

Abstract
Autophagy is an important physiological process in the heart, and alterations in autophagic activity can exacerbate or mitigate injury during various pathological processes. Methods to assess autophagy have changed rapidly as the field of research has expanded. As with any new field, methods and standards for data analysis and interpretation evolve as investigators acquire experience and insight. The purpose of this review is to summarize current methods to measure autophagy, selective mitochondrial autophagy (mitophagy), and autophagic flux. We will examine several published studies where confusion arose in in data interpretation, in order to illustrate the challenges. Finally we will discuss methods to assess autophagy in vivo and in patients.
Keywords: autophagy, mitophagy, cardiac, physiology, methodology, fluorescence microscopy
OVERVIEW OF AUTOPHAGY
Autophagy emerged as a cardiac research topic starting in 2005 with the recognition of its role in hibernating myocardium1 and in the protective response to BNIP32; in the ensuing 8 years, the number of publications on cardiac autophagy have increased ten-fold. Thus, there is a substantial need to have reliable methods to measure the process of autophagy. Autophagy is an essential housekeeping function responsible for eliminating unwanted protein aggregates or organelles.3, 4 Autophagy encompasses three primary pathways for lysosomal degradation: macroautophagy (the methodological focus of this review), microautophagy, and chaperone-mediated autophagy. Microautophagy is a non-selective form of degradation in which lysosomal membranes directly engulf cytoplasm along the periphery of the organelle. Glycogen and other endosomal components are examples of material that is degraded by microautophagy. Chaperone-mediated autophagy targets cytosolic proteins and translocates them straight across the lysosomal membrane to be degraded. Proteins targeted by chaperone-mediated autophagy must contain a specific targeting motif that is recognized by HSPA8/HSC70 which subsequently directs the cargo for degradation. Ultimately, all three processes depend upon lysosomal function, failure of which carries pathological consequences. Genetic defects in lysosomal function such as Danon disease and Pompe disease, are recognized to result in cardiomyopathy due to glycogen accumulation5–7. While lysosomal function is essential to macroautophagy—the focus of this review—we do not cover specific methods to assess lysosomal function.
Macroautophagy involves the engulfment of a portion of the cytoplasm by a double-membrane phagophore which elongates around both selectively and non-selectively targeted cargo. After phagophore closure, the newly-formed vesicle, now termed the autophagosome, fuses with the lysosome forming the autolysosome. In the acidic milieu of the lysosome, pH-dependent hydrolases degrade the proteins, lipids, nucleic acids, and carbohydrates of the cargo. The phagophore membrane is thought to arise from multiple membrane sources including the endoplasmic reticulum (ER). In one model, phagophore initiation begins with the distention of the ER into a phosphatidylinositol-3-phosphate (PtdIns3P)-rich structure called the omegasome. This process is initiated with the recruitment of the ULK (unc-51-like kinase) complex and the class III PdIns-3-kinase complex which are targeted to the omegasome via ATG13 and ATG14L1 respectively. From the omegasome emerges the phagophore membrane at which point WIPI2 and ATG16L1 are recruited. ATG12 is conjugated to ATG5 through the action of ubiquitin ligase-like enzymes ATG7 and ATG10; the covalently linked ATG5-ATG12 duo complexes with ATG16L1, enabling the processing of microtubule-associated protein 1A/1B-light chain 3 (LC3). The precursor form of LC3 is truncated by the cysteine protease ATG4 to expose the C-terminal glycine of LC3 to form LC3-I. This in turn is conjugated to the amino group of phosphatidylethanolamine (PE) (forming membrane-associated LC3-II) by the action of E1 and E2-like enzymes ATG7 and ATG3. Once the LC3-II-decorated phagophore closes around its target, the ATG12-ATG5-ATG16L complex is released from the membrane. When the autophagosome fuses with the lysosome, LC3-II on the outer face of the autolysosome is released by the action of ATG4; however, LC3 on the inner face is retained and eventually degraded by lysosomal enzymes.
EXAMPLES OF CONFUSING AUTOPHAGY STUDIES AND CONFOUNDING VARIABLES
To highlight the need for appropriate standards for measuring autophagy and interpreting results, we will first present some recent autophagy studies in which the results were contradictory, incomplete, or subject to alternative interpretation, leading to controversial and possibly incorrect conclusions.
Difficulties in interpreting autophagic changes in the setting of diet-induced obesity
At a recent meeting, contradictory results were reported by two groups using mCherry-LC3 transgenic mice, in which the fusion protein is expressed under the control of the αMHC promoter. Abel’s group observed an increase in the number of mCherry-labeled puncta in mice fed a high-fat diet, whereas Mentzer’s group observed a decrease [Fig.1]. Diet composition differed: Mentzer’s group used the D12492 60% fat diet, whereas Abel’s group used a western diet of 40% fat and increased sucrose. Mice were enrolled at 5–6 wk of age and maintained for 15 wk in the Mentzer study while the Abel study enrolled mice at 8 wk and maintained them on the diet for 12 wk. Another important difference was the genetic background of the mice. The Mentzer lab used mCherry-LC3 mice in the FVBN background, whereas Abel’s group backcrossed the line into the C57BL/6 background. Interestingly, both groups arrived at the same conclusion, that the obesogenic diet impaired cardiac autophagy. Abel’s group used chloroquine to show that autophagic flux was attenuated. They noted elevated p62/SQSTM1 that did not increase further when flux was inhibited with chloroquine. Mentzer’s group also observed elevated p62/SQSTM1 in the high fat diet group, and similarly concluded that autophagic flux was impaired. This illustrates the difficulties in determining autophagic activity based on “snapshot” measurements of LC3 puncta or western blot and emphasizes the need to assess flux directly (e.g. by chloroquine) or indirectly (by p62/SQSTM1).
Figure 1. Autophagic puncta in hearts of mCherry-LC3 mice on chow or high fat diet (HFD).
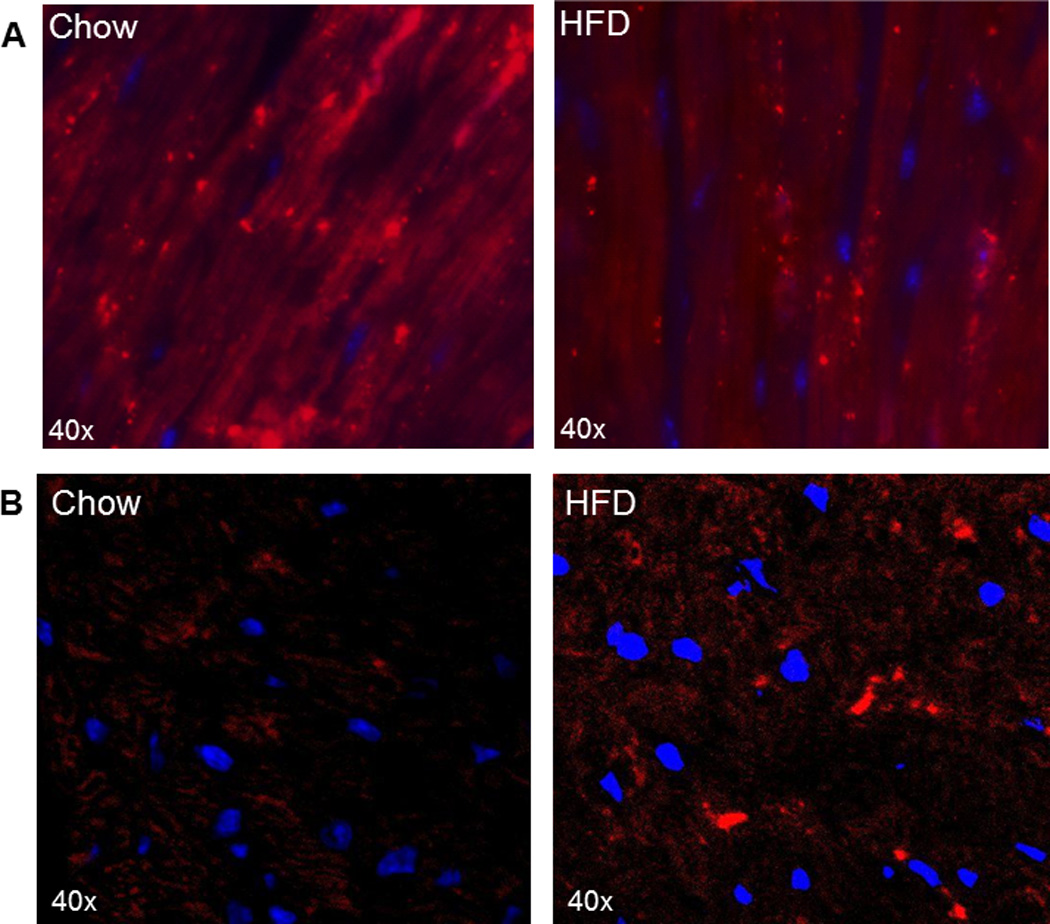
A. Representative heart sections of mice from the Mentzer study after 15 wk of chow or Teklad D12492 high fat diet. Cardiac sections (5–6 µm) were made using a cryostat, mounted on glass slides and nuclei stained with Hoechst 33342. Images were acquired with a Nikon TE300 fluorescence microscope equipped with a 60× Plan Apo objective with excitation/emission wavelengths of 560/630 nm. (Unpublished images generously provided by Dr. Bruce Ito and Dr. Robert Mentzer.) B. Representative heart sections of mice from the Abel study after 12 wk of chow or western diet. Images were taken with Zeiss LSM 510 confocal microscope using x63 immersion oil objective with excitation/emission wavelengths of 543/613 nm. (Unpublished images generously provided by Dr. Bharat Jaishy and Dr. E. Dale Abel.)
Becn1 haploinsufficient mice
Deletion of Atg5 results in death in the newborn period unless nutritional support is provided during the first 12–24 hr of postnatal life8, but surviving pups are relatively normal, although they develop heart failure and other abnormalities with time. Subsequent studies revealed an alternative autophagic pathway independent of ATG5 and ATG7 but dependent on BECN1, ULK1, and RAB99. BECN1, which participates in both pathways, is essential, as the deletion of the corresponding gene is embryonic lethal10. Subsequently, BECN1 haploinsufficient mice were used as a model of attenuated autophagy in a number of studies; however, BECN1 also regulates autophagosome-lysosome fusion and when partnered with KIAA0226/Rubicon, can suppress fusion11, 12. These complexities have led to a number of contradictory findings: Becn1+/− mice showed less hypertrophy in response to aortic banding, which was used as evidence that autophagy contributed to pathologic hypertrophy13. Becn1+/− mice are observed to have smaller infarcts after ischemia/reperfusion injury, leading the authors to conclude that BECN1-dependent autophagy contributes to reperfusion injury14. In contrast, chloramphenicol administered to pigs before ischemia or at reperfusion profoundly reduces infarct size and is associated with a substantial upregulation of BECN115. Thus, contradictory roles for BECN1 and autophagy emerged from these studies, largely dependent on the interpretation of LC3 autophagic markers. In the study by Hill’s group of Becn1+/− mice subjected to aortic banding, they observed fewer autophagosomes in heart tissue and concluded that this indicated decreased autophagy13. However, they did not measure autophagic flux; increased autophagic flux would also be consistent with their observation. Subsequent work by Diwan’s group using neonatal rat ventricular myocytes showed that BECN1 knockdown results in accelerated autophagic flux16, presumably by reducing the BECN1-KIAA0226 impediment to autophagosome-lysosome fusion11, 12. A criticism of Diwan’s work is that it is limited to cell culture. However a study by Xu et al. examined autophagic flux in normal and diabetic mice, comparing wild type and BECN1 haploinsufficient mice17. They measured LC3 levels in the absence and presence of the lysosomal inhibitor Bafilomycin A1 and reported flux as the difference in LC3-II content between BAF and vehicle, reaching the conclusion that flux was not different between Becn1+/− and WT mice. However, when flux is calculated as the fold increase over vehicle control as originally described by Tanida et al.18, they might have reached a different conclusion that would have supported the idea that BECN1 can slow autophagic flux. These controversies over interpretation of the data and the functional effects of BECN1 are reflected in prior work with OVE26 diabetic mice19 (BECN1 suppresses flux) and other studies with the Becn1+/− mice20 (BECN1 deficiency results in insufficient autophagy initiation). Whether diminished autophagy initiation or accelerated flux is the dominant effect may depend upon the tissue and the context of pathologic stress. Taken together, these studies exemplify the difficulties associated with using BECN1 to modulate autophagy and the challenges associated with interpreting results.
If the dominant effect of BECN1 in the heart is to slow autophagic flux, then it becomes necessary to revisit the conclusions reached by Hill13 and Sadoshima14: BECN1 haploinsufficiency might have accelerated autophagic flux to reduce hypertrophy and infarct size in their models. This illustrates the importance of measuring autophagic flux correctly and emphasizes the profound limitations of static measurement of LC3.
As BECN1 also has the potential to regulate apoptosis through interaction with Bcl-2, its downregulation might benefit the heart through reduction of apoptosis. To examine this, Diwan’s group used chloroquine and found that suppression of autophagic flux was sufficient to exacerbate autophagy after hypoxia/reoxygenation. They found that the cell death was prevented by cyclosporine A but not that pan-caspase inhibitor ZVAD, suggesting that the mitochondrial permeability transition pore was responsible for triggering necrotic cell death16. Other studies have shown the importance of clearing damaged mitochondria to reduce ischemia/reperfusion injury2, 15, 21–23. Postconditioning was also shown to restore autophagic flux during reperfusion, whereas inhibition of flux (with chloroquine) abolished the protective effects of sevoflurane postconditioning24.
HDAC inhibitors
Histone deacetylases regulate an expanding number of pathways, and substrate specificity is restricted according to the enzyme subtype25. In 2011, Hill’s group reported that trichostatin A (TSA), an inhibitor of class I and II HDACs, reversed cardiac hypertrophy due to pressure overload by downregulating autophagy26. In their study, autophagy measurements were limited to detection of autophagic puncta and LC3 western blots. No flux measurements or assessment of p62/SQSTM1 were performed on the hearts, although they did assess flux in neonatal rat ventricular myocytes treated with TSA. A careful inspection of their results (their supplemental data figure S1) reveals that autophagic flux was enhanced in TSA-treated cardiomyocytes (lysosomal blockade resulted in a 2.1-fold increase in LC3-II in controls vs. a 2.8-fold increase in TSA-treated cells). Confusingly, the authors concluded that because the absolute levels of LC3-II were lower in the TSA group, its effects were due to suppression of autophagy, stating that “suppression of autophagic flux contributes to the salutary effects of HDAC inhibitor therapy”. It should be noted that if autophagic flux was suppressed, LC3-II levels would not increase after lysosomal blockade. The fact that the levels nearly triple after lysosomal blockade suggests that the low level of LC3-II seen in the TSA group (with or without lysosomal blockade) is due to brisk autophagic flux.
In 2013, the same group used a related HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA; vorinostat), to show that HDAC inhibition increased autophagic flux and decreased infarct size in a rabbit model of I/R injury27. Using mice expressing tandem RFP-GFP-LC3, they observed that SAHA increased autophagic flux. The effect of SAHA on autophagic flux was confirmed in cell culture in which flux was assessed by the gold standard of comparing LC3-II in the presence and absence of lysosomal blockade with bafilomycin. The requirement for autophagy in SAHA’s beneficial effects was further confirmed by downregulating ATG7, which abolished the protective effects of SAHA. These two studies again illustrate the challenges in interpreting LC3 results and also support the use of the tandem RFP-GFP-LC3 construct for evaluating autophagic flux. Importantly, they provided further evidence to support the role of autophagy through knockdown of a key factor in the canonical autophagy pathway, ATG7 (we have used dominant negative ATG5 with good effect28). As noted above, BECN1 can have paradoxical effects on autophagy due to its interaction with KIAA0226/Rubicon and suppression of autophagosome-lysosome fusion.
Lessons learned from analysis of these studies
These publications have been widely-cited and have influenced our understanding of autophagy. However, the failure to measure flux and the controversy over interpretation of results have resulted in confusion about the role and significance of autophagy. The purpose of our rather detailed dissection of those publications is to point out the challenges associated with analyzing autophagy, to encourage investigators to use proper methodology in assessing autophagy, to call for a thoughtful reevaluation of the conclusions of those studies, and to exhort readers to exercise caution when reading new publications.
Several things emerge from this discussion. Static levels of LC3 or scoring of autophagic puncta is an incomplete assessment of autophagy without assessment of flux (directly, through lysosomal blockade, or indirectly, inferred from a decrease in p62/SQSTM1). The most definitive way to demonstrate a role for autophagy is to knock down ATG7 or inhibit ATG5. BECN1 has paradoxical effects and a knockdown of this protein may be confusing when assessing the role of autophagy in a particular organ or disease process. While not discussed in detail in this review, it should be noted that the transcriptional regulation of autophagy has not been closely correlated with functional autophagic flux: mRNA may be increased when autophagy is impaired, and mRNA levels may be stable even when autophagic flux is active. However, mRNA upregulation is part of the longer-term autophagic response. There is a need for additional methods to assess autophagy in tissues, and to be able to image autophagy noninvasively.
Additional variables that may confound autophagy results
Impact of mouse strain on autophagy
In addition to the difference in autophagy noted in the mCherry-LC3 transgenics that may be due to strain differences (FVBN vs. C57BL/6), a comparison of cardiac autophagy in C57BL/6 and BALB/c mice conducted by Phyllis Linton’s group revealed substantial differences in autophagy (unpublished data). While many upstream signals driving autophagy were increased in BALB/c mice compared to C57BL/6 mice at 3mo of age, the BALB/c mice had increased aggregate-associated p62/SQSTM1, higher levels of protein carbonylation, and lower levels of LC3-I and -II. Autophagic flux was impaired, evidenced by a failure to increase LC3-II after chloroquine treatment and by the presence of elevated levels of p62/SQSTM1 and ubiquitinated proteins. Strain differences have been noted by others as well29, 30.
Circadian effects
Many investigators have noted a significant diurnal variation in autophagy31–34: we observed that LC3-II levels increased by 40% between 10am and 2pm in hearts of FVBN mice maintained on a 12 h light/dark schedule (lights off at 6pm) (Ito, Mentzer, and Gottlieb, unpublished data). The magnitude of this increase is comparable to that achieved by fasting; thus it is important to design experiments to avoid circadian effects.
Effects of sex on autophagy
Examination of autophagy (LC3-II levels) in hearts of C57Bl/6 mice revealed that females showed 50% more LC3-I and BECN1 under basal conditions than male animals (unpublished data, Michael Gurney and Phyllis-Jean Linton). Sex differences in autophagy in various tissues have been reported by several labs35, 36,6, 35, 37–40. Response to rapamycin also differed according to sex, suggesting sex-related differences in the MTOR signaling pathway41, 42.
METHODS TO MEASURE AUTOPHAGY AND MITOPHAGY
Measurement of autophagy in cells and tissues
Western blot of LC3 and p62/SQSTM1
Measuring protein expression remains the mainstay method for investigating the process of autophagy. Until recently, the paradigm for measuring autophagy was to examine LC3-II and LC3-I via western blot. LC3-II is consistently associated with autophagosomes and for that reason is a useful indicator of autophagosome initiation43–46. Conveniently, lipidation of LC3 alters its conformation such that it has faster mobility in SDS-PAGE. While some groups have used the LC3-II/LC3-I ratio as an indication of autophagic activity, this is unreliable and has been generally discounted47. While LC3-I levels can rise in response to an autophagic stimulus, if flux is rapid, levels may remain stable or actually decrease, if lipidation outstrips expression of LC3 or recycling of LC3-II off of autolysosomes by ATG4. Furthermore, LC3-II levels can rise with autophagy, but can rise even more if lysosomal degradation of autophagosomes is impaired. Depending on the particular dynamics of a given cell responding to stress, the ratio can be up, down, or unchanged. LC3-II normalized to a protein loading control is used for flux measurements.
The autophagy receptor protein p62/SQSTM1 can serve as a surrogate marker of increased autophagy when levels are diminished, although caveats are appropriate here as well. It is important to understand that p62/SQSTM1 can be present in cells or tissues as a freely soluble form in the cytosol, or associated with detergent-insoluble ubiquitinated protein aggregates. The normal response to an autophagic stimulus involves an early increase in p62/SQSTM1 expression in the cytosol, followed by clearance of p62/SQSTM1 associated with aggregates or other cargo. While not indicated in every instance, assessment of soluble and detergent-insoluble p62/SQSTM1 pools can provide valuable information48. Like LC3, western blot analysis of p62/SQSTM1 is only a snapshot of a dynamic process. For that reason, measurement of LC3 and p62/SQSTM1 in the presence or absence of lysosomal blockade with chloroquine or bafilomycin A1 provides essential information about the rate of transit of autophagosome cargo through lysosomal degradation (autophagic flux)49–52. Table I indicates the expected changes in LC3 and p62/SQSTM1 under basal conditions, during initiation of autophagy, at equilibrium upregulated autophagy, when autophagic precursors are limited, and in the setting of impaired flux (acute and chronic). Also shown are the expected effects of lysosomal blockade in each scenario. Availability of precursors can be limiting if protein synthesis is suppressed, e.g., during ischemia. In that case, brisk autophagic flux is reflected by a substantial depletion of available factors53. Besides LC3 and p62/SQSTM1, other autophagy-related factors including ATG16L1, BECN1, and the ATG5-ATG12 complex, commonly rise in parallel with LC3-II and provide supportive evidence, particularly if LC3 blots are of poor quality (see technical concerns below).
Table 1.
Expected changes in common autophagy markers in various scenarios (snapshot and after lysosomal blockade (+CQ)). “+” indicates relative intensity of the band on western blot. Note that in chronic flux blockade, there is feedback inhibition of autophagy initiation, possibly mediated by Beclin 116.
| Basal | Early Atg | Equilibrium Atg |
Precursors Limited |
Early Flux Blockade |
Chronic Flux Blockade |
|
|---|---|---|---|---|---|---|
| LC3-I | ++ | ++ | ++ | − | ++ | + |
| LC3-I + CQ | ++ | +++ | +++ | + | ++ | + |
| LC3-II | + | ++ | +++ | + | +++ | + |
| LC3-II +CQ | ++ | +++ | ++++ | +++ | +++ | + |
| P62/SQSTM1 | + | +++ | + | − | +++ | ++++ |
| P62/SQSTM1 + CQ | ++ | ++++ | +++ | ++ | +++ | ++++ |
Immunostaining of LC3 puncta
Detection of autophagosomes by immunostaining of LC3 in fixed cells or tissues is technically challenging because of the high background from LC3-I unassociated with autophagosomes; this can be improved by gentle permeabilization of cells (e.g., with digitonin or saponin) to extract free LC3-I without affecting membrane-associated LC3-II54. This method is technically challenging and has not been widely accepted, however.
Technical concerns specific to LC3
LC3 is a fragile low-abundance protein that can be lost from frozen tissue samples with repetitive freeze-thawing, a concern that diminishes for samples stored in SDS-PAGE sample buffer. Heart tissue should be snap-frozen in liquid nitrogen and stored at −80°C until it can be processed. We have compared RIPA buffer: [50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40]); a Triton X-100-based detergent extraction buffer [50 mM Tris-HCl pH=7.4, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton-X 100]; and RIPA solubilization of tissue ground to a powder at liquid nitrogen temperatures, and have concluded that the most reliable protocol for solubilization of LC3 from frozen heart tissue is with RIPA followed quickly by centrifugation at 1000× g to remove nuclei. After removing an aliquot for protein determination, the solubilized sample should be promptly boiled in sample buffer with fresh 2-mercaptoethanol (2ME) and stored frozen at −80°C until used for gel loading (fresh 2ME may be added back). LC3 binds more avidly to PVDF than nitrocellulose (this differential binding is particularly notable for human heart samples), and is readily lost from membranes that are stripped and reprobed. For that reason, LC3 should be the first protein probed for on a membrane. Because of its low molecular weight, transfer time should be kept short (e.g. 150 mA for 2hr). Antibodies to LC3 are available from a number of sources and vary with respect to isoforms recognized, species specificity, and avidity for LC3-I vs. LC3-II. We have found the antibody against LC3A/B [cat#4108, Cell Signaling Technology] to be quite reliable for rodent heart tissue (rat and mouse) as well as for porcine and human heart samples.
Cell signaling markers of autophagy induction
Autophagy initiation is controlled by two major cell signaling pathways. The AKT/MTOR signaling axis is a well-recognized negative regulator of autophagy, while AMPK is known to promote initiation. These opposing signaling pathways converge on ULK1 to dictate the fate of autophagy. ULK1 is important for the nucleation of autophagosomes. Phosphorylation of ULK1 by AMPK on Ser317, 555 and 777 promotes autophagy55, 56. In contrast, phosphorylation of ULK1 at Ser757 by MTOR inhibits autophagy initiation56. Markers of MTOR activity include MTOR phosphorylation at Ser2448, phosphorylation of ribosomal subunit RPS6 at Ser235/236, and phosphorylation of the translation repressor protein EIF4E-BP1 at Ser65 and Thr70.
mRNA markers of autophagy
Measurement of mRNA for autophagy proteins can provide additional insights, but the presence of upregulation at the mRNA level does not necessarily correlate with protein expression or functional autophagy. It can, however, reflect an intact signaling pathway for the induction of autophagy.
Measuring mitophagy
The specific targeting of mitochondria for autophagic disposal during I/R insult is an important element of cardioprotection21, 22. Because mitochondrial autophagy is a major pathway for turnover of mitochondria, it is important to be able to monitor mitophagy. Examining mitochondrial content via western blot or mitochondrial DNA to nuclear DNA ratio (mtDNA:nucDNA) must be paired with the same measurement in the presence of lysosomal blockade with chloroquine or bafilomycin A1 in order to infer mitophagy. It is important to assess markers in outer mitochondrial membrane (TOMM70 or VDAC) as well as inner membrane/matrix (COX4I1/COX IV or ACO2/aconitase). This is because outer mitochondrial membrane proteins are also subject to degradation by the ubiquitin proteasome system57. An observed decrease in mitochondrial mass that is prevented by lysosomal blockade is indicative of mitophagy.
Use of fluorescent reporters to assess mitophagy
The coral-derived fluorescent protein Keima, changes its fluorescence properties over the pH range of 4–8; when targeted to the mitochondrial matrix (mitoKeima), it serves to report on delivery of mitochondria to the lysosome58. MitoKeima holds great promise for monitoring mitochondrial autophagy in cells, and a transgenic reporter mouse would have considerable utility. A somewhat different approach was pioneered by our lab: we targeted fluorescent Timer protein to the mitochondrial matrix (MitoTimer). Newly-synthesized protein fluoresces green, but the conformation matures over 24–48 h to a more stable red fluorescent conformation. This allows detection of mitochondrial biogenesis (green MitoTimer) and mitophagy (decrease in red MitoTimer if new protein is not being made)59, 60. Using constitutive expression of MitoTimer electroporated into mouse skeletal muscle, Laker et al. showed that exercise increases turnover of mitochondria (biogenesis and mitophagy) whereas a high-fat diet slows turnover61. The advent of a transgenic MitoTimer mouse will doubtless provide additional insights.
Methods to assess autophagy in intact animals
Measuring autophagic flux in vivo with lysosomal blockade
This method to measure flux introduces a pharmacological blockade that prevents lysosomal/autophagosomal fusion or prevents lysosomal-mediated enzymatic degradation. A number of agents are suitable to achieve lysosomal blockade: bafilomycin A1, chloroquine, and ammonium chloride raise the intralysosomal pH, preventing autophagosome fusion with the lysosome51, 62–65, while protease inhibitors such as pepstatin A, E-64d, and leupeptin inhibit lysosomal proteases47, 66, 67 and microtubule inhibitors such as vinblastine prevent trafficking of autophagosomes and lysosomes68. For in vivo studies, chloroquine (10–50 mg/kg i.p.)48, 69, leupeptin (20–40 mg/kg i.p.)67, or bafilomycin A1 (2.5 mg/kg i.p. every 12 h)70 have been used to block autophagic flux. Mice subjected to lysosomal blockade as well as untreated comparators are sacrificed for tissue harvest at a specified time (usually 2–4 h) after blockade. Lysosomal inhibition results in accumulation of autophagosomes that would have progressed through the pathway during that period. Differential accumulation of autophagosomes is assessed by microscopy, or autophagosomal LC3-II is measured by western blot66. Intact autophagic flux is indicated by an increase in autophagosome puncta or in LC3-II on western blot compared to paired animals without lysosomal blockade and is calculated as a ratio rather than the difference (according to Tanida18). Little work has been done to establish whether the magnitude of increase is proportional to the rate of autophagic flux. We have found that the receptor protein p62/SQSTM1 is a fairly reliable surrogate marker of autophagic flux: when flux is intact, p62/SQSTM1 levels should decrease, whereas when flux is absent, p62/SQSTM1 levels will rise. This is helpful when interpreting results of samples obtained before and after an intervention, such as atrial biopsies obtained at the beginning and end of aortic cross-clamp53.
Measuring autophagic flux ex vivo in tissue samples
Ana Maria Cuervo’s group71 has developed a method to measure autophagic flux in small chunks of liver tissue which are minced finely and then divided between two wells (with or without lysosomal inhibitors) and then incubated at 37°C for 1–2 h with occasional swirling. The minced tissue is then recovered, homogenized, and processed for western blotting. We have begun preliminary work to adapt this to heart tissue and are optimistic that this ex vivo flux assay can provide reliable information about the level of autophagy and the presence/absence of autophagic flux in small tissue samples. This approach will eliminate the need for paired animals treated +/− lysosomal inhibitors, and will also make it possible to monitor flux in freshly-obtained biopsies from human tissues.
Imaging of autophagic puncta in transgenic mice
Transgenic mice expressing a fluorescent protein fused to LC3 have been used to monitor autophagy in a number of studies, although as noted above, it is essential to monitor flux either through comparison with mice in which lysosomal function has been inhibited, or inferred from changes in p62/SQSTM1. Cryosections are preferable for preserving fluorescence of the fusion proteins. Puncta can be scored in various ways: as the number per unit area, or as the percent of area occupied by fluorescent puncta (useful when puncta are so numerous that it is difficult to count individual units). Because autophagosomes are typically more numerous in the perinuclear zone, scoring will be more consistent in sections where myocytes are longitudinally arrayed. Normalizing puncta number (or area) to nuclei may be preferable to normalizing to area. It should be noted that transgenics expressing LC3 fusion proteins may also develop protein aggregates which can easily be mistaken for puncta. It is important to select a line in which expression is relatively low to reduce the occurrence of aggregates. Although the transgene is not under control of a physiologic promoter, the abundance of the fusion protein changes rapidly in response to physiologic cues such as exercise, fasting, or ischemic preconditioning23, suggesting that post-translational regulation dominates. Proteasomal inhibition increased the abundance of mCherry-LC3 (C Perry, unpublished observations), consistent with findings reported in astrocytes72. Several additional caveats should be noted when using LC3 fusion proteins for assessing autophagy. LC3B, which is preferentially detected by one of the commonly-used antibodies (rabbit-anti-LC3AB antibody, Cell Signaling [4108S]), and which was used for the fusion proteins, is a member of the ATG8 gene family, which has 7 members in humans: MAP1LC3A, B, B2, C, GABARAP, GABARAPL1, and GABARAPL2/GATE-16. There are tissue-specific differences in expression of ATG8 family members: GABARAPL2 and GABARAPL1 are expressed predominantly in the central nervous system, whereas GABARAP is more heavily expressed in endocrine glands, and LC3C is predominantly expressed in the lung. Different family members may be responsible for specific roles in protein trafficking and selective target engulfment, and they may be upregulated in response to different cues73. It has been suggested that GABARAPL1 interacts with STBD1 (starch binding domain 1) and therefore may play a role in autophagic degradation of glycogen granules (glycophagy). Thus monitoring LC3B (including LC3B fusion proteins) may be only part of the story. Non-canonical autophagy (independent of LC3B and ATG5) shares some autophagy machinery and often responds to similar cues74.
LC3 fusion proteins can also be monitored by western blotting: conveniently, the initial degradation of GFP-LC3 in the lysosome gives rise to a fragment that can be readily detected with antibody to GFP75.
Use of tandem GFP-RFP-LC3 to assess flux
This approach utilizes a tandem fluorescent-tagged LC3 protein, monomeric red fluorescent protein (or mCherry) coupled to green fluorescent protein-LC3 (mRFP/mCherry-GFP-LC3), to report autophagy induction and flux76. A similar approach can be achieved by crossing GFP-LC3 and mCherry-LC3 transgenic mice77, although theoretical disadvantages of the double-transgenic have been noted20. GFP fused to LC3 loses fluorescence rapidly in the acidic environment of the lysosome, while mRFP or mCherry retains fluorescence somewhat longer in the lysosome. Thus autophagosomes fluoresce in both green and red channels, but after fusion with lysosomes, the green fluorescence is lost, leaving only red fluorescence47, 76. Samples may be fixed; however, it has been suggested that some fixation protocols may artifactually restore green fluorescence by neutralizing the acidic autolysosome47 Sadoshima’s group embedded heart slices in Tissue-Tek OCT compound, and 10-micron cryosections were air-dried and fixed in ethanol before counterstaining with DAPI and mounting. Since this fails to preserve lysosomal pH, differences in red vs. green fluorescence may either be a function of proteolytic degradation of GFP in the lysosome, which must proceed at a faster rate than degradation of mRFP or mCherry or may be lost upon neutralization47. To determine flux, the relative abundance of red-only vs. red+green puncta are compared. The presence of red-only puncta indicates intact autophagic flux, although here again, there is little information as to whether the relative ratio of red-only to red+green puncta is a quantitative index of flux. A key advantage of this method is that it allows an estimation of flux in a single animal, rather than having to use paired animals (+/− chloroquine) for each data point. This was used to study autophagic flux after ischemia/reperfusion20.The disadvantage is that it is limited to the transgenic mouse line and as such is not available for concurrent use in other mouse lines or other species.
Optical imaging of fluorescent proteins
We hoped to use cardiac-restricted mCherry-LC3 to image autophagy in the heart. mCherry was chosen because its fluorescence properties were better suited to spectral imaging than GFP. We used the Caliper Life Sciences Spectrum In Vivo Imaging System to detect an increase in photon flux from the baseline of 3.19±0.72 to a mean of 3.93±1.10 at 4 h after i.p. administration of rapamycin (2 mg/kg) and chloroquine (10 mg/kg) (p<0.01, n=14) [Fig. 2]. This approach raised hope that it would be possible to image autophagy in vivo. However, to date, there has not been widespread adoption of such an imaging methodology to monitor autophagy. The technique works best in young (small) mice (e.g., 6 wk of age), and requires thorough depilation to minimize autofluorescence.
Figure 2. Optical imaging of autophagy in hearts of mCherry-LC3 mice.
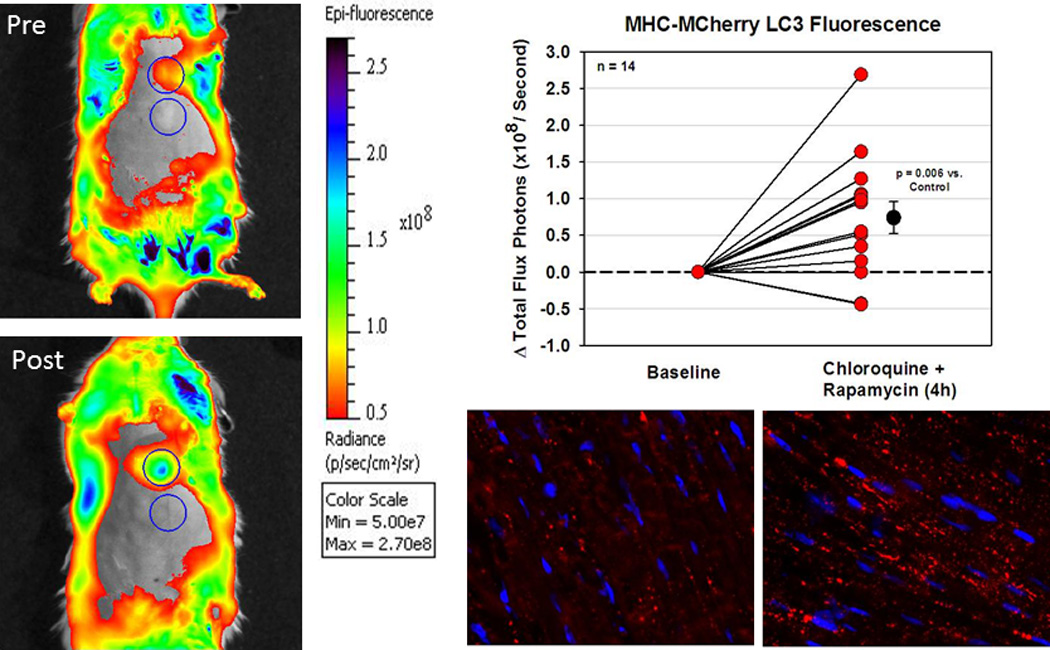
A. Mice were imaged at baseline (Pre) and 4 h after rapamycin and chloroquine administration (Post), using a protocol of 3 acquisitions of 15 sec each. Representative images from a single mouse are shown.
B. Graph shows the change in fluorescence from baseline to 4 h after rapamycin and chloroquine administration (n=14 mice; each point represents the average of 3 acquisitions).
C. Cryosections of typical hearts of mCherry-LC3 mice under fed (left) and fasted (right) conditions, showing the typical increase in total fluorescence as well as the increase in number of fluorescent red puncta (autophagosomes). Nuclei are stained blue with DAPI. (Images from A and B are reprinted with permission from Circulation Research Abstract P066: Imaging Autophagy in Living Mice v109:AP066, 2011. Unpublished images from C provided by Dr. Chengqun Huang and Dr. Roberta A. Gottlieb.)
Reporting nanoparticles
Increased activity of the autophagy-associated protease ATG4 can be detected using fluorescent nanoparticles in which a peptide substrate of ATG4 was labeled with FITC and a quenching agent (BHQ1) and assembled onto a hydrophobically modified glycol chitosan shell loaded with a red fluorescent lysosomotropic dye (Lysolite Red)78. Activity of ATG4 cleaved the peptide, thereby de-quenching the FITC. The purpose of the lysosomal dye was to account for direct (ATG4-independent) lysosomal degradation of nanoparticles (indicated by colocalization of red and green signals). As the autophagy-independent component is quite small compared to the autophagy-dependent pathway, it may be possible to adapt this approach to in vivo imaging. Although this approach was described in 2011, its use for in vivo imaging has not been published.
A related approach was used to monitor the lysosomal compartment, which expands during autophagy79. A cathepsin-activatable near infrared fluorochrome (CAF-680) was used to demonstrate an 8-fold increase in fluorescence 2 h after rapamycin administration followed by ischemia/reperfusion. The signal was localized to lysosomes as documented by colocalization with LAMP2 in immunostained cryosections. Interestingly, Feraheme (Ferumoxytol) nanoparticles that are fluorescently tagged with CyAl5.5 are taken up via endocytosis at a much faster rate when autophagy is stimulated with rapamycin, suggesting that it may be possible to infer autophagic activity from retention of such nanoparticles, which can be readily imaged by MRI.
Delayed enhancement after injection of chelated Gd MRI contrast agents (late gadolinium enhancement, LGE) has been noted in a variety of conditions and has been attributed to slower clearance from tissue due to fibrosis or the presence of macrophages that take up the agent. However, many of the pathological processes that give rise to fibrosis and LGE are also associated with increased autophagy, including scleroderma80, 81, heart failure82, 83, radiation-induced myocardial injury84, 85, Friedreich ataxia86, 87, and Fabry disease88, 89. It is possible that some instances of LGE are due to upregulation of autophagy rather than fibrosis or macrophage activity, but this will need to be formally tested.
Assessment of autophagy in human subjects
Detection of autophagy in humans is restricted to biochemical or microscopy analysis of biopsies of the tissue of interest or analysis of peripheral blood, thus severely limiting our ability to understand its role in human disease or to develop effective therapies. Kassiotis et al. reported a decrease in autophagic markers in hearts of patients who received a left ventricular assist device; they inferred that autophagy was an adaptive response in the failing heart, and that mechanical unloading relieved the stress responsible for triggering autophagy90. Garcia et al. studied autophagy markers by western blot and electron microscopy of atrial appendage tissue obtained during coronary artery bypass graft surgery (CABG); they noted that postoperative atrial fibrillation was more common in patients with evidence of impaired autophagic flux91. Jahania and colleagues detected autophagic flux in atrial tissue in patients undergoing cardiopulmonary bypass based on changes in p62/SQSTM1 and other autophagy proteins, and found that the magnitude of p62/SQSTM1 clearance was inversely related to mortality and morbidity risk scores53. However, Gedik et al. examined autophagy markers (by western blot) in left ventricle of patients who underwent remote ischemic preconditioning prior to CABG and saw no evidence of enhanced autophagy compared to non-preconditioned patients, nor did they detect evidence of an autophagic response during early reperfusion in either group92. A study conducted by Verma and colleagues examined autophagy gene expression by PCR and western blotting for autophagy proteins and regulators AMPK and MTOR93. They identified upregulation of mRNAs for autophagy components including LC3B, ATG4A, ATG4C, and ATG4D, as well as WIPI1, GABARAP, and GABARAPL2; they also observed an increase in LC3-II from beginning to end of cardiopulmonary bypass, and an increase in phospho-AMPK and a drop in protein levels of MTOR. Interestingly, they did not see a drop in p62/SQSTM1 levels. The results differ widely between these various studies, making it difficult to establish specific guidelines to apply to interpreting results, or even to generalize about the findings. While the studies of atrial tissue noted changes in autophagy, this was not the case for ventricular biopsies, although since no study compared atrial and ventricular samples, it is not possible to know whether the human left ventricle is unresponsive compared to the right atrium or whether the procedural differences in that study accounted for the lack of change in autophagy markers. More work will be needed in order to resolve these questions, but the difficulties in obtaining human heart tissue pose substantial obstacles to a comprehensive study.
Systemic conditions that affect autophagy, such as drug exposure, fasting, or inflammation might be expected to have parallel effects in multiple tissues. That rationale has led some investigators to examine autophagy in peripheral blood leukocytes. Autophagy markers were assessed in CD4 lymphocytes of males subjected to different exercise regimens or sedentary controls after 5 weeks of training; they found that exercise conditioning (especially high intensity interval training) triggers autophagy and suppresses apoptosis in lymphocytes when the subjects undergo a hypoxic exercise challenge94. The authors did not go so far as to suggest that the autophagic response in lymphocytes might reliably report on autophagy in other organs, but two studies by another group found a significant reduction in autophagy markers (by western blotting) in leukocytes of patients with coronary artery disease95 or acute myocardial infarction96 compared to healthy controls. However, none of these studies correlated autophagy markers in lymphocytes with those in heart tissue, so it remains to be seen whether peripheral blood cells represent a reliable reflection of autophagic activity in the heart. Dysregulation of autophagy was also noted by the same group in peripheral leukocytes of patients with sporadic Parkinson disease97, suggesting that underlying abnormalities that affect autophagy in the brain may be paralleled in leukocytes.
Recent advances in flow cytometry-based approaches to measuring autophagy hold promise for broader use of leukocyte autophagy as an index of organism-wide autophagic competence98, 99. These approaches are based on flow cytometric image analysis of LC3-decorated autophagosomes; however, given the parallel upregulation of lysosomal activity, cathepsin-based fluorogenic reagents or lysosomotropic dyes may provide an adequate readout if appropriate controls and validation studies are included.
CONCLUSIONS
Autophagy is increasingly recognized to play an important role in mitigating or exacerbating various cardiovascular diseases; hence it is important to be able to measure it accurately and to understand the significance of the findings. Given the highly dynamic nature of autophagy, it is essential to be able to account for autophagic flux. Current methods are imperfect, but newer technologies are emerging and there is hope that noninvasive imaging modalities may be developed for in vivo animal and human investigations.
Acknowledgments
SOURCES OF FUNDING
RAG holds the Dorothy and E. Phillip Lyon Chair in Molecular Cardiology in honor of Clarence M. Agress, MD. This work was funded in part by NIH P01 HL112730 (RAG).
Abbreviations
- PE
phosphatidylethanolamine
- TSA
trichostatin A
- HDAC
histone deacetylase
- SAHA
suberoylanilide hydroxamic acid (vorinostat)
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- 2ME
2-mercaptoethanol
- RIPA
radioimmunoprecipitation assay buffer
- PVDF
polyvinylidine difluoride
- FITC
fluorescein isothiocyanate
- BHQ1
Black Hole Quencher-1
- MRI
magnetic resonance imaging
- LGE
late gadolinium enhancement
Footnotes
Disclosures
RAG is a consultant for Takeda Pharmaceuticals and is a cofounder of TissueNetix, Inc. The other authors have no potential conflicts of interest to disclose.
REFERENCES
- 1.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamacher-Brady A, Brady NR, Gottlieb RA, Gustafsson AB. Autophagy as a protective response to bnip3-mediated apoptotic signaling in the heart. Autophagy. 2006;2:307–309. doi: 10.4161/auto.2947. [DOI] [PubMed] [Google Scholar]
- 3.Levine B, Klionsky DJ. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Developmental cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishino I, Fu J, Tanji K, et al. Primary lamp-2 deficiency causes x-linked vacuolar cardiomyopathy and myopathy (danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 6.Reichelt ME, Mellor KM, Curl CL, Stapleton D, Delbridge LM. Myocardial glycophagy - a specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol. 2013;65:67–75. doi: 10.1016/j.yjmcc.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Takikita S, Myerowitz R, Zaal K, Raben N, Plotz PH. Murine muscle cell models for pompe disease and their use in studying therapeutic approaches. Mol Genet Metab. 2009;96:208–217. doi: 10.1016/j.ymgme.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 9.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of atg5/atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 10.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. The Journal of clinical investigation. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong Y, Wang QJ, Yue Z. Atg14l and rubicon: Yin and yang of beclin 1-mediated autophagy control. Autophagy. 2009;5:890–891. doi: 10.4161/auto.9162. [DOI] [PubMed] [Google Scholar]
- 12.Sun Q, Zhang J, Fan W, Wong KN, Ding X, Chen S, Zhong Q. The run domain of rubicon is important for hvps34 binding, lipid kinase inhibition, and autophagy suppression. J Biol Chem. 2011;286:185–191. doi: 10.1074/jbc.M110.126425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of amp-activated protein kinase and beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 15.Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM, Jr, Gottlieb RA, Przyklenk K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation. 2010;122:S179–184. doi: 10.1161/CIRCULATIONAHA.109.928242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia-reperfusion injury. Circulation. 2012 doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077–18092. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous lc3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 19.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic ove26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andres AM, Hernandez G, Lee P, Huang C, Ratliff EP, Sin J, Thornton CA, Damasco MV, Gottlieb RA. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by parkin and p62/sqstm1. PLoS One. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, Jr, Gottlieb RA. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res. 2010;3:365–373. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang YL, Yao YT, Fang NX, Zhou CH, Gong JS, Li LH. Restoration of autophagic flux in myocardial tissues is required for cardioprotection of sevoflurane postconditioning in rats. Acta pharmacologica Sinica. 2014;35:758–769. doi: 10.1038/aps.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olsen CA, Ghadiri MR. Discovery of potent and selective histone deacetylase inhibitors via focused combinatorial libraries of cyclic alpha3beta-tetrapeptides. Journal of medicinal chemistry. 2009;52:7836–7846. doi: 10.1021/jm900850t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (hdac) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie M, Kong Y, Tan W, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson AB, Mentzer RM, Jr, Gottlieb RA. Autophagy is required for preconditioning by the adenosine a1 receptor-selective agonist ccpa. Basic Res Cardiol. 2009;104:157–167. doi: 10.1007/s00395-009-0006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martyniszyn L, Szulc-Dabrowska L, Boratynska-Jasinska A, Badowska-Kozakiewicz AM, Niemialtowski MG. In vivo induction of autophagy in splenocytes of c57bl/6 and balb/c mice infected with ectromelia orthopoxvirus. Polish journal of veterinary sciences. 2013;16:25–32. doi: 10.2478/pjvs-2013-0004. [DOI] [PubMed] [Google Scholar]
- 30.Pinheiro RO, Nunes MP, Pinheiro CS, D'Avila H, Bozza PT, Takiya CM, Corte-Real S, Freire-de-Lima CG, DosReis GA. Induction of autophagy correlates with increased parasite load of leishmania amazonensis in balb/c but not c57bl/6 macrophages. Microbes and infection / Institut Pasteur. 2009;11:181–190. doi: 10.1016/j.micinf.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 31.Bray MS, Young ME. Diurnal variations in myocardial metabolism. Cardiovasc Res. 2008;79:228–237. doi: 10.1093/cvr/cvn054. [DOI] [PubMed] [Google Scholar]
- 32.Ma D, Li S, Molusky MM, Lin JD. Circadian autophagy rhythm: A link between clock and metabolism? Trends in endocrinology and metabolism: TEM. 2012;23:319–325. doi: 10.1016/j.tem.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfeifer U, Scheller H. A morphometric study of cellular autophagy including diurnal variations in kidney tubules of normal rats. J Cell Biol. 1975;64:608–621. doi: 10.1083/jcb.64.3.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young ME, Razeghi P, Cedars AM, Guthrie PH, Taegtmeyer H. Intrinsic diurnal variations in cardiac metabolism and contractile function. Circ Res. 2001;89:1199–1208. doi: 10.1161/hh2401.100741. [DOI] [PubMed] [Google Scholar]
- 35.Campesi I, Straface E, Occhioni S, Montella A, Franconi F. Protein oxidation seems to be linked to constitutive autophagy: A sex study. Life sciences. 2013;93:145–152. doi: 10.1016/j.lfs.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Olivan S, Calvo AC, Manzano R, Zaragoza P, Osta R. Sex differences in constitutive autophagy. BioMed research international. 2014;2014:652817. doi: 10.1155/2014/652817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C, Hu LX, Dong T, Wang GQ, Wang LH, Zhou XP, Jiang Y, Murao K, Lu SQ, Chen JW, Zhang GX. Apoptosis and autophagy contribute to gender difference in cardiac ischemia-reperfusion induced injury in rats. Life sciences. 2013;93:265–270. doi: 10.1016/j.lfs.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 38.Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 2011;71:1710–1720. doi: 10.1158/0008-5472.CAN-10-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le TY, Ashton AW, Mardini M, Stanton PG, Funder JW, Handelsman DJ, Mihailidou AS. Role of androgens in sex differences in cardiac damage during myocardial infarction. Endocrinology. 2014;155:568–575. doi: 10.1210/en.2013-1755. [DOI] [PubMed] [Google Scholar]
- 40.Lista P, Straface E, Brunelleschi S, Franconi F, Malorni W. On the role of autophagy in human diseases: A gender perspective. J Cell Mol Med. 2011;15:1443–1457. doi: 10.1111/j.1582-4934.2011.01293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fok WC, Chen Y, Bokov A, Zhang Y, Salmon AB, Diaz V, Javors M, Wood WH, 3rd, Zhang Y, Becker KG, Perez VI, Richardson A. Mice fed rapamycin have an increase in lifespan associated with major changes in the liver transcriptome. PLoS One. 2014;9:e83988. doi: 10.1371/journal.pone.0083988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller RA, Harrison DE, Astle CM, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13:468–477. doi: 10.1111/acel.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barth S, Glick D, Macleod KF. Autophagy: Assays and artifacts. J Pathol. 2010;221:117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanida I, Waguri S. Measurement of autophagy in cells and tissues. Methods Mol Biol. 2010;648:193–214. doi: 10.1007/978-1-60761-756-3_13. [DOI] [PubMed] [Google Scholar]
- 45.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menzies FM, Moreau K, Puri C, Renna M, Rubinsztein DC. Measurement of autophagic activity in mammalian cells. Current protocols in cell biology / editorial board, Juan S. Bonifacino … [et al.] 2012;Chapter 15(Unit 15):16. doi: 10.1002/0471143030.cb1516s54. [DOI] [PubMed] [Google Scholar]
- 47.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gurney MA, Huang C, Ramil JM, Ravindran N, Andres AM, Sin J, Linton PJ, Gottlieb RA. Measuring cardiac autophagic flux in vitro and in vivo. Methods Mol Biol. 2015;1219:187–197. doi: 10.1007/978-1-4939-1661-0_14. [DOI] [PubMed] [Google Scholar]
- 49.Verschooten L, Barrette K, Van Kelst S, Rubio Romero N, Proby C, De Vos R, Agostinis P, Garmyn M. Autophagy inhibitor chloroquine enhanced the cell death inducing effect of the flavonoid luteolin in metastatic squamous cell carcinoma cells. PLoS One. 2012;7:e48264. doi: 10.1371/journal.pone.0048264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Egger ME, Huang JS, Yin W, McMasters KM, McNally LR. Inhibition of autophagy with chloroquine is effective in melanoma. J Surg Res. 2013;184:274–281. doi: 10.1016/j.jss.2013.04.055. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin a1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, h-4-ii-e cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 52.Wu YC, Wu WK, Li Y, Yu L, Li ZJ, Wong CC, Li HT, Sung JJ, Cho CH. Inhibition of macroautophagy by bafilomycin a1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem Biophys Res Commun. 2009;382:451–456. doi: 10.1016/j.bbrc.2009.03.051. [DOI] [PubMed] [Google Scholar]
- 53.Jahania SM, Sengstock D, Vaitkevicius P, Andres A, Ito BR, Gottlieb RA, Mentzer RM., Jr Activation of the homeostatic intracellular repair response during cardiac surgery. J Am Coll Surg. 2013;216:719–726. doi: 10.1016/j.jamcollsurg.2012.12.034. discussion 726-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaminskyy V, Abdi A, Zhivotovsky B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy. 2011;7:83–90. doi: 10.4161/auto.7.1.13893. [DOI] [PubMed] [Google Scholar]
- 55.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ulk1 is regulated via opposing phosphorylation by ampk and mtor. Autophagy. 2011;7:643–644. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim J, Kundu M, Viollet B, Guan KL. Ampk and mtor regulate autophagy through direct phosphorylation of ulk1. Nature cell biology. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by parkin is critical for mitophagy. Human molecular genetics. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chemistry & biology. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 59.Hernandez G, Thornton C, Stotland A, Lui D, Sin J, Ramil J, Magee N, Andres A, Quarato G, Carreira RS, Sayen MR, Wolkowicz R, Gottlieb RA. Mitotimer: A novel tool for monitoring mitochondrial turnover. Autophagy. 2013;9:1852–1861. doi: 10.4161/auto.26501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferree AW, Trudeau K, Zik E, Benador IY, Twig G, Gottlieb RA, Shirihai OS. Mitotimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy. 2013;9:1887–1896. doi: 10.4161/auto.26503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, Zhang M, Royal MA, Hoehn KL, Driscoll M, Adler PN, Wessells RJ, Saucerman JJ, Yan Z. A novel mitotimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem. 2014;289:12005–12015. doi: 10.1074/jbc.M113.530527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seglen PO, Reith A. Ammonia inhibition of protein degradation in isolated rat hepatocytes. Quantitative ultrastructural alterations in the lysosomal system. Exp Cell Res. 1976;100:276–280. doi: 10.1016/0014-4827(76)90148-8. [DOI] [PubMed] [Google Scholar]
- 63.Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin a1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4:849–950. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- 64.Poole B, Ohkuma S. Effect of weak bases on the intralysosomal ph in mouse peritoneal macrophages. J Cell Biol. 1981;90:665–669. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawai A, Uchiyama H, Takano S, Nakamura N, Ohkuma S. Autophagosome-lysosome fusion depends on the ph in acidic compartments in cho cells. Autophagy. 2007;3:154–157. doi: 10.4161/auto.3634. [DOI] [PubMed] [Google Scholar]
- 66.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous lc3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 67.Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, Choi AM. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy. 2011;7:629–642. doi: 10.4161/auto.7.6.15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munafo DB, Colombo MI. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. Journal of cell science. 2001;114:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 69.Perry CN, Kyoi S, Hariharan N, Takagi H, Sadoshima J, Gottlieb RA. Novel methods for measuring cardiac autophagy in vivo. Methods Enzymol. 2009;453:325–342. doi: 10.1016/S0076-6879(08)04016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tian Z, Wang C, Hu C, Tian Y, Liu J, Wang X. Autophagic-lysosomal inhibition compromises ubiquitin-proteasome system performance in a p62 dependent manner in cardiomyocytes. PLoS One. 2014;9:e100715. doi: 10.1371/journal.pone.0100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaushik S, Cuervo AM. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 2009;452:297–324. doi: 10.1016/S0076-6879(08)03619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Janen SB, Chaachouay H, Richter-Landsberg C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia. 2010;58:1766–1774. doi: 10.1002/glia.21047. [DOI] [PubMed] [Google Scholar]
- 73.Shpilka T, Weidberg H, Pietrokovski S, Elazar Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biology. 2011;12:226. doi: 10.1186/gb-2011-12-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 75.Ni H-M, Bockus A, Wozniak AL, Jones K, Weinman S, Yin X-M, Ding W-X. Dissecting the dynamic turnover of gfp-lc3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged lc3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 77.Terada M, Nobori K, Munehisa Y, Kakizaki M, Ohba T, Takahashi Y, Koyama T, Terata Y, Ishida M, Iino K, Kosaka T, Watanabe H, Hasegawa H, Ito H. Double transgenic mice crossed gfp-lc3 transgenic mice with alphamyhc-mcherry-lc3 transgenic mice are a new and useful tool to examine the role of autophagy in the heart. Circ J. 2010;74:203–206. doi: 10.1253/circj.cj-09-0589. [DOI] [PubMed] [Google Scholar]
- 78.Choi KM, Nam HY, Na JH, Kim SW, Kim SY, Kim K, Kwon IC, Ahn HJ. A monitoring method for atg4 activation in living cells using peptide-conjugated polymeric nanoparticles. Autophagy. 2011;7:1052–1062. doi: 10.4161/auto.7.9.16451. [DOI] [PubMed] [Google Scholar]
- 79.Chen HH, Mekkaoui C, Cho H, Ngoy S, Marinelli B, Waterman P, Nahrendorf M, Liao R, Josephson L, Sosnovik DE. Fluorescence tomography of rapamycin-induced autophagy and cardioprotection in vivo. Circulation. Cardiovascular imaging. 2013;6:441–447. doi: 10.1161/CIRCIMAGING.112.000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schicchi N, Valeri G, Moroncini G, Agliata G, Salvolini L, Gabrielli A, Giovagnoni A. Myocardial perfusion defects in scleroderma detected by contrast-enhanced cardiovascular magnetic resonance. La Radiologia medica. 2014 doi: 10.1007/s11547-014-0419-7. [DOI] [PubMed] [Google Scholar]
- 81.Frech T, De Domenico I, Murtaugh MA, Revelo MP, Li DY, Sawitzke AD, Drakos S. Autophagy is a key feature in the pathogenesis of systemic sclerosis. Rheumatology international. 2014;34:435–439. doi: 10.1007/s00296-013-2827-8. [DOI] [PubMed] [Google Scholar]
- 82.Wong TC, Piehler KM, Zareba KM, et al. Myocardial damage detected by late gadolinium enhancement cardiovascular magnetic resonance is associated with subsequent hospitalization for heart failure. Journal of the American Heart Association. 2013;2:e000416. doi: 10.1161/JAHA.113.000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ren SY, Xu X. Role of autophagy in metabolic syndrome-associated heart disease. Biochimica et biophysica acta. 2014 doi: 10.1016/j.bbadis.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Umezawa R, Ota H, Takanami K, Ichinose A, Matsushita H, Saito H, Takase K, Jingu K. Mri findings of radiation-induced myocardial damage in patients with oesophageal cancer. Clinical radiology. 2014 doi: 10.1016/j.crad.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 85.Sridharan V, Tripathi P, Sharma S, Moros EG, Zheng J, Hauer-Jensen M, Boerma M. Roles of sensory nerves in the regulation of radiation-induced structural and functional changes in the heart. International journal of radiation oncology, biology, physics. 2014;88:167–174. doi: 10.1016/j.ijrobp.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang ML, Sivagurunathan S, Ting S, Jansson PJ, Austin CJ, Kelly M, Semsarian C, Zhang D, Richardson DR. Molecular and functional alterations in a mouse cardiac model of friedreich ataxia: Activation of the integrated stress response, eif2alpha phosphorylation, and the induction of downstream targets. Am J Pathol. 2013;183:745–757. doi: 10.1016/j.ajpath.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 87.Raman SV, Phatak K, Hoyle JC, Pennell ML, McCarthy B, Tran T, Prior TW, Olesik JW, Lutton A, Rankin C, Kissel JT, Al-Dahhak R. Impaired myocardial perfusion reserve and fibrosis in friedreich ataxia: A mitochondrial cardiomyopathy with metabolic syndrome. Eur Heart J. 2011;32:561–567. doi: 10.1093/eurheartj/ehq443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Satoh H, Sano M, Suwa K, Saitoh T, Nobuhara M, Saotome M, Urushida T, Katoh H, Hayashi H. Distribution of late gadolinium enhancement in various types of cardiomyopathies: Significance in differential diagnosis, clinical features and prognosis. World journal of cardiology. 2014;6:585–601. doi: 10.4330/wjc.v6.i7.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chevrier M, Brakch N, Celine L, Genty D, Ramdani Y, Moll S, Djavaheri-Mergny M, Brasse-Lagnel C, Annie Laquerriere AL, Barbey F, Bekri S. Autophagosome maturation is impaired in fabry disease. Autophagy. 2010;6:589–599. doi: 10.4161/auto.6.5.11943. [DOI] [PubMed] [Google Scholar]
- 90.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120:S191–S197. doi: 10.1161/CIRCULATIONAHA.108.842252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garcia L, Verdejo HE, Kuzmicic J, Zalaquett R, Gonzalez S, Lavandero S, Corbalan R. Impaired cardiac autophagy in patients developing postoperative atrial fibrillation. The Journal of thoracic and cardiovascular surgery. 2012;143:451–459. doi: 10.1016/j.jtcvs.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 92.Gedik N, Thielmann M, Kottenberg E, Peters J, Jakob H, Heusch G, Kleinbongard P. No evidence for activated autophagy in left ventricular myocardium at early reperfusion with protection by remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. PLoS One. 2014;9:e96567. doi: 10.1371/journal.pone.0096567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Singh KK, Yanagawa B, Quan A, Wang R, Garg A, Khan R, Pan Y, Wheatcroft MD, Lovren F, Teoh H, Verma S. Autophagy gene fingerprint in human ischemia and reperfusion. The Journal of thoracic and cardiovascular surgery. 2014;147:1065.e1061–1072.e1061. doi: 10.1016/j.jtcvs.2013.04.042. [DOI] [PubMed] [Google Scholar]
- 94.Weng T-P, Huang S-C, Chuang Y-F, Wang J-S. Effects of interval and continuous exercise training on cd4 lymphocyte apoptotic and autophagic responses to hypoxic stress in sedentary men. PLoS ONE. 2013;8:e80248. doi: 10.1371/journal.pone.0080248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu G, Wei G, Huang J, Pang S, Liu L, Yan B. Decreased gene expression of lc3 in peripheral leucocytes of patients with coronary artery disease. European Journal of Clinical Investigation. 2011;41:958–963. doi: 10.1111/j.1365-2362.2011.02486.x. [DOI] [PubMed] [Google Scholar]
- 96.Wu G, Liu L, Huang J, Pang S, Wei G, Cui Y, Yan B. Alterations of autophagic–lysosomal system in the peripheral leukocytes of patients with myocardial infarction. Clinica Chimica Acta. 2011;412:1567–1571. doi: 10.1016/j.cca.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 97.Wu G, Wang X, Feng X, Zhang A, Li J, Gu K, Huang J, Pang S, Dong H, Gao H, Yan B. Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic parkinson's disease. Brain Res. 2011;1394:105–111. doi: 10.1016/j.brainres.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 98.Chan LL, Shen D, Wilkinson AR, Patton W, Lai N, Chan E, Kuksin D, Lin B, Qiu J. A novel image-based cytometry method for autophagy detection in living cells. Autophagy. 2012;8:1371–1382. doi: 10.4161/auto.21028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eng KE, Panas MD, Karlsson Hedestam GB, McInerney GM. A novel quantitative flow cytometry-based assay for autophagy. Autophagy. 2010;6:634–641. doi: 10.4161/auto.6.5.12112. [DOI] [PubMed] [Google Scholar]