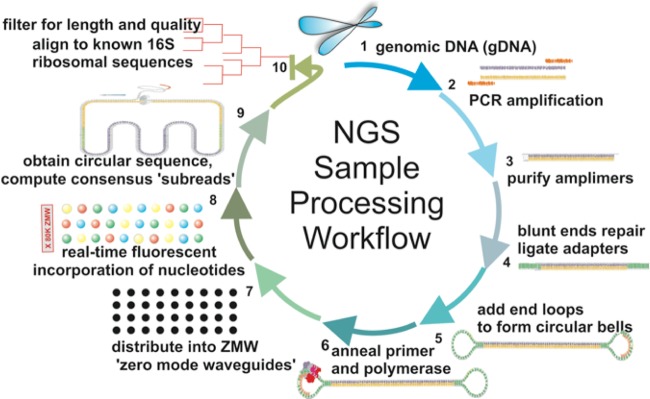
NGS analytical workflow. Deep bronchial aspirates from intubated patients in the intensive care unit were analyzed by the standard hospital microbiological workup, and the remaining samples were analyzed by next-generation sequencing to compare the diagnostic methods. Aspirates were liquified with N-acetylcysteine and centrifuged to pellet cells. Pellets containing both bacteria and human cells were chemically/enzymatically disrupted to isolate genomic DNA (gDNA) (step 1), which was then PCR amplified using primers for nearly full-length 16S (step 2). The purified amplimers (step 3) were ligated into circular sequencing loops (steps 4, 5, and 6) and then distributed into zero-mode waveguides on the Pacific Biosystems single molecule sequencer (step 7). The incorporation of individual fluorescent nucleotides is recorded in real-time at 3 to 4 bases per second, often making multiple passes across the amplimer insert (step 8). Multiple passes across the 16S insert were aligned to create a consensus read (step 9), which was then classified for its bacterial origin using multiple analytical strategies (step 10).
Amplifications were carried out on an ABI 2700 thermocycler using a hot-start AmpliTaq Gold 360 DNA polymerase master mix with GC enhancer (Life Technologies). The optimal loading of 500 ng of gDNA was used for the majority of samples, although many samples amplified well with 100 to 250 ng. PCRs were conducted with 40 cycles of 95°C for 15 s, 40°C for 30 s, and 72°C for 90 s. There was a final 7-min extension step at 72°C, after which the samples were held at 4°C until processed. The presence of amplimers at the expected 1,466-bp size was confirmed by gel electrophoresis on a 1% agarose gel stained with ethidium bromide (EtBr) and by an Agilent 2100 bioanalyzer using a 7,500-bp DNA chip (Agilent Technologies). The gel-resolved amplimers were not used for NGS due to an adverse effect of EtBr and UV light on the DNA integrity. Rather, the PCR products were purified with Agencourt AMPure XP magnetic beads (Beckman-Coulter). Purified PCR products were quantified with an optical density at 260 or 280 nm (OD260/280) (NanoDrop) and fluorescence staining (Qubit). All PCR amplification reactions that did not produce detectable amplimers of the expected sizes were repeated with different amounts of gDNA, and if these failed, the samples were excluded from NGS. In cases of insufficient starting quantities for library preparation, amplimers were pooled from multiple reactions.
Next-generation sequencing.
Purified 16S amplimers (200 to 750 ng) were prepared for long-read single-molecule sequencing (Pacific Biosystems) using a DNA Prep kit 2.0 (250 bp to <3 kb). Briefly, the purified bacterial PCR amplimers were blunt ligated to a common adapter sequence and a hairpin loop at each end to create a circular loop with the amplimer/adapter in the middle. The circular “bells” were bound with a sequencing primer and polymerase (PacBio C2 chemistry) and then diluted into individual zero-mode waveguides (ZMWs), which act as single molecule wells for sequencing. The incorporation of fluorescently labeled nucleotides at approximately 3 to 4 bp/s is recorded by digital video and then deconvolved into sequences reported in PacBio native bas.h5 and standard FastQ formats.
NGS alignment and bacterial classification.
The raw SMRT reads were processed through the PacBio SMRT Portal pipeline to filter out (i) short reads of <100, (ii) reads with no insert, (iii) trimming of adapter sequences, and (iv) low-complexity or poor-quality reads. Without prior knowledge of the clinical microbiology results, microbial diversity characterization was performed using multiple tools, including (i) PathoScope (34, 35), (ii) RDP naive Bayesian classifier (36), (iii) mothur-based rDnaTools application (37), (iv) SMRT Portal (38) (Pacific Biosciences), and (v) Geneious R7 (39) (Biomatters, NZ) software.
PathoScope analysis was performed by mapping reads against a bacterial 16S rRNA data set derived from “The All-Species Living Tree” Project (LTP), supplemented with human sequences (35, 40, 41). Bowtie 2 was used to map reads using default settings except “--very-sensitive-local −k 100 --score-min L,20,1.0” parameters (42).
Statistical analysis.
The descriptive statistics and the resulting numbers of species and subspecies were analyzed in Microsoft Excel using the built-in statistical tools. Statistical comparisons between groups were calculated using the Student t test, and correlations between quantitative measures were calculated as Pearson r coefficients.
Nucleotide sequence accession numbers.
The raw data files with sequences from each patient were submitted in bas.h5 format to the NCBI Short Read Archive (SRA) under the accession numbers SRP028704 and SRP031650.
RESULTS
Clinical parameters.
A total of 61 patients had residual samples that were sufficient to be processed (>1 ml). No detectable gDNA was isolated from 17 samples, resulting in 44 gDNA samples (72%) that were suitable for PCR amplification of 16S. Subject characteristics for this cohort are shown in Table 1, according to their grouping by the clinical diagnosis of low risk of infection (CPIS <5) versus high risk of infection (CPIS ≥5). The two groups did not differ in demographic parameters, such as age or gender, or with respect to most clinical parameters, except the fraction of inspired oxygen (FiO2), which was higher in the high-risk group (P = 0.03, uncorrected). Ten subjects (23%) were not receiving an antibiotic regimen at the time of aspirate sampling. Clinically, the most common bacteria identified by standard microbiology are often normal gastrointestinal/oropharyngeal flora, Staphylococcus aureus, Pseudomonas aeruginosa, and Enterobacteriaceae, that can colonize the lungs of intubated patients. Other bacteria that were cultured included Streptococcus pneumoniae, Haemophilus influenzae, Acinetobacter baumannii, Moraxella catarrhalis, and Stenotrophomonas maltophilia. Complete clinical and genomic information for all subjects is available in Table SA1 in the supplemental material.
Isolation and amplification metrics.
From the 44 DNA-positive samples, 27 samples produced sufficient PCR amplimers (>200 ng) for NGS (61%), and some 16S DNA sequence was obtained from 100% of PCR-positive samples, for an overall diagnostic success rate of 44% of the 61 samples obtained. The 1,466-bp PCR product covers 95% of the 1,542-bp prokaryotic 16S sequence. Some PCRs also yielded a minor band at ∼300 bp, which was determined from the DNA sequence reads to correspond to human 18S ribosomal DNA.
Sequencing metrics.
On SMRT sequencing, a typical run involved 75,153 potentially productive ZMWs per flow cell. However, depending on the loading efficiency and other factors, on average, 11,790 mapped consensus reads were obtained (standard error of the mean [SEM], 1,868; range, 9 to 36,684). Those reads, however, were quite long, with an average length of 3,810 bp, >2 full-length single-pass subreads of the 1,466-bp insert. The accuracy of the sequencing of the individual subreads, as measured against the reference E. coli is only 83.1%, but this increases markedly when the subreads are combined to produce a consensus sequence, which is 99.8% accurate compared to the E. coli reference. The accuracy issues are complicated by the fact that a typical bacterium contains multiple copies of 16S ribosomal DNA, with E. coli K-12, for instance, apparently having 7 copies with very high similarity. The spike-in reference sequence was used as another test of accuracy; the apparent accuracy of the sequencing, on a single pass, is likewise 81.98%, which was comparable to that observed in the 27 patient samples (84.17% mapped subread accuracy). The consensus accuracy, which combines multiple passes of the same insert, was >99.5% with a minimum coverage (QV) of >100 (for a sample-by-sample coverage report, see Table SA1 in the supplemental material).
Alignment strategies.
These long reads of PCR amplimers differ from other NGS reads, which are typically <200 bp in length, and thus the alignment approach needed to be reconsidered. To identify an optimal analytical workflow, two different taxonomy assignment pipelines were employed: PathoScope and SMRT Portal. For both PathoScope and SMRT Portal, the 16S reference file was derived from LTP version 111, to which the human 18S sequences were added to account for the 300-bp PCR product. The total number of quality mapped subreads was 1,127,682 for all 27 sequenced samples (mean, 43,372; standard deviation, 52,590; minimum, 4,611; maximum, 219,863). Thus, the combination of read depth coverage of >1,000 per identified pathogen and accuracy of consensus reads >99.5%, generated substantial overall confidence in the results. The number of bacterial species identified was highly correlated between the aligners (r = 0.68, P < 0.001) (Fig. 2), but PathoScope consistently produced a tighter diagnosis (3.55 versus 11.03 species/sample, P < 0.001). To account for chimeric sequences, all ATCC standards and patients' sample sequences were run through the Database Enabled Code for Ideal Probe Hybridization Employing R (DECIPHER) web application (University of Wisconsin-Madison) (43), and the results demonstrated a very low presence of chimeric sequences in patients' samples (see Table SA1 in the supplemental material).
FIG 2.
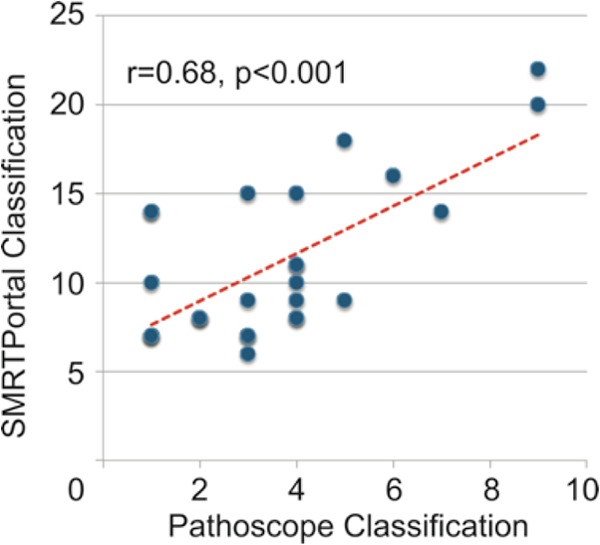
Effect of the alignment strategy on classification of 16S ribosomal reads. Consensus reads, built from multiple subreads of the sequencing loop, were aligned to the LTP111 16S bacterial reference library using either PathoScope (x axis) or SMRT Portal (y axis). Each data point is a single patient's lung aspirate DNA sequences processed as described in Fig. 1. For each alignment strategy, the number of bacterial species utilizing >1% of the total bacterial reads was plotted.
Effect of read length on classification.
With few exceptions, most microbiome characterization to date has been based on shorter variable region PCR amplifications, followed by Sanger sequencing or NGS. Within the 16S open reading frame (ORF), there are several relatively invariant “constant” regions, which are useful as primer sites, and then there are 9 variable regions (V1 to V9, shown schematically in Fig. 3), which can be used for the assignment of the sequence to a particular bacterial taxon, genus, species, and often, subspecies, with the V1 to V3 and V7 to V9 having the greatest utility in classification (44). By sequencing essentially the entire 16S ORF, there was no need to choose just one variable region. To determine whether the long reads, as used here, had any beneficial effect on microbiome characterization, we conducted in silico experiments in which the long-read sequences were used to create simulated short reads spanning the 16S gene. The results indicate that the shorter reads increased the apparent microbiome complexity of the sample, as determined by the identified number of genera per sample (SEM): short reads of 5.6 (± 0.74) versus long reads of 3.7 (± 0.44) (Fig. 3), probably due to shorter fragments detecting spurious homologies with conserved regions of 16S. Thus, the long reads produced a narrower diagnosis of the lung bacterial diversity, which is an expected benefit of combining the diagnostic strengths of different 16S regions (44).
FIG 3.

Effect of long versus short reads on classification. (A) Schematic representation of the long-read PCR amplimer, PCR primers, known variable regions, and in silico short reads of 16S. (B) Bacterial classifications by PathoScope of individual 16S consensus reads were compared when the input sequences were either intact full-length reads or the same reads were split into 100-bp fragments prior to alignment and classification.
Effect of sequence read number and quality on classification. (i) Sequence read number.
Despite a large variation in sequence reads between patients (9 to 36,684 mapped consensus reads/patient), there was not a positive relationship between read quantity and species identified (r = −0.11), potentially due to the very low microbiome diversity in these samples.
(ii) Sequence read quality.
It is plausible that the sequence quality of individual subreads would be related to the classification accuracy. Conversely, the error rate might not contribute to a significant misclassification rate because (i) the errors should be random, (ii) consensus reads minimize subread errors, (iii) only errors in variable regions should result in misclassification, and (iv) errors in a single variable region might be insufficient to misclassify the entire read. These assumptions have to be tempered with the knowledge that existing aligner/classifiers are optimized for short reads and have not been optimized for long-read technologies. Comparisons of the Roche 454 and Illumina technologies confirmed that the read lengths and error rates exerted significant influence on the bacterial classifications in intestinal microbiota (45).
An in silico experiment was conducted in which known bacteria were sequenced and then the effect of sequence quality on bacterial classification was examined. Using all quality reads, we found that PathoScope correctly identified 5 of 6 known bacteria with high confidence, with minor ambiguities in discriminating E. coli from Shigella and P. aeruginosa from P. otitidis, which have nearly identical 16S sequences. With the example of S. aureus, the consensus reads were further aligned and classified in SMRT Portal. Then the assemblies were used to plot the number of sequences per assembly, and the percentage of low-quality (LQ) sequences (Q of <20). The results clearly show that the major classifications of S. aureus and the closely related Staphylococcus simiae (Fig. 4), which as the top 3 assemblies account for about 70% of reads, had essentially no consensus reads of low quality. However, as the number of reads per assembly drops and the classifications diverge from the correct classification, the number of LQ reads increases rapidly, demonstrating that LQ reads are associated with misdiagnosis.
FIG 4.
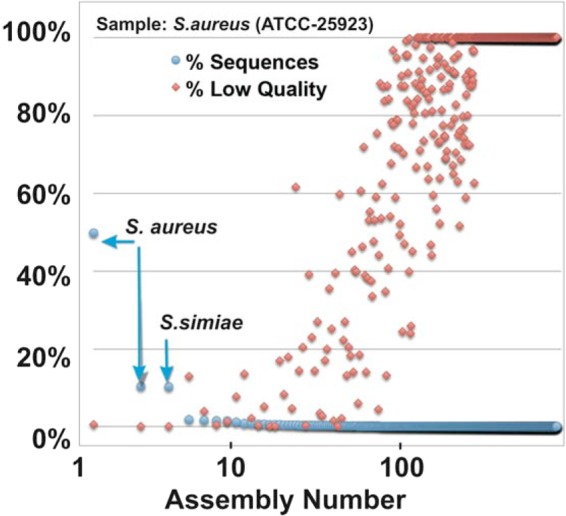
Effect of sequence quality on bacterial classification. A known strain of S. aureus (ATCC) was processed through the NGS workflow, and the consensus long reads (n = 20,291) were aligned to the LTP111 bacterial reference file using SMRT Portal to understand the relationship between sequence quality and bacterial classification. Using a Bowtie 2 algorithm, 850 assemblies were built, ranging from 10,092 reads (assembly 1, S. aureus) to as few as 2 reads per assembly. For each assembly, the blue circles indicate the percentages of reads assembled (% Sequences), and the red diamonds indicate the percentages of those reads of low quality (% Low Quality).
Effect of NGS on diagnostic accuracy.
Figure 5 shows a typical taxonomic classification for a patient with a clinically diagnosed pulmonary infection with Enterobacter. From an initial set of 914 quality-filtered reads, 479 (52%) were aligned to the “bacteria” classification, which was then further refined with 180 reads as “Proteobacteria” and “Gammaproteobacteria” of which 174 reads matched “enterobacteria,” and thus the detected predominant sequences agreed with the clinical diagnosis. However, interestingly, of the 174 reads, 123 matched “unclassified Enterobacteriaceae,” while 45 were matched to Raoultella, thereby suggesting that a significant part of the infectious community was the less commonly diagnosed Raoultella ornithinolytica. The fact that >40% (45× coverage) of enterobacterial reads matched Raoultella ornithinolytica suggests that it is not likely to be a random sequencing error.
FIG 5.
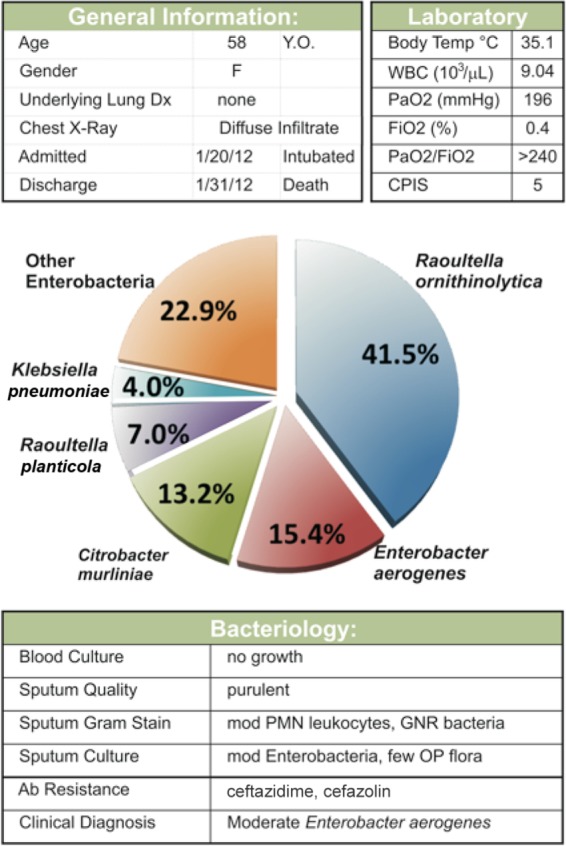
Typical taxonomic classification of bacteria in a lung aspirate. The pulmonary aspirate from a female ICU patient was characterized by clinical criteria (top) and by next-generation sequencing (NGS) of 16S amplimers from the genomic DNA (Bottom Panels). While the clinical and NGS diagnosis both include “Enterobacter” as a pathogen, the NGS diagnosis detects significant quantities of Raoultella, Citrobacter, and Klebsiella. Y.O., years old; F, female; WBC, white blood cells; PNM, polymorphonuclear; GNR, Gram-negative rod; OP, oral and oropharynx; Ab, antibiotic.
In general, by comparing the NGS results obtained by PathoScope (long reads) with the microbiologic result obtained by standard culture of the aspirate, a high degree of concordance was observed. In the 27 patients with productive NGS results, 20 patients had a predominant bacterial pathogen identified on plate agar culture in the microbiological lab (clinical diagnosis). In those 20 samples, there was an exact match to the predominant NGS bacterial species in 12 specimens (60%), while 17 specimens (85%) had a match to one of the major bacterial species identified by NGS, with only 3 cases (15%) showing clear nonconcordance between the clinical and NGS results. In the remaining 7 patients with no microbiological pathogen identified in the hospital lab, the aspirate showed either no growth in culture, the presence of yeasts, or various amounts of normal opportunistic oropharyngeal flora, while NGS still identified several bacteria species (see Table SA1 in the supplemental material).
Differentiation of bacterial communities from low-risk versus high-risk patients.
From an analysis of the 44 patient samples obtained, neither the yield of gDNA nor the yield of 16S PCR product differed between the low-risk patients (CPIS <5) relative to that of the high-risk patients (CPIS >5) (Table 1). A working hypothesis was that patients exhibiting lower CPIS were more likely to have mild commensal infections, which might have greater bacterial diversity than serious pathogenic infections with a single organism. In the 27 patient samples on which NGS was completed, the number of NGS-identifiable bacterial genera tended to be higher in patients with low CPIS, but the correlation was weak overall (r = −0.23, P > 0.1; see Fig. SA1 in the supplemental material). Thus, while the gDNA yield, PCR positivity, and bacterial diversity via NGS did not clearly distinguish between low-risk and high-risk infections, as assessed by the CPIS, NGS was quite valuable in determining the precise identities of the bacteria (Fig. 6).
FIG 6.

Differentiation of bacterial communities from low-risk versus high-risk patients. NGS sequencing results for pulmonary aspirates were classified with PathoScope using long reads. For each patient (x axis), the percentage of the reads assigned to each bacterial genus is plotted on the y axis. The patients are grouped by their CPIS, a clinical index of the severity of the pulmonary distress, often used to predict the risk of bacterial infection in intensive care patients.
DISCUSSION
The diagnosis of pulmonary infections is a crucial component of the management of critically ill patients, and, thus, any improvements are likely to have real-world clinical benefits. The present studies demonstrate the proof of principle that clinically obtained pulmonary aspirates are generally amenable for molecular diagnosis by next-generation sequencing. From 16S PCR amplimers, these results reflect the first long-read NGS of clinically relevant bacterial communities. The results indicate that contrary to our common usage of singular terms such as “infectious agent” or “bacterial pathogen,” bacterial lung infections, like any microbiome, appear to have one or more dominant bacteria, but also contain potentially important coconspirators that might modulate growth, virulence, biofilm formation, quorum sensing, and antibiotic resistance.
The present results should not be interpreted as an analysis of the normal lung microbiome, because these were intubated ICU patients with suspected pneumonia. In approximately 80% of the cases, the clinical management of the patient required antibiotic therapy prior to obtaining of the aspirate sample, and this would have significantly altered the bacterial community. Furthermore, as is often the case, these ICU patients had other medical complications, including asthma, emphysema, and chronic obstructive pulmonary disease (COPD), which prior studies have established as factors associated with microbiome changes (22, 46).
In the course of these studies, we recognized certain strengths and weaknesses to this approach, which can inform future studies and clinical applications in this area. Among the strengths of this approach is the significant improvement in the accuracy of the diagnosis that is offered by long-read sequencing-based approaches. As long-read technologies improve with respect to the number of reads they produce, it will be increasingly feasible to conduct PCR-directed, multitargeted sequencing of pathogens for identification and antibiotic resistance and eventually long-read full microbial genomes. A second major advantage of the NGS approach is that it does not require culture of the microbes, which should shorten diagnostic times and increase the range of diagnosable microbes. Overall, a strength of this study design is that it employed real-world clinical samples collected under the true conditions in which an accurate diagnosis is necessary. Finally, the accuracy of the DNA diagnosis, coupled with sophisticated analytical tools, will allow for more accurate and comprehensive monitoring of pathogen outbreaks, which will improve therapeutic efforts and improve the tracking of pathogens in health care facilities and the community.
However, it is important to recognize both the theoretical and actual limitations of this methodology. The results represent a pilot study with a modest number of observations, which might limit the comparison of our findings against established clinical parameters, such as the CPIS. It is also plausible that a certain subset of patients with apparent infections would not be diagnosed by this method. Several factors can be identified. First, it is quite possible that in some patients with a true infection, the pulmonary aspirate would not collect relevant pathogens. This might be especially true in infections caused by pathogens that are highly encapsulated, are strongly adherent, or form robust biofilms. Second, the isolation of bacteria from the aspirate might be improved by some type of affinity separation, rather than relying on centrifugation alone. A further challenge is that the highly variable nature of the clinical samples makes it difficult to standardize the analysis. Some samples are essentially “clots,” probably neutrophil extracellular traps (NETS), which isolate and attack bacteria with neutrophil-derived peptides, such as defensins (47). While an essential part of innate immunity, NETs make reliable and quantitative recovery of pathogens more difficult. It is likely that any clinical implementation will benefit from more rapid, sensitive, and automated preparatory steps. Certainly a relevant factor in apparent “DNA-negative” samples is that many patients are started on antibiotics as soon as there is a suspicion of pneumonia.
A second set of concerns pertains to the PCR amplification step. First, it is desirable to sequence all DNA in the aspirate, so that any pathogen type, whether viral, bacterial, or fungal is identified, but the presence of human genomic DNA might consume the majority of reads and sufficient microbial reads for accurate diagnosis might not be produced. This problem can be overcome by generating a larger number of reads and then excluding the human reads from analysis using bioinformatics tools. A second potential concern in any type of PCR-based diagnosis is amplification bias (45). The universal primers have minor mismatches against specific bacteria that might reduce their annealing efficiency (33), although under these PCR conditions, there was successful amplification of a broad range of bacterial pathogens. While degenerate primers, which are pools of primers with different specificities, can be employed, one still confronts the problem of whether the abundance of the primers matches the relative abundance of the target bacterial sequences. The ideal solution is an amplification-independent methodology, but this requires improvements mentioned above to enrich for the bacteria, so that the human sequences do not overwhelm the bacterial reads. Alternatively, our preliminary studies with “shotgun” sequencing of pulmonary aspirates, suggests that if the initial read number is large, then the human reads can be filtered to produce a diagnosis of the microbes, including fungal and viral sequences (S. K. Hilton, E. Castro-Nallar, M. Pérez-Losada, I. Toma, T. A. McCaffrey, E. P. Hoffman, M. O. Siegel, G. L. Simon, W. E. Johnson, and K. A. Crandall, unpublished data).
Overall, the success or failure of this general strategy will likely depend upon three major factors: (i) the reliable isolation of pathogens from complex biological samples, (ii) the generation of large numbers of long reads, and (iii) the speed with which the sequencing and computational analysis can be completed. While sequencing technologies are increasing in speed, down to hours per run, most of the time-intensive steps are involved in bacterial separation, DNA isolation, PCR amplification, and preparatory steps prior to sequencing. Despite the apparent speed of the actual sequencing, only about 1 h per sample, the sample preparatory work presently consumes days of DNA purification, amplification, blunting, ligation, and library preparation prior to sequencing. We estimate that with current methods, including the computational time and interpretation, a minimum of 48 h per sample would be required to produce a laboratory diagnosis. Technical improvements with real-time sequencing of unmodified DNA via nanopores, for instance, might significantly shorten diagnostic times and increase the effective read lengths for fast, accurate diagnosis of pathogens.
Supplementary Material
ACKNOWLEDGMENTS
This publication was supported by a Pilot grant award from the CTSI-CN at Children's National Medical Center/George Washington University (award UL1RR031988/UL1TR000075 from the NIH National Center for Advancing Translational Sciences). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. We are also grateful to the Abramson family for their ongoing generosity in the financial support for the project. The studies were also supported by a Gill Fellowship to A.K.
The outstanding efforts of Brian Ensor, Sean Connelly, and Warren Santner for advancing research supercomputing, culminating in the Colonial One supercomputing cluster, are gratefully recognized. The contributions of Adam Wong and Tim Wickberg (GWU) and Brett Bowman (Pacific Biosystems) for bioinformatics support in the installation and use of aligners on Colonial One High Performance Computing cluster is greatly appreciated.
Keith Crandall and Eduardo Castro-Nallar have ownership in a commercial startup company, NextGen Diagnostics, Inc., that uses next-generation sequence data for pathogen diagnostics.
Author contributions are as follows. M.O.S., G.L.S., T.A.M., L.S.C., and E.P.H. conceived and designed the studies. L.S.C., M.O.S, J.K., A.Y., and R.A. collected samples and clinical data. I.T. developed the protocol and with A.Y. and A.K. isolated, purified, and amplified the DNA. I.T. prepared sequencing templates and with L.D. and J.D. conducted SMS sequencing. K.A.C., E.C.-N,, M.P.-L., S.H., I.T., A.Y. and T.A.M. conducted alignment and taxonomic assignment. T.A.M., I.T., and M.O.S., wrote the manuscript with all authors contributing.
Footnotes
Published ahead of print 20 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.01678-14.
REFERENCES
- 1.Safdar N, Dezfulian C, Collard HR, Saint S. 2005. Clinical and economic consequences of ventilator-associated pneumonia: a systematic review. Crit. Care Med. 33:2184–2193. 10.1097/01.CCM.0000181731.53912.D9. [DOI] [PubMed] [Google Scholar]
- 2.Ashraf M, Ostrosky-Zeichner L. 2012. Ventilator-associated pneumonia: a review. Hosp. Pract. (1995) 40:93–105. 10.3810/hp.2012.02.950. [DOI] [PubMed] [Google Scholar]
- 3.Heyland DK, Cook DJ, Griffith L, Keenan SP, Brun-Buisson C. 1999. The attributable morbidity and mortality of ventilator-associated pneumonia in the critically ill patient. The Canadian Critical Trials Group. Am. J. Respir. Crit. Care Med. 159:1249–1256. 10.1164/ajrccm.159.4.9807050. [DOI] [PubMed] [Google Scholar]
- 4.Luyt CE, Brechot N, Combes A, Trouillet JL, Chastre J. 2013. Delivering antibiotics to the lungs of patients with ventilator-associated pneumonia: an update. Expert Rev. Anti Infect. Ther. 11:511–521. 10.1586/eri.13.36. [DOI] [PubMed] [Google Scholar]
- 5.Dupont H, Mentec H, Sollet JP, Bleichner G. 2001. Impact of appropriateness of initial antibiotic therapy on the outcome of ventilator-associated pneumonia. Intensive Care Med. 27:355–362. 10.1007/s001340000640. [DOI] [PubMed] [Google Scholar]
- 6.Aryee A, Price N. Antimicrobial stewardship—can we afford to do without it? Br. J. Clin. Pharmacol., in press. 10.1111/bcp.12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.American Thoracic Society, Infectious Diseases Society of America. 2005. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am. J. Respir. Crit. Care Med. 171:388–416. 10.1164/rccm.200405-644ST. [DOI] [PubMed] [Google Scholar]
- 8.Blot F, Raynard B, Chachaty E, Tancrede C, Antoun S, Nitenberg G. 2000. Value of gram stain examination of lower respiratory tract secretions for early diagnosis of nosocomial pneumonia. Am. J. Respir. Crit. Care Med. 162:1731–1737. 10.1164/ajrccm.162.5.9908088. [DOI] [PubMed] [Google Scholar]
- 9.Fagon JY, Chastre J, Wolff M, Gervais C, Parer-Aubas S, Stephan F, Similowski T, Mercat A, Diehl JL, Sollet JP, Tenaillon A. 2000. Invasive and noninvasive strategies for management of suspected ventilator-associated pneumonia. A randomized trial. Ann. Intern. Med. 132:621–630. 10.7326/0003-4819-132-8-200004180-00037. [DOI] [PubMed] [Google Scholar]
- 10.Tetenta S, Metersky ML. 2011. Tracheal aspirate Gram stain has limited sensitivity and specificity for detecting Staphylococcus aureus. Respirology 16:86–89. 10.1111/j.1440-1843.2010.01855.x. [DOI] [PubMed] [Google Scholar]
- 11.Demers AM, Boulle A, Warren R, Verver S, van Helden P, Behr MA, Coetzee D. 2010. Use of simulated sputum specimens to estimate the specificity of laboratory-diagnosed tuberculosis. Int. J. Tuberc. Lung Dis. 14:1016–1023. [PubMed] [Google Scholar]
- 12.Van Dalfsen JM, Stapp JR, Phelps C, Stewart P, Burns JL. 2002. Comparison of two culture methods for detection of tobramycin-resistant gram-negative organisms in the sputum of patients with cystic fibrosis. J. Clin. Microbiol. 40:26–30. 10.1128/JCM.40.1.26-30.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrett-Connor E. 1971. The nonvalue of sputum culture in the diagnosis of pneumococcal pneumonia. Am. Rev. Respir. Dis. 103:845–848. [DOI] [PubMed] [Google Scholar]
- 14.Mondi MM, Chang MC, Bowton DL, Kilgo PD, Meredith JW, Miller PR. 2005. Prospective comparison of bronchoalveolar lavage and quantitative deep tracheal aspirate in the diagnosis of ventilator associated pneumonia. J. Trauma 59:891–895; discussion 895–896. 10.1097/01.ta.0000188011.58790.e9. [DOI] [PubMed] [Google Scholar]
- 15.Shariatzadeh MR, Marrie TJ. 2009. Does sputum culture affect the management and/or outcome of community-acquired pneumonia? East Mediterr. Health J. 15:792–799. [PubMed] [Google Scholar]
- 16.Johansson N, Kalin M, Tiveljung-Lindell A, Giske CG, Hedlund J. 2010. Etiology of community-acquired pneumonia: increased microbiological yield with new diagnostic methods. Clin. Infect. Dis. 50:202–209. 10.1086/648678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beck JM, Young VB, Huffnagle GB. 2012. The microbiome of the lung. Transl. Res. 160:258–266. 10.1016/j.trsl.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson MR, Naccache SN, Samayoa E, Biagtan M, Bashir H, Yu G, Salamat SM, Somasekar S, Federman S, Miller S, Sokolic R, Garabedian E, Candotti F, Buckley RH, Reed KD, Meyer TL, Seroogy CM, Galloway R, Henderson SL, Gern JE, DeRisi JL, Chiu CY. 2014. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N. Engl. J. Med. 370:2408–2417. 10.1056/NEJMoa1401268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stressmann FA, Rogers GB, Klem ER, Lilley AK, Donaldson SH, Daniels TW, Carroll MP, Patel N, Forbes B, Boucher RC, Wolfgang MC, Bruce KD. 2011. Analysis of the bacterial communities present in lungs of patients with cystic fibrosis from American and British centers. J. Clin. Microbiol. 49:281–291. 10.1128/JCM.01650-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawanami T, Fukuda K, Yatera K, Kido M, Mukae H, Taniguchi H. 2011. A higher significance of anaerobes: the clone library analysis of bacterial pleurisy. Chest 139:600–608. 10.1378/chest.10-0460. [DOI] [PubMed] [Google Scholar]
- 21.Rogers GB, Hart CA, Mason JR, Hughes M, Walshaw MJ, Bruce KD. 2003. Bacterial diversity in cases of lung infection in cystic fibrosis patients: 16S ribosomal DNA (rDNA) length heterogeneity PCR and 16S rDNA terminal restriction fragment length polymorphism profiling. J. Clin. Microbiol. 41:3548–3558. 10.1128/JCM.41.8.3548-3558.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang YJ, Lynch SV. 2011. The emerging relationship between the airway microbiota and chronic respiratory disease: clinical implications. Expert Rev Respir. Med. 5:809–821. 10.1586/ers.11.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flanagan JL, Brodie EL, Weng L, Lynch SV, Garcia O, Brown R, Hugenholtz P, DeSantis TZ, Andersen GL, Wiener-Kronish JP, Bristow J. 2007. Loss of bacterial diversity during antibiotic treatment of intubated patients colonized with Pseudomonas aeruginosa. J. Clin. Microbiol. 45:1954–//,962. 10.1128/JCM.02187-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, Cooper J, Sin DD, Mohn WW, Hogg JC. 2012. The lung tissue microbiome in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 185:1073–1080. 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kakizaki E, Ogura Y, Kozawa S, Nishida S, Uchiyama T, Hayashi T, Yukawa N. 2012. Detection of diverse aquatic microbes in blood and organs of drowning victims: first metagenomic approach using high-throughput 454-pyrosequencing. Forensic Sci. Int. 220:135–146. 10.1016/j.forsciint.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. 2012. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One 7:e47305. 10.1371/journal.pone.0047305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabrera-Rubio R, Garcia-Nunez M, Seto L, Anto JM, Moya A, Monso E, Mira A. 2012. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J. Clin. Microbiol. 50:3562–3568. 10.1128/JCM.00767-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tunney MM, Einarsson GG, Wei L, Drain M, Klem ER, Cardwell C, Ennis M, Boucher RC, Wolfgang MC, Elborn JS. 2013. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am. J. Respir. Crit. Care Med. 187:1118–1126. 10.1164/rccm.201210-1937OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L, Jablonski K, Kleerup E, Lynch SV, Sodergren E, Twigg H, Young VB, Bassis CM, Venkataraman A, Schmidt TM, Weinstock GM, Lung HIVMP. 2013. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am. J. Respir. Crit. Care Med. 187:1067–1075. 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. 2011. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 184:957–963. 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perera J, Arachchi DM. 1999. The optimum relative centrifugal force and centrifugation time for improved sensitivity of smear and culture for detection of Mycobacterium tuberculosis from sputum. Trans. Roy. Soc. Trop. Med. Hyg. 93:405–409. 10.1016/S0035-9203(99)90135-9. [DOI] [PubMed] [Google Scholar]
- 32.Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 1991. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173:697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74:2461–2470. 10.1128/AEM.02272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francis OE, Bendall M, Manimaran S, Hong C, Clement NL, Castro-Nallar E, Snell Q, Schaalje GB, Clement MJ, Crandall KA, Johnson WE. 2013. PathoScope: species identification and strain attribution with unassembled sequencing data. Genome Res. 23:1721–1729. 10.1101/gr.150151.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Byrd A, Perez-Rogers J, Manimaran S, Castro-Nallar E, Toma I, McCaffrey T, Siegel M, Benson G, Crandall K, Johnson W. 2014. Clinical PathoScope: rapid alignment and filtration for accurate pathogen identification in clinical samples using unassembled sequencing data. BMC Bioinformatics 15:262. 10.1186/1471-2105-15-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145. 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fichot E, Norman RS. 2013. Microbial phylogenetic profiling with the Pacific Biosciences sequencing platform. Microbiome 1:10. 10.1186/2049-2618-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munoz R, Yarza P, Ludwig W, Euzeby J, Amann R, Schleifer KH, Glockner FO, Rossello-Mora R. 2011. Release LTPs104 of the All-Species Living Tree. Syst. Appl. Microbiol. 34:169–170. 10.1016/j.syapm.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 41.Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer KH, Ludwig W, Glockner FO, Rossello-Mora R. 2008. The All-Species Living Tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst. Appl. Microbiol. 31:241–250. 10.1016/j.syapm.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright ES, Yilmaz LS, Noguera DR. 2012. DECIPHER, a Search-Based Approach to Chimera Identification for 16S rRNA Sequences. Appl. Environ. Microbiol. 78:717–725. 10.1128/AEM.06516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar PS, Brooker MR, Dowd SE, Camerlengo T. 2011. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS One 6:e20956. 10.1371/journal.pone.0020956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Claesson MJ, Wang Q, O'Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O'Toole PW. 2010. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38:e200. 10.1093/nar/gkq873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang YJ, Kim E, Cox MJ, Brodie EL, Brown R, Wiener-Kronish JP, Lynch SV. 2010. A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. Omics 14:9–59. 10.1089/omi.2009.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng OZ, Palaniyar N. 2013. NET balancing: a problem in inflammatory lung diseases. Front. Immunol. 4:1. 10.3389/fimmu.2013.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.