

Abstract
Tumor microenvironment has a major role in cancer progression and resistance to treatment. The bone marrow (BM) is a dynamic network of growth factors, cytokines and stromal cells, providing a permissive environment for leukemogenesis and progression. Both BM stroma and leukemic blasts promote angiogenesis, which is increased in acute lymphoblastic leukemia and acute myeloid leukemia. Growth factors like vascular endothelial growth factor (VEGF), basic fibroblast growth factor and angiopoietins are the main proangiogenic mediators in acute leukemia. Autocrine proleukemic loops have been described for VEGF and angiopoietin in hematopoietic cells. Interactions of stromal cells and extracellular matrix with leukemic blasts can also generate antiapoptotic signals that contribute to neoplastic progression and persistence of treatment-resistant minimal residual disease. High expression of CXC chemokine ligand 4 (CXCR4) by leukemic blasts and activation of the CXCR4–CXCL12 axis is involved in leukemia progression and disruption of normal hematopoiesis. Leukemia-associated bone microenvironment markers could be used as prognostic or predictive indicators of disease progression and/or treatment outcome. Studies related to bone microenvironment would likely provide a better understanding of the treatment resistance associated with leukemia therapy and design of new treatments.
Keywords: acute leukemia, microenvironment, angiogenesis, bone marrow, endosteal niche, endovascular niche
Introduction
The importance of tumor microenvironment for cancer progression is becoming widely recognized in recent years. Interaction of cancer cells with their immediate stromal microenvironment overcoming the physiological barrier function of stromal cells synergizes growth, angiogenesis and initiation of an invasive and metastatic phenotype of the cancer cell.
Unlike many epithelial (mucosal and non-mucosal) organs, normal bone marrow (BM) does not appear to be organized in well-defined epithelial and stromal compartments separated by a basement membrane. However, BM is loosely composed of hematolymphoid cells set within a milieu surrounded by a mesenchymal landscape. The hematolymphoid compartment consists of a dominant hematopoietic compartment and a less prominent lymphoid compartment. The lymphoid compartment (consisting of B, T and NK cells) is less prominent during normal hematopoiesis but increases in senility and in specific marrow diseases. This review will focus on the hematopoietic compartment that consists of hematopoietic stem cells (HSCs), erythroid, myeloid, monocytic/macrophage and megakaryocytic precursors and their maturing elements. The stromal or mesenchymal compartment composed of a common hematopoietic and mesenchymal progenitor cells, endothelial progenitor cells (EPC), BM stromal cells and the extracellular matrix (ECM). The BM is a dynamic microenvironment with high concentration of growth factors and cytokines necessary for hematopoiesis, making it a highly permissive zone for cancer cell homing and survival. This is supported by the fact that solid tumors very frequently home and metastasize to BM. It is possible that the same factors that enhance survival of metastatic cancers promote leukemogenesis, enhance blast survival and make them resistant to treatment within the marrow microenvironment.
Bone marrow micro niches
There have been various attempts to define bone microenvironments that nurture and determine stem cell fate. Two such stem cell ‘niches’ are well described: the vascular niche and the endosteal niche. The granulocytes and megakaryocytes thrive at vascular endothelial niches whereas the endosteal niche serves as stem cell reserve.1 The endosteal niche is defined by osteoblasts and its proximal stromal cells.2 The cell type that is a common denominator for both the niches is the CXC chemokine ligand 12 (CXCL12) abundant reticular (CAR) cell. The CAR cells surround sinusoidal endothelial cells or are located close to the endosteum. Hematopoietic cells around the endosteal niche are in direct contact with these CAR cells. The importance of CXCL12 is shown in animal studies where deletion of the receptor for receptor for CXCL12 (CXCR4) results in severe reduction in stem cell numbers in adult mice.
As opposed to the endosteal niche, whose primary function is to maintain stem cell reserves, the vascular niche is important in maintaining a boundary between immature cells and peripheral blood circulation. Somewhat similar to the basement membrane, in its barrier function, the vascular niche is responsible for regulating the release of only the most mature hematopoietic element such as mature non-nucleated red blood cells (RBCs), platelets and mature granulocytes. In diseases such as primary myelofibrosis, the vascular niche is altered, and this can lead to release of immature precursors (‘intrasinusoidal hematopoiesis’) into circulation. Thus in these disorders, a defective release mechanism and a faulty vascular niche contribute to circulating leukemic blasts (with or without proliferation of abnormal precursors).3 This is also illustrated in Figure 1, where immature erythroid cells (nucleated RBCs and blasts) are present within vascular sinusoids in a case of primary myelofibrosis.
Figure 1. Endovascular niche.
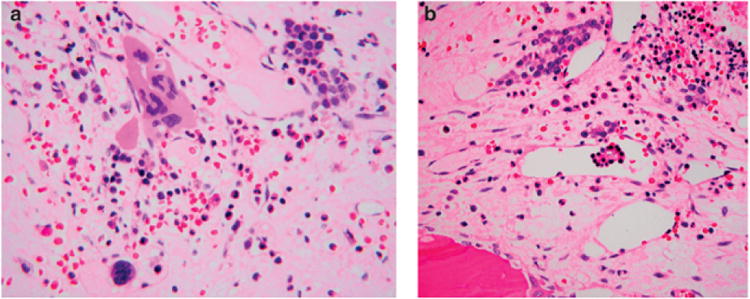
(a) The endovascular niche is an area immediately surrounding vessels and vascular sinusoids. The endovascular niche is involved in regulation of hematopoietic elements, especially granulocytic and megakaryocytes. In many myeloid disorders (leukemia, myelodysplastic syndromes or myeloproliferative neoplasms) the vascular microvessel density is increased within the marrow stroma. A case of primary myelofibrosis is illustrated above, where there are abnormal clusters of megakaryocytes and increased sinusoids that are dilated. As shown in the lower (left) part of the photomicrograph, abnormal megakaryocytes are sometimes found within the vascular sinusoid (intra-sinusoidal hematopoiesis). This is not seen in a normal endovascular niche, where megakaryocytes are inhibited from crossing over the endothelial barrier. Normally, megakaryocytic cytoplasmic processes penetrate the endothelium and release platelets as a shower (thrombopoiesis). (b) In this case of advanced myeloproliferative neoplasm, because of an abnormal endovascular niche, immature elements gain access to peripheral blood through vascular sinusoids, as illustrated above. This includes myeloblasts resulting in a leukemic phase. Illustrated above is a cluster of immature erythroid cells within vascular sinusoids. Leukemia and tumor microenvironment
Neoangiogenesis and acute leukemia
Neoangiogenesis, the formation of new blood vessels, is an essential contributor to growth and malignant progression of solid tumors. In fact, in solid malignancies, failure of vascularization results in a tumor size that is limited to a few millimeters in size.4 A delicate balance exists between pro- and antiangiogenic factors. Tilting this balance favoring neoangiogenesis and proangiogenic factors (for example, upregulating vascular endothelial growth factor (VEGF)) in itself contributes to neoplastic progression.5 The role of angiogenesis in hematological malignancies has remained controversial until recently,6 but many compelling evidences are emerging to support its role in leukemogenesis, leukemic progression and treatment.7
Increased vascularization is not only seen in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) but also in various preleukemic disorders, such as yelodysplastic syndromes (MDS) and myeloproliferative neoplasms. Not only is there increased microvessel density (MVD) at diagnosis in adult AML,8 but there is also a marked change in vessel density in response to chemo-inductive therapies, and differentiation therapies (all-trans retinoic acid (ATRA) in promyelocytic leukemia).9–11 Similarly, recent studies have shown the effect of antiangiogenic agents such as bortezomib (Velcade) in certain preleukemic disorders such as primary myelofibrosis. An ex vivo study of BM biopsy specimens with bortezomib resulted in reduction in MVD; however, antiangiogenic therapy has not shown any effect in human primary myeloproliferative neoplasms or in leukemia.
Leukemic blasts12 and BM microenvironment13 contribute equally to neoangiogenesis by secretion of different angiogenic growth factors and mediators. Given the complex interaction of various factors (pro- and antiangiogenic), stromal and hematopoietic cells involved and different phenotypes of acute leukemia, a measurable correlation between angiogenic mediators, angiogenic assays10 and BM microvascular density has been elusive and perhaps contributed to the controversies surrounding angiogenesis and hematopoietic neoplasms.
In AML, the well-documented blast-derived proangiogenic factors are VEGF and angiopoietins. VEGF, the major proangiogenic factor in AML, acts as an autocrine and paracrine growth factor in some AMLs that express the receptor VEGF-R2.14,15 Clinical studies have also suggested the potential prognostic value of VEGF levels independent of blast counts for survival in some high-risk AML.16 Similarly, expression of angiopoietins, another group of vascular growth factors, and their receptor Tie2 has been demonstrated on leukemic cells.17 Other mediators of angiogenesis without a strongly documented correlation with MVD are basic fibroblast growth factor (bFGF),18 interleukin (IL)-6 and IL-8.19 Like VEGF, most of these cytokines and growth factors have proleukemic autocrine or paracrine actions.20
A proangiogenic phenotype with higher MVD is observed in ALL, although the profile of involved angiogenic factors seems to be different of that from AML. As stated by Folkman's group21 and confirmed by others, elevation of bFGF with normal VEGF levels is found in most patients with childhood ALL. As stated above, after chemotherapy-induced remission, vascular density reverts to normal.16 The biological relevance of ALL angiogenesis has been demonstrated in an NOD/SCID murine model of human ALL, in which plasma collected from BM promoted proliferation, migration and the formation of capillary-like structures by BM endothelial cells. These studies also revealed a cross-talk between endothelial and leukemic cells, in which BM endothelium promoted leukemia cell survival through modulation of apoptosis signaling pathways (overexpression of bcl-2 and bcl-xL).22
Matrix metalloproteinases (MMP) are involved in regulation of angiogenesis and tumor progression and are produced by both stromal and leukemic cells. An abnormal pattern of expression of MMP has been described in AML and B-ALL, in which levels of MMP-2 and -9 are lower than in normal BM samples.23,24 Heparanase, another enzyme that degrades basement membranes, is expressed in AML,25 although its functional importance for angiogenesis in AML has not been investigated. Downregulation of heparanase expression has been reported in acute promyelocytic leukemia, the leukemia that responds well to differentiation therapies such as ATRA, with normalization of marrow vasculature (French–American–British, FAB: M3).11, 26
An imbalance of microenvironmental factors toward proangiogenesis initiates recruitment and growth of endothelial cells, necessary for neoangiogenesis. Malignant myeloid cells share similar adhesion molecules and genetic abnormalities with endothelial cells, thus suggesting either a common cell of origin or a similar microenvironmental regulation for cells in these two different compartments.27 Another study demonstrated a higher level of circulating endothelial cells, among half of which additionally shared the genetic abnormality with the leukemic clone.28 One group of preleukemic syndromes (MDS) also has similar abnormalities.29 Interestingly, the EPC seem to share similar features with monoblasts in FAB M5 leukemia. Monoblasts were found to express endothelial markers such as podocalyxin in 20% of patients with M5 leukemia. They were also characterized by a higher expression of VEGF-R, Tie-2, and a higher production of VEGF-A and angiopoietin-1. When leukemic monoblasts from these patients were cultured in the presence of VEGF and Ang1, they differentiated into endothelial cells.30,31 However, the biological and in vivo relevance of this ‘blast-to-endothelial’ transformation is still unclear.
Myelodysplastic syndromes, as briefly discussed above are preleukemic, clonal HSC disorders resulting from ineffective maturation with a high risk of progression to acute leukemia.32 Alteration of microenvironment is readily appreciated in a subset of MDS cases. Normally, myeloid stem cells are localized close to the bony trabeculae—the endosteal niche around osteoblasts. This niche is specifically important in maintaining the stem cell reserve. The stem cells seldom form 1- to 2-cell-thick areas in the paratrabecular endosteal niche. In MDS, the immature precursors are often found in the interstitium in aggregates (see Figure 2). These are described as abnormal localization of immature precursors (ALIPs). An angiogenesis switch has been proposed as one of the mechanisms of the progression of MDS to acute leukemia. However, although an increased microvascular density has been observed in most studies of MDS, there are conflicting data about its increase33,34 or not35,36 during the transformation to overt acute leukemia. In a recent analysis of this issue, vascular density and expression of basic FGF, angiopoietins, VEGFR2 and Tie2 were lower in MDS transformed to leukemia than in de novo AML, suggesting a certain independence of angiogenesis in the late phase of leukemic evolution. An increase in transforming growth factor-β (TGF-β) expression was also found in this setting, which correlated with suppression of angiogenesis.37 These differences might be therapeutically relevant and partially explain the resistance of this group of secondary leukemias (acute leukemia arising from multilineage dysplasia: AML-MLD) to chemotherapy.
Figure 2. Endosteal niche and abnormal localization of immature precursors (ALIPS).
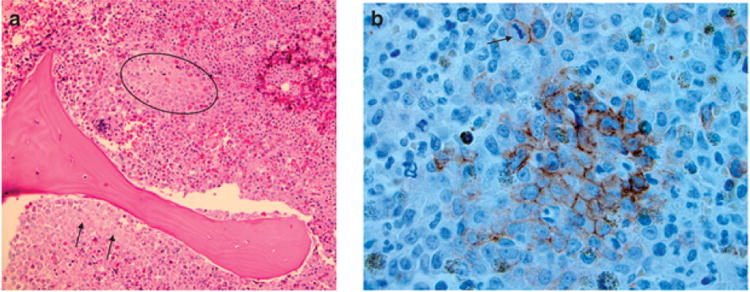
(a) Often seen in myelodysplastic syndrome ALIPS refers to a cluster of immature precursors away from its normal microenvironment (circle). Illustrated here is the localization of a cluster of myeloid precursors away from the normal endosteal niche. (b) CD117 stains highlight this cluster of immature precursors. A mitotic figure is also seen above the cluster (arrow).
Stromal cells, growth factors, cytokines and acute leukemia
Bone marrow stromal cells are composed of fibroblasts, adipocytes, endothelial cells and osteoblasts. They are the major source of growth factors and cytokines in normal and in pathological conditions (Figure 3). The stromal cell composition is altered in AML, contributing to differences in microenvironment, and includes a reduction in fibroblasts and adipocytes. However, it is not known if alteration in microenvironment resulted in acute leukemia or is a secondary change.38 Genetic alterations in BM mesenchymal stromal cells are also a subject of controversy. Chromosomal changes (numerical and structural) resembling those of leukemic cells have been described in stromal cells in a majority of 50% patients with MDS or AML. They do not appear to be clonal abnormalities and in some studies these changes were distinct from those in the leukemic cells.39 It is not yet clear if phenotypic changes like those observed in cancer-associated fibroblasts40 are also present in leukemic BM fibroblasts.
Figure 3. Interactions between leukemic blasts and bone marrow stromal cells.

Leukemic blasts and stromal cells produce vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF) and other proangiogenic mediators, which also have protumoral paracrine and autocrine effects. Integrins and CXCR4 are fundamental for blast–stroma adhesion and mediate blasts ‘homing’ and persistence of residual disease after treatment. Different mechanisms have been described for acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL), the latter being more dependent on bFGF than on VEGF as proangiogenic mediator. ALL microenvironment is also richer in interleukin mediators and in asparaginase synthetase, another mechanism of resistance to treatment.
The effect of acute leukemic blast on the BM microenvironment is shown from its direct effect on hematopoiesis. Leukemic cells profoundly disrupt hematopoietic stroma creating abnormal malignant niches by the secretion of stem cell factor. These niches compete for CD34 + homing and finally cause an abnormal progenitor cell function, even in early phases of the disease;41 one such effect is the ALIPs, described above.
The leukemic blast–stromal interaction effect is both ways: thus stromal cells also actively contribute to leukemic blast growth and progression. The effect of stromal cells on leukemic blasts, contrary to the other, appears to be recapitulating normal physiological cell–cell adhesion through adhesive receptors like integrins in normal hematopoietic progenitors.42 Leukemic blast integrins (for example, β1 and β2 in AML)43,44 interact with stromal ligands such as VCAM-1, or fibronectin in the ECM. This adhesive interaction is necessary for leukemic blast survival and proliferation. Reciprocal activation of ILK/Akt pathway, in leukemic blasts and stromal cells, appears to be critical for this adhesion-driven AML blast survival.45 ALL blasts on the other hand show constitutive expression of different adhesive integrins LFA-1 (αLβ2; T-ALL) and VLA-4 (α4β1; B-ALL).46,47 Elegant in vitro models demonstrate the supportive effects of BM stromal layers for leukemic B cells.48 Adhesion of blasts is also dependent on interactions between membrane surface receptors and the fibronectin produced by stromal cells. Overexpression of β1-integrins, especially VLA-4, on AML blasts enhances adhesion to fibronectin contributing to therapy resistance and persistent minimal residual disease.49 BM stem cells within the microenvironment further support leukemic blasts survival by preventing apoptosis, and this is mediated by upregulation of Bcl2 and Bclx proteins.50 In addition to adhesive cell-cell and ECM––cell interactions, growth factors and cytokines elaborated by BM stromal cells enhance leukemia growth and progression through autocrine and paracrine effects. This allows a bidirectional cross talk between AML blasts and BM fibroblasts in which the latter stimulate neoplastic proliferation and angiogenesis.51 The exact role of the different growth factors and cytokines in leukemic cell survival is not well known, but probably they form complex and dynamic networks in which leukemic blasts and stromal cells complement each other's cytokine expression, as shown in co-culture models of ALL.52 Interleukins have a key function in survival of neoplastic T-ALL blasts. The CXCR4-CXCL12 axis participates in the upregulation of IL-8 by leukemic cells, through nuclear factor-κB and JNK/AP-1,53 and the high levels of IL-8 could enhance angiogenesis in the BM microenvironment.54 IL-7 is also an important mediator in the early induction of survival by BM stroma on T-ALL cells.55 Similar effects of IL-7 and IL-3 have been demonstrated for B-ALL.56 As previously described, the autocrine and paracrine loops established by angiogenic factors (VEGF, angiopoietins) and their receptors in AML also support blast survival and proliferation.57 Other factors secreted by stromal cells, such as hepatocyte growth factor (HGF) may also contribute to migration and proliferation of AML blasts.50,58
Chemokines and their receptors control cell trafficking and homing of hematopoietic progenitors in BM and in other organs.59 In particular, CXCL12, previously known as SDF-1 (stromal-derived factor 1), is the main cytokine produced by BM stromal cells. The interaction between CXCL12 and its receptor CXCR4 specifically mediates migration of normal hematopoietic cells to BM niches. A similar role for CXCR4 in homing and migration of leukemic blasts has been demonstrated in ALL, where blast homing is dependent on its expression.60 Conversely, CXCR4 expression on AML blasts and its dependence on CXCR4/CXCL12 for homing is much more variable.61
Only recently the connections between the CXCR4/CXCL12 pathway in leukemia and hypoxia in the BM have begun to be unveiled. BM microenvironment is thought to be hypoxic.62 Control of CXCL12 and CXCR4 by HIF-1a within the HSC niche has been suggested as a mechanism of homing for HSCs.63 Low (suprahypoxic) oxygenation level has also been demonstrated in leukemic BM,64 although its relevance for leukemia progression is not as clear as in solid tumors as reduced proliferation of leukemic cells was found in association with hypoxia.65 A recently published study has shed some light into this process, showing a direct link between the BM low oxygen levels and the high CXCR4 expression on AML blasts. Because CXCR4 promotes leukemia cell survival and adhesion, it is thought that microenvironmental hypoxia contributes directly to leukemic progression.66 The exact role of hypoxia and HIF-1α (tightly related to angiogenesis in other tumors)67 in regulation of angiogenesis within the BM is still unknown. Currently, studies are mostly restricted to human leukemic cells and cell lines and there are no convincing human-tissue-based studies to prove that hypoxia is involved in microenvironmental regulation. Markers such as HIF-1alpha; may be useful in predicting/prognosticating recurrence or response to treatment.
As previously mentioned, the phosphoinositide 3-kinase/Akt signaling cascade is a key pathway in the interactions between stromal cells and leukemic blasts, especially those related to β1-integrin receptors and CXCR4–CXCL12 axis, but also in blasts' autocrine loops involving VEGF and angiopoietin.68,49
Stromal cues promote lineage-specific differentiation in leukemia. In a human cord blood model of MLL-AF9 leukemia transplanted into immunodeficient mice, microenvironmental alterations result in multipotent leukemia stem cells to switch lineages. In this elegant study, AML, ALL and biphenotypic leukemia were generated by simply changing the cytokine medium and/or the mouse strain, thus demonstrating the importance of microenvironmental signals for lineage commitment.69 Further demonstration of the possibility of modification of leukemic cells behavior by microenvironment and most specifically by fibroblasts comes from studies showing differentiation toward a less malignant phenotype in a co-culture model with cell lines and patient-derived AML cells.70 In one of these works, fibroblast clustering produced high levels of HGF/SF and other mediators (IL-1β, IL-6, IL-8, LIF, granulocytemonocyte colony-stimulating factor) and these paracrine signals were able to induce growth inhibition and differentiation of leukemic cells. Interestingly, only leukemic cell lines lacking c-Met showed this response, thus exemplifying both the differences with epithelial tumors and the probable phenotype specificity of leukemic blasts–stroma interactions.71
Interaction of leukemic cells and immune cells of the surrounding stroma is complex but mostly biased to an inhibition of immune response by leukemic cells. For example, direct cell interactions of AML blasts and soluble factors of leukemic microenvironment potently inhibit T-cell response.72 Indoleamine 2,3-dioxygenase, a potent immunosuppressor, is not only expressed in AML blasts but it also induces the generation of CD4 + CD25 + Treg populations, which contributes to immune escape.73
Normal and pathologic BM is in contact with the endosteal surface of bone, which is covered by osteoblasts. The osteoblast, a specialized cell sharing many characteristics with fibroblast, is the main constituent of one of the two types of hematopoietic niches: the osteoblastic niche, characterized by high levels of bone morphogenetic protein, osteopontin and angiopoietin-1.74 In vitro experiments with co-culture of normal osteoblasts have shown that osteoblasts may also support leukemic blasts growth by direct contact and by stimulating blasts to produce proangiogenic IL-8.75
Clinical relevance of microenvironment in acute leukemia
Prognostic value of microenvironment-related markers
Emerging data on the relevance of microenvironment for acute leukemia progression have been accompanied by a growing amount of studies looking for stroma-specific markers as prognostic and predictive factors. Genetic polymorphisms, like those of VEGF76 and CXCL1277 have shown prognostic value in small case-control studies, although they have not been validated in adequately sized cohorts. Gene expression profiles with prognostic relevance have been developed for acute leukemia,78 but to the best of our knowledge no stromal-based genetic signatures, like those developed for lymphomas79 and other solid tumors,80 are known.
Proangiogenic factors levels have been the objective of most published works to date. Several unicentric studies have demonstrated a correlation between vascular density, levels of proangiogenic factors and survival in both AML17,18,81 and ALL.21,24 Selected studies suggest the utility of angiogenic markers for prediction of response to chemotherapy or allogeneic transplantation,82 but currently there are no validated clinical studies for their utility in a clinical setting.
Implication of bone marrow microenvironment in treatment resistance
Bone marrow microenvironment has been demonstrated to participate in resistance of acute leukemia to chemotherapy. Virtually all mechanisms reviewed here contribute to protection of leukemic cells from the effects of treatment. Specifically both direct cell adhesion-mediated and soluble-factor-mediated drug resistance have been described.83 Accordingly, a higher protection of AML blasts from apoptosis by stromal cells has been observed in chemotherapy-resistant cell lines.50 Different soluble mediators are involved in these processes. Using ALL cell lines, induction of TIEG1 (TGF-β inducible early gene 1) in leukemic cells was demonstrated after exposure to TGF-β and BMP-6 and after interaction with BM stroma cells. This resulted in inhibition of proliferation and protection from chemotherapy, thus suggesting a role for these two mediators in stroma-mediated escape from treatment.84 High levels of asparagine synthetase in BM-derived mesenchymal cells are also one of the mechanisms participating in the resistance of ALL to asparaginase.85 Participation of VEGF-C and other angiogenic factors in resistance mechanisms has also been proposed.86 Hypothesized mechanisms probably include autocrine and paracrine loops, blast homing promotion by endothelial cells and stimulation of endothelium-driven blast survival through antiapoptotic signals.22
Adhesion of leukemic blasts to stromal cells and fibronectin promotes persistence of minimal residual disease and protection from drug-induced cell death. Thus, it is not surprising the demonstration of the major role of CXCR4–CXCL12 axis, essential for BM homing, in persistence of AML minimal residual disease and in development of resistance to chemotherapy.61 The same processes participate in resistance to new tyrosine kinase inhibitors, as demonstrated by the effectiveness of CXCR4 inhibitors in overcoming the protection of stroma on treatment-induced apoptosis of leukemic cells.87,88
New microenvironment targets for leukemia treatment
The progressive understanding of the contributions of BM microenvironment to acute leukemia progression is currently permitting the rational development of new therapeutic approaches.7,89 Although a complete description of these new targeted therapies is beyond the scope of this review, some of the most promising agents are those blocking the interactions of leukemic cells with stromal cells or stroma-derived growth factors. Inhibitors of CXCR4–CXCL12 axis90,91 and agents targeting the ILK/Akt or the PI3K/Akt pathways92,93 are currently the object of intense study. Integrin inhibitors are also being evaluated as possible preventive agents against cell adhesionmediated drug resistance.49 Furthermore, a better understanding of microenvironment-driven resistance to treatment may also allow a finer tuning of current treatment. For example, in vitro exposure of stroma to chemotherapy can decrease the protection against spontaneous and chemotherapy-induced apoptosis that BM exerts on AML cells,94 thus providing a possible synergistic antitumoral effect. We speculate that the relatively leukemic-blast-free milieu established after induction chemotherapy may be important for directing normal hematopoietic reconstitution or persistence of minimal residual disease. However, dissecting out the different in vivo effects of treatment on stromal cells and on leukemic blast and their therapeutic relevance will probably be a difficult task and a more comprehensive understanding of the specific effects of the different drugs on stromal cells in vitro and in vivo is needed before accomplishing it. To this regard, although normal BM microenvironment damage and altered hematopoiesis have always been a concern for chemotherapy, the same could not be true for the combination of stromal-targeted therapies with conventional treatment as demonstrated by some experiments showing low BM toxicity with the use of CXCR4 or β1 -integrin-blocking agents.95,49
Last but not least, the considerable amount of clinical and experimental data supporting a role of angiogenesis in leukemia progression, and the demonstration of the possibility of in vivo modulation of acute leukemia progression by angiogenesis-related factors,96,97,98 also offers the possibility of using antiangiogenic drugs for the treatment of human leukemia. Consequently, angiogenesis-targeted drugs previously shown to be effective in solid neoplasms are now being tested in patients with refractory acute leukemia.99
In conclusion, interactions of leukemic cells with BM microenvironment stromal cells and soluble mediators are fundamental for blasts survival, disease progression and resistance to treatment. Although rapid advances in the understanding of these processes are being made, we need a clearer picture of the relative contribution and importance of each of the interactions and of the stromal cells involved in them; clearly, more relevant in vivo models will be necessary to dissect the role of each stromal component. In fact, of all the components, it is still entirely unclear if there is a single most relevant stromal cell-are the CAR cells capable of having a vital role in the absence of its other stromal counterparts? Or interactions between the stromal elements are important in regulating the niche in health and disease? Key areas for future research are the disruption of normal hematopoietic niche by leukemic blasts, the cellular mechanisms and signaling pathways involved in persistence of minimal residual disease after treatment, and the contribution of the angiogenic switch to leukemogenesis and leukemia progression. Exploitation of leukemic microenvironment for the design of newly targeted therapies is a promising strategy for ameliorating the prognosis of these patients and for limiting the toxicity and resistance development associated to conventional treatments. Blocking of proangiogenic pathways, manipulation of the CXCR4–CXCL12 axis and anti-integrin drugs are among the most interesting areas for therapeutic development.
Acknowledgments
This work was primarily supported by National Institutes of Health Grants DK62987, DK55001, AA13913, DK61688 and CA125550; Champalimaud Foundation, Stop and Shop Pediatric Brain Cancer Fund and funds from the Department of Medicine for the Division of Matrix Biology at Beth Israel Deaconess Medical Center. FA was supported by Grant BAE-90058 from the Health Research Fund, Ministry of Health, Spain; and by Health Service from Comunidad Autonoma of Murcia, Spain.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Quesenberry PJ, Aliotta JM. The paradoxical dynamism of marrow stem cells: considerations of stem cells, niches and microvesicles. Stem Cell Rev. 2008;4:137–147. doi: 10.1007/s12015-008-9036-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raaijmakers MHGP, Scadden DT. Evolving concepts on the micro environmental niche for hematopoietic stem cells. Curr Opin Hematol. 2008;15:301–306. doi: 10.1097/MOH.0b013e328303e14c. [DOI] [PubMed] [Google Scholar]
- 3.Lataillade JJ, Pierre-Louis O, Hasselbalch HC, Uzan G, Jasmin C, Martyré MC, et al. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence Blood. 2008;112:3026–3035. doi: 10.1182/blood-2008-06-158386. [DOI] [PubMed] [Google Scholar]
- 4.Folkmann J, Kalluri R. Cancer without disease. Nature. 2008;427:787. doi: 10.1038/427787a. [DOI] [PubMed] [Google Scholar]
- 5.Nyberg P, Salo T, Kalluri R. Tumor microenvironment and angiogenesis. Front Biosci. 2008;13:6537–6553. doi: 10.2741/3173. [DOI] [PubMed] [Google Scholar]
- 6.Ribatti D. Is angiogenesis essential for the progression of hematological malignancies or is it an epiphenomenon? Leukemia. 2009;23:433–434. doi: 10.1038/leu.2008.381. [DOI] [PubMed] [Google Scholar]
- 7.Zhou J, Mauerer K, Farina L, Gribben JG. The role of microenvironment in hematological malignancies and implication for therapy. Front Biosci. 2005;10:1581–1596. doi: 10.2741/1642. [DOI] [PubMed] [Google Scholar]
- 8.Hussong JW, Rodgers, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood. 2005;95:309–313. [PubMed] [Google Scholar]
- 9.Padró T, Ruiz S, Bieker R, Bürger H, Steins M, Kienast J, et al. Increased angiogenesis in the marrow of patients with acute myeloid leukemia. Blood. 2000;15:2637–2644. [PubMed] [Google Scholar]
- 10.De Bont ES, Rosati S, Jacobs S, Kamps WA, Vellenga E. Increased bone marrow vascularization in patients with acute myeloid leukaemia: a possible role for vascular endothelial growth factor. Br J Hematol. 2001;113:296–304. doi: 10.1046/j.1365-2141.2001.02722.x. [DOI] [PubMed] [Google Scholar]
- 11.Kini AR, Peterson LA, Tallman MS, Lingen MW. Angiogenesis in acute promyelocytic leukemia: induction by vascular endothelial growth factor and inhibition by all-trans retinoic acid. Blood. 2001;97:3919–3924. doi: 10.1182/blood.v97.12.3919. [DOI] [PubMed] [Google Scholar]
- 12.Padró T, Bieker R, Ruiz S, Steins M, Retzlaff S, Bürger H, et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia. 2002;16:1302–1310. doi: 10.1038/sj.leu.2402534. [DOI] [PubMed] [Google Scholar]
- 13.Litwin C, Leong KG, Zapf R, Sutherland H, Naiman SC, Karsan A. Role of the microenvironment in promoting angiogenesis in acute leukemia. Am J Hematol. 2002;70:22–30. doi: 10.1002/ajh.10092. [DOI] [PubMed] [Google Scholar]
- 14.Dias S, Hattori K, Heissig B, Zhu Z, Wu Y, Witte L, et al. Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathway is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci USA. 2001;98:10857–10862. doi: 10.1073/pnas.191117498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiedler W, Graeven U, Ergün S, Verago S, Kilic N, Stockschläder M, et al. Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood. 1997;89:1870–1875. [PubMed] [Google Scholar]
- 16.Aguayo A, Estey E, Kantarjian H, Mansouri T, Gidel C, Keating M, et al. Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood. 1999;94:3717–3721. [PubMed] [Google Scholar]
- 17.Loges S, Heil G, Bruweleit M, Schoder V, Butzal M, Fischer U, et al. Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol. 2005;23:1109–1117. doi: 10.1200/JCO.2005.05.058. [DOI] [PubMed] [Google Scholar]
- 18.Bieker R, Padro T, Kramer J, Steins M, Kessler T, Retzlaff S, et al. Overexpression of basic fibroblast growth factor and autocrine stimulation in acute myeloid leukemia. Cancer Res. 2003;63:7241–7246. [PubMed] [Google Scholar]
- 19.Neegard HF, Iversen N, Bowitz-Lothe IM, Sandset PM, Steinvisk B, Ostenstad B, et al. Increased bone marrow microvascular density in haematological malignancies is associated with differential regulation of angiogenic factors. Leukemia. 2009;23:162–169. doi: 10.1038/leu.2008.255. [DOI] [PubMed] [Google Scholar]
- 20.Bruserud O, Glenjen N, Ryningen A. Effects of angiogenic regulators on in vitro proliferation and cytokine secretion by native human acute myelogenous leukemia blasts. Eur J Haematol. 2003;71:9–17. doi: 10.1034/j.1600-0609.2003.00080.x. [DOI] [PubMed] [Google Scholar]
- 21.Perez-Atayde AR, Sallan SE, Tedrow U, Connors S, Allred E, Folkman J. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am J Pathol. 1997;150:815–821. [PMC free article] [PubMed] [Google Scholar]
- 22.Veiga P, Costa LF, Sallan SE, Nadler LM, Cardoso AA. Leukemiastimulated bone marrow endothelium promotes leukemia cell survival. Exp Hematol. 2006;34:610–621. doi: 10.1016/j.exphem.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Travaglino E, Benatti C, Malcovatti L, Della Porta MG, Galli A, Bonetti E, et al. Biological and clinical relevance of matrix metalloproteinases 2 and 9 in acute myeloid leukaemias and myelodysplastic syndromes. Eur J Haematol. 2008;80:216–226. doi: 10.1111/j.1600-0609.2007.01012.x. [DOI] [PubMed] [Google Scholar]
- 24.Aref S, Salama O, Shamaa S, El-Refaie M, Mourkos H. Angiogenesis factor pattern differs in acute lymphoblastic leukemia and chronic lymphocytic leukemia. Hematology. 2007;12:319–324. doi: 10.1080/10245330701340759. [DOI] [PubMed] [Google Scholar]
- 25.Bitan M, Polliack A, Zecchina G, Nagler A, Friedmann Y, Nadav L, et al. Heparanase expression in human leukemias is restricted to acute myeloid leukemias. Exp Hematol. 2002;30:34–41. doi: 10.1016/s0301-472x(01)00766-4. [DOI] [PubMed] [Google Scholar]
- 26.Eshel R, Benz-Zaken O, Vainas O, Nadir Y, Minucci S, Polliack A, et al. Leukomogenic factors downregulate heparanase expression in acute myeloid leukemia cells. Biochem Biophys Res Commun. 2005;335:1115–1122. doi: 10.1016/j.bbrc.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Watarai M, Miwa H, Shikami M, Sugamura K, Wakabayashi M, Satoh A, et al. Expression of endothelial cell-associated molecules in AML cells. Leukemia. 2002;16:112–119. doi: 10.1038/sj.leu.2402326. [DOI] [PubMed] [Google Scholar]
- 28.Rigolin GM, Mauro E, Ciccone M, Fraulini C, Sofritti O, Castoldi G, et al. Neoplastic circulating endothelial-like cells in patients with acute myeloid leukaemia. Eur J Haematol. 2007;78:365–373. doi: 10.1111/j.1600-0609.2007.00839.x. [DOI] [PubMed] [Google Scholar]
- 29.Della Porta MG, Malcovati L, Rigolin GM, Rosti V, Bonetti E, Travaglino E, et al. Immunophenotypic, cytogenetic and functional characterization of circulating endothelial cells in myelodysplastic syndromes. Leukemia. 2008;22:530–537. doi: 10.1038/sj.leu.2405069. [DOI] [PubMed] [Google Scholar]
- 30.Rehman J, Li J, Orschell CM, March KL. Peripheral blood endothelial progenitor cells are derived from monocyte/macrophage and secrete angiogenic growth factors. Circulation. 2003;107:1164–1169. doi: 10.1161/01.cir.0000058702.69484.a0. [DOI] [PubMed] [Google Scholar]
- 31.Riccioni R, Calzolari A, Biffoni M, Senese M, Riti V, Petrucci E, et al. Podocalyxin is expressed in normal and leukemic monocytes. Blood Cells Mol Dis. 2006;37:218–225. doi: 10.1016/j.bcmd.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JCY, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer. 2007;7:118–129. doi: 10.1038/nrc2047. [DOI] [PubMed] [Google Scholar]
- 33.Wimazal F, Krauth MT, Vales A, Bohm A, Agis H, Sonneck K, et al. Immunohistochemical detection of vascular endothelial growth factor (VEGF) in the bone marrow in patients with myelodysplastic syndromes: correlation between VEGF expression and the FAB category. Leuk Lymphoma. 2007;47:451–460. doi: 10.1080/10428190500353083. [DOI] [PubMed] [Google Scholar]
- 34.Korkolopoulou P, Apostolidou E, Pavlopoulos PM, Kavantzas N, Vyniou N, Thymara I, et al. Prognostic evaluation of the microvascular network in myelodysplastic syndromes. Leukemia. 2001;15:1369–1376. doi: 10.1038/sj.leu.2402220. [DOI] [PubMed] [Google Scholar]
- 35.Lundberg LG, Hellstrom-Lindberg E, Kanter-Lewensohn L, Lerner R, Palmblad J. Angiogenesis in relation to clinical stage, apoptosis and prognostic score in myelodysplastic syndromes. Leukemia Res. 2006;30:247–253. doi: 10.1016/j.leukres.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Campioni D, Punturieri M, Bardi A, Moretti S, Tammiso E, Lanza F, et al. In vitro evaluation of bone marrow angiogenesis in myelodysplastic syndromes: a morphological and functional approach. Leukemia Res. 2004;28:9–17. doi: 10.1016/s0145-2126(03)00123-1. [DOI] [PubMed] [Google Scholar]
- 37.Keith T, Araki Y, Ohyagi M, Hasegawa M, Yamamoto K, Kurata M, et al. Regulation of angiogenesis in the bone marrow of myelodysplastic syndromes transforming to overt leukaemia. Br J Haematol. 2007;137:206–215. doi: 10.1111/j.1365-2141.2007.06539.x. [DOI] [PubMed] [Google Scholar]
- 38.Mayani H. Composition and function of the hemopoietic microenvironment in human myeloid leukemia. Leukemia. 1995;10:1041–1047. [PubMed] [Google Scholar]
- 39.Blau O, Hofmann WK, Baldus CD, Thiel G, Serbent V, Schümann E, et al. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol. 2007;35:221–229. doi: 10.1016/j.exphem.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 41.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–1865. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- 42.Oostendorp RA, Dörmer P. VLA-4 mediated interactions between normal human hematopoietic progenitors and stromal cells. Leuk Lymphoma. 1997;24:423–435. doi: 10.3109/10428199709055581. [DOI] [PubMed] [Google Scholar]
- 43.Bradstock KF, Gottlieb DJ. Interaction of acute leukemia cells with the bone marrow microenvironment: implications for control of minimal residual disease. Leuk Lymphoma. 1995;18:1–16. doi: 10.3109/10428199509064917. [DOI] [PubMed] [Google Scholar]
- 44.Bendall LJ, Daniel A, Kortlepel K, Gottlieb DJ. Bone marrow adherent layers inhibit apoptosis of acute myeloid leukemia. Exp Hematol. 1994;22:1252–1260. [PubMed] [Google Scholar]
- 45.Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, Moqueen T, Priebe W, et al. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res. 2007;67:684–694. doi: 10.1158/0008-5472.CAN-06-3166. [DOI] [PubMed] [Google Scholar]
- 46.Winter SS, Sweatman JJ, Lawrence MB, Rhoades TH, Hart AL, Larson RS. Enhanced T-lineage acute lymphoblastic leukaemia cell survival on bone marrow stroma requires involvement of LFA-1 and ICAM-1. Br J Haematol. 2001;115:862–871. doi: 10.1046/j.1365-2141.2001.03182.x. [DOI] [PubMed] [Google Scholar]
- 47.Damiano JS, Cress AE, Hazlehurst LA, Shitl AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 48.Campana D, Coustan-Smith E, Manabe A, Kumagai M, Murti KG, Silvennoinen O, et al. Human B-cell progenitors and bone marrow microenvironment. Hum Cell. 1996;9:317–322. [PubMed] [Google Scholar]
- 49.Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158–1165. doi: 10.1038/nm909. [DOI] [PubMed] [Google Scholar]
- 50.Konopleva M, Konoplev S, Hu W, Zaritskey AY, Afanasiev BV, Andreeff M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia. 2002;16:1713–1724. doi: 10.1038/sj.leu.2402608. [DOI] [PubMed] [Google Scholar]
- 51.Ryningen A, Wergeland L, Glenjen N, Gjertsen BT, Bruserud O. In vitro crosstalk between fibroblasts and native human acute myelogenous leukemia (AML) blasts via local cytokine network results in increased proliferation and decreased apoptosis of AML cells as well as increased levels of proangiogenic Interleukin 8. Leuk Res. 2005;29:185–196. doi: 10.1016/j.leukres.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Wu S, Korte A, Kebelmann-Betzing C, Gessner R, Henze G, Seeger K. Interaction of bone marrow stromal cells with lymphoblasts and effects of prednisolone on cytokine expression. Leuk Res. 2005;29:63–72. doi: 10.1016/j.leukres.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 53.Scupoli MT, Donadelli M, Cioffi F, Rossi M, Perbellini O, Malpeli G, et al. Bone marrow stromal cells and the upregulation Leukemia and tumor microenvironment of interleukin-8 production in human T-cell acute lymphoblastic leukemia trough the CXCL12/CXCR4 axis and the NF-kB and JNK/AP-1 pathways. Hematologica. 2008;93:524–532. doi: 10.3324/haematol.12098. [DOI] [PubMed] [Google Scholar]
- 54.Li A, Varney ML, Valasek J, Godfrey M, Dave BJ, Singh RK. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis. 2005;8:63–71. doi: 10.1007/s10456-005-5208-4. [DOI] [PubMed] [Google Scholar]
- 55.Scupoli MT, Perbellini O, Krampera M, Vinante F, Cioffi F, Pizzolo G. Interleukin 7 requirement for survival of T-cell acute lymphoblastic leukemia and human thymocytes on bone marrow stroma. Hematologica. 2007;92:264–266. doi: 10.3324/haematol.10356. [DOI] [PubMed] [Google Scholar]
- 56.Juarez J, Baraz R, Gaundar S, Bradstock K, Bendall L. Interaction of interleukin-7 and interleukin-3 with the CXCL12-induced proliferation of B-cell progenitor acute lymphoblastic leukemia. Haematologica. 2007;92:450–459. doi: 10.3324/haematol.10621. [DOI] [PubMed] [Google Scholar]
- 57.Bellamy WT, Richter L, Sirjani D, Roxas C, Glinsmann-Gibson B, Frutiger Y, et al. Vascular endothelial growth factor is an autocrine promoter of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood. 2001;97:1427–1434. doi: 10.1182/blood.v97.5.1427. [DOI] [PubMed] [Google Scholar]
- 58.Weimar IS, Voermans C, Bourhis JH, Miranda N, van der Berk PC, Nakamura T, et al. Hepatocyte growth factor/scatter factor (HGJF/SF affects proliferation and migration of myeloid leukemic cells. Leukemia. 1998;12:1195–1203. doi: 10.1038/sj.leu.2401080. [DOI] [PubMed] [Google Scholar]
- 59.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 60.Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 62.Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood. 2002;99:394. doi: 10.1182/blood.v99.1.394. [DOI] [PubMed] [Google Scholar]
- 63.Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in bone marrow according to regional hypoxia. Proc Natl Acad Sci USA. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mortensen BT, Jensen PO, Helledie N, Iversen PO, Ralfkiaer E, Larsen J, et al. Changing bone marrow micro-environment during development of acute myeloid leukaemia in rats. Br J Haematol. 1998;102:458–464. doi: 10.1046/j.1365-2141.1998.00801.x. [DOI] [PubMed] [Google Scholar]
- 65.Jensen PO, Mortensen BT, Hodgkiss RJ, Iversen PO, Christensen IJ, Helledie N, et al. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif. 2000;33:381–395. doi: 10.1046/j.1365-2184.2000.00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fiegl M, Samudio I, Clise-Dwyer K, Burks J, Mnjoyan Z, Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood. 2009;113:1504–1512. doi: 10.1182/blood-2008-06-161539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maxwell The HIF pathway in cancer. Semin Cell Dev Biol. 2005;16:523–530. doi: 10.1016/j.semcdb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 68.Martelli AM, Nyakern M, Tabellini G, Bortul R, Tazzari PL, Evangelisti C, et al. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006;20:911–928. doi: 10.1038/sj.leu.2404245. [DOI] [PubMed] [Google Scholar]
- 69.Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13:483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pramanik K, Trüpschuch S, Greiner A, Ruthardt M, Henschler R, Müller AM. The aorta-gonad-mesonephros-derived stroma cell line DAS104-4 induces differentiation of leukemic cells. Leuk Res. 2008;32:781–789. doi: 10.1016/j.leukres.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 71.Kankuri E, Babusikova O, Hlubinova K, Salmenpera P, Boccaccio C, Lubitz W, et al. Fibroblast nemosis arrests growth and induces differentiation of human leukemia cells. Int J Cancer. 2008;122:1243–1252. doi: 10.1002/ijc.23179. [DOI] [PubMed] [Google Scholar]
- 72.Buggins AGS, Milojkovic D, Arno MJ, Lea NC, Mufti GJ, Thomas NS, et al. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kB, c-Myc, and pRb pathways. J Immunol. 2001;167:6021–6030. doi: 10.4049/jimmunol.167.10.6021. [DOI] [PubMed] [Google Scholar]
- 73.Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25into CD25+T regulatory cells. Blood. 2007;109:2871–2877. doi: 10.1182/blood-2006-07-036863. [DOI] [PubMed] [Google Scholar]
- 74.Yin T, Linheng L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–1201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bruserud O, Ryningen A, Wergeland L, Glenjen NI, Gjertsen BT. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica. 2004;89:391–402. [PubMed] [Google Scholar]
- 76.Kim DH, Lee NY, Lee MH, Sohn SK, Do YR, Park JY. Vascular endothelial growth factor (VEGF) gene (VEGFA) polymorphism can predict the prognosis in acute myeloid leukaemia patients. Br J Haematol. 2008;140:71–79. doi: 10.1111/j.1365-2141.2007.06887.x. [DOI] [PubMed] [Google Scholar]
- 77.Dommange F, Cartron G, Espanel C, Gallay N, Domenech J, Benboubker L, et al. CXCL12 polymorphism and malignant cell dissemination/tissue infiltration in acute myeloid leukemia. FASEB J. 2006;20:1913–1915. doi: 10.1096/fj.05-5667fje. [DOI] [PubMed] [Google Scholar]
- 78.Wouters BJ, Löwenberg B, Delwel R. A decade of genome-wide gene expression profiling in acute myeloid leukemia: flashback and prospects. Blood. 2009;113:291–298. doi: 10.1182/blood-2008-04-153239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signature in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. 2009;15:68–74. doi: 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- 81.Aguayo A, Kantarjian HM, Estey EH, Giles FJ, Verstovsek S, Manshouri T, et al. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer. 2002;95:1923–1930. doi: 10.1002/cncr.10900. [DOI] [PubMed] [Google Scholar]
- 82.Kümpers P, Koenecke C, Hecker H, Hellpap J, Horn R, Verhagen W, et al. Angiopoietin-2 predicts disease-free survival after allogeneic stem cell transplantation in patients with high-risk myeloid malignancies. Blood. 2008;112:2139–2148. doi: 10.1182/blood-2007-12-130021. [DOI] [PubMed] [Google Scholar]
- 83.Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–2526. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 84.Dosen-Dahl G, Munthe F, Nygren MK, Stubberud H, Hystad ME, Rian E. Bone marrow stroma cells regulate TIEG1 expression in acute lymphoblastic leukemia cells: role of TGF beta/BMP-6 and TIEG1 in chemotherapy escape. Int J Cancer. 2008;123:2759–2766. doi: 10.1002/ijc.23833. [DOI] [PubMed] [Google Scholar]
- 85.Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. 2007;117:1049–1057. doi: 10.1172/JCI30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dias S, Choy MK, Rafii S. Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood. 2002;99:2179–2184. doi: 10.1182/blood.v99.6.2179. [DOI] [PubMed] [Google Scholar]
- 87.Zeng Z, Samudio IJ, Munsell M, An J, Huang Z, Estey E, et al. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther. 2006;5:3113–3121. doi: 10.1158/1535-7163.MCT-06-0228. [DOI] [PubMed] [Google Scholar]
- 88.Zeng Z, Shi YX, Samudio IJ, Wang R, Ling X, Frolova, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–6224. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown VI, Seif AE, Reid GS, Teachey DT, Grupp SA. Novel molecular and cellular therapeutic targets in acute lymphoblastic leukemia and lymphoproliferative disease. Immunol Res. 2008;42:84–105. doi: 10.1007/s12026-008-8038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 91.Juarez J, Dela Pena A, Baraz R, Hewson J, Khoo M, Cisterne A, et al. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia. 2007;21:1249–1257. doi: 10.1038/sj.leu.2404684. [DOI] [PubMed] [Google Scholar]
- 92.Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, Moqueen T, Priebe W, et al. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res. 2007;67:684–694. doi: 10.1158/0008-5472.CAN-06-3166. [DOI] [PubMed] [Google Scholar]
- 93.Papa V, Tazzari PL, Chiarini F, Cappellini A, Ricci F, Billi AM, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia. 2008;22:147–160. doi: 10.1038/sj.leu.2404980. [DOI] [PubMed] [Google Scholar]
- 94.Moshaver B, van der Pol MA, Westra AH, Ossenkoppele GJ, Zweegman S, Schuurhuis GJ. Chemotherapeutic treatment of bone marrow stromal cells strongly affects their protective effect on acute myeloid leukemia cell survival. Leuk Lymphoma. 2008;49:134–148. doi: 10.1080/10428190701593636. [DOI] [PubMed] [Google Scholar]
- 95.Tavor S, Petit I, Porozov S, Avigdor A, Leider-Trejo L, Shemtov N, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64:2817–2824. doi: 10.1158/0008-5472.can-03-3693. [DOI] [PubMed] [Google Scholar]
- 96.Schuch G, Machluf M, Bartsch G, Nomi M, Richard H, Atala A, et al. In vivo administration of vascular endothelial growth factor (VEGF) and its antagonist, soluble neuropilin-1, predicts a role of VEGF in the progression of acute myeloid leukemia in vivo. Blood. 2002;100:4622–4628. doi: 10.1182/blood.V100.13.4622. [DOI] [PubMed] [Google Scholar]
- 97.Schuch G, Oliveira-Ferrer L, Loges S, Laack E, Bokemeyer C, Hossfeld DK, et al. Antiangiogenic treatment with endostatin inhibits progression of AML in vivo. Leukemia. 2005;19:1312–1317. doi: 10.1038/sj.leu.2403824. [DOI] [PubMed] [Google Scholar]
- 98.Iversen PO, Sorensen DR, Benestad HB. Inhibitors of angiogenesis selectively reduce the malignant cell load in rodent models of human myeloid leukemias. Leukemia. 2002;16:376–381. doi: 10.1038/sj.leu.2402376. [DOI] [PubMed] [Google Scholar]
- 99.Li WW, Hutnik M, Gehr G. Antiangiogenesis in haematological malignancies. Br J Hematol. 2008;143:622–631. doi: 10.1111/j.1365-2141.2008.07372.x. [DOI] [PubMed] [Google Scholar]