

Abstract
Chromosome fragmentation is a hallmark of apoptosis, conserved in diverse organisms. In mammals, caspases activate apoptotic chromosome fragmentation by cleaving and inactivating an apoptotic nuclease inhibitor. We report that inactivation of the C. elegans dcr-1 gene, which encodes the Dicer ribonuclease important for processing of small RNAs, compromises apoptosis and blocks apoptotic chromosome fragmentation. DCR-1 was cleaved by the CED-3 caspase to generate a C-terminal fragment with deoxyribonuclease activity, which produced 3’ hydroxyl DNA breaks on chromosomes and promoted apoptosis. Thus caspase-mediated activation of apoptotic DNA degradation is conserved. DCR-1 functions in fragmenting chromosomal DNA during apoptosis, in addition to processing of small RNAs, and undergoes a protease-mediated conversion from a ribonuclease to a deoxyribonuclease.
One of the hallmarks of apoptosis is fragmentation of chromosomal DNA at internucleosomal regions, which generates DNA fragments differing by approximately 180 bp and contributes to the irreversible cell killing process (1, 2). Multiple deoxyribonucleases (DNases) have been implicated in mediating apoptotic DNA fragmentation, including 40 kDa DNA fragmentation factor (DFF40), also known as caspase activated deoxyribonuclease (CAD), endonuclease G, deoxyribonuclease II (DNase II), and several other nucleases (3, 4). DFF40 is a component of a DNA fragmentation factor complex, which also contains an inhibitory subunit, DFF45 (also known as ICAD, inhibitor of CAD)(5–8). DFF45 serves both as a chaperone for the proper folding of DFF40 and a cognate inhibitor that holds DFF40 activity in check in normal cells. During apoptosis, the cleavage of DFF45 by activated caspases such as caspase-3 and caspase-7 results in the release and activation of DFF40 (5–8). The activated DFF40 nuclease then associates with chromosomal proteins such as histone H1, HMG (high mobility group) proteins, and topoisomerase II to promote cleavage of internucleosomal DNA, generating 3’ hydroxyl DNA breaks that can be detected by the TUNEL (TdT-mediated dUTP nick end labeling) assay (6, 9, 10). In mice deficient in either DFF40 or DFF45, which is needed for proper folding of DFF40, chromosome fragmentation fails to occur during apoptosis (11, 12).
A mitochondrial nuclease, endonuclease G (endoG), mediates residual apoptotic DNA fragmentation observed in DFF45 deficient cells (13). EndoG is released from mitochondria during apoptosis and translocates into the nucleus to facilitate chromosome fragmentation (13). EndoG is a member of a conserved family of endonucleases, including the C. elegans CPS-6 (CED-3 Protease Suppressor) nuclease (14, 15). In C. elegans animals deficient in cps-6, TUNEL-stained nuclei accumulate in mutant embryos and developmental cell death is delayed or even inhibited in sensitized genetic backgrounds such as animals partially deficient for the CED-3 caspase (14), suggesting that CPS-6 is important for apoptotic DNA degradation and progression of apoptosis. In addition to cps-6, seven cell-death related nucleases (named CRN nucleases) have been identified in a candidate-based RNAi screen in C. elegans (16). RNAi mediated depletion of six of these crn nuclease genes (crn-1 to crn-5 and cyp-13) results in similar cell death defects, including accumulation of TUNEL-stained nuclei in RNAi-treated embryos and inhibition of apoptosis in sensitized genetic backgrounds (16). Biochemical analysis reveals that some of these CRN nucleases interact with CPS-6 and with one another to promote step-wise DNA degradation, first turning the 3’ OH DNA nicks into small gaps through their exonuclease activities and then creating double-stranded DNA breaks via a gap-dependent endonuclease activity (16, 17). Two C. elegans DNase II homologues, NUC-1 and CRN-6, function in a later stage to complete degradation of chromosomal DNA (16, 18), as loss of either gene or both also results in accumulation of TUNEL-stained nuclei but does not affect the activation or the kinetics of apoptosis (14, 18). In all instances, loss of any one or a combination of the nine C. elegans apoptotic nucleases results in accumulation of TUNEL-stained nuclei, indicating that these nucleases function in resolving 3' OH DNA breaks generated during apoptosis and that at least one additional C. elegans nuclease, presumably a functional analog of DFF40, is activated during apoptosis to generate 3’ OH DNA breaks. Because no candidate DFF40 or DFF45 homologue is predicted to be encoded by the C. elegans genome (4), it remains enigmatic how apoptotic DNA degradation is initiated in C. elegans. We report that this role is played by the C. elegans Dicer ribonuclease.
Dicer is a highly conserved ribonuclease that plays a central role in processing double-stranded RNA (dsRNA) to generate small RNAs important for various gene silencing events (19–26). It is an RNase III enzyme that processes precursor dsRNA into small duplex RNA species of approximately 21 to 25 nucleotides, with 5' phosphate and 3' hydroxyl termini (22, 27–30). These small RNAs are then unwound and incorporated into various silencing complexes such as the RNA-induced silencing complex (RISC) that direct sequence-specific cleavage or translational repression of complementary mRNA (24–26, 31–34). Multiple proteins have been identified to serve as Dicer co-factors or interactors to mediate various gene silencing events, including proteins in the Argonaute family that contain PAZ (Piwi, Argo and Zwille) and PIWI domains (32–34), RDE-1 (RNAi Defective, also an Argonaute protein), RDE-4 and its fly homologue R2D2, DRH-1 and DRH-2 (Dicer Related Helicases)(31, 35, 36), ERI-1 (Enhanced RNAi), and RRF-3 (RNA-dependent RNA polymerase Family)(26, 37, 38). Dicer enzymes typically contain a DExH-box helicase domain, a PAZ domain, two tandem RNase III domains (RNase IIIa and IIIb), and a dsRNA-binding domain (39, 40). The intramolecular dimerization of the two RNase III domains of Dicer generates the dicing activity (29) and the distance between the PAZ and RNase III domains determines the length of small RNA fragments processed (41). Dicer is important for multiple cellular processes, including viral defense, chromatin remodeling, genome rearrangement, developmental timing, and stem cell maintenance (21, 23, 42–47). These cellular functions likely are attributed to the generation of small interfering RNAs (siRNAs), micro RNAs (miRNAs), and other small RNAs (34, 40). We uncovered a previously uncharacterized role of C. elegans Dicer (DCR-1) in activating the apoptotic DNA degradation process that is dependent on generation of a new DNase activity through cleavage by the cell death protease CED-3 and that is independent of Dicer’s actions in gene silencing in C. elegans.
dcr-1 is required for initiating apoptotic DNA degradation in C. elegans
To identify nucleases that function in apoptotic DNA degradation in C. elegans, and in particular, nucleases that generate 3’ hydroxyl DNA breaks during apoptosis, we performed RNAi on cps-6(sm116) mutant animals, which contain an average of 16 TUNEL-stained cells in early stage embryos (Fig. 1, A and B), and looked for RNAi knockdown of nucleases that reduced the number of TUNEL-stained cells in cps-6(sm116) embryos (16, 17). We found that dcr-1 RNAi reduced the number of TUNEL signals in cps-6(sm116) embryos (Fig. 1A). Given that dcr-1 RNAi also reduces the efficiency of RNA interference, we examined whether two deletion mutations in dcr-1 (ok247 and pk1351) caused a stronger reduction in TUNEL staining in the cps-6(sm116) mutant. Because dcr-1 loss-of-function (lf) mutants are sterile, a balancer, hT2(qIs48), was used to maintain the dcr-1(lf) heterozygous strain. qIs48 is a transgene integrated into the hT2 chromosome and carries a Pmyo-2gfp reporter that directs strong GFP expression in the pharynx in most developmental stages. dcr-1(lf) homozygous embryos derived from the dcr-1(lf)/hT2 mother can thus be identified as embryos that do not express GFP. We performed TUNEL and anti-GFP double staining to score the number of TUNEL-stained cells in dcr-1(lf) embryos (48). Although dcr-1(ok247) and dcr-1(pk1351) homozygous embryos had comparable numbers of TUNEL-stained cells to those seen in dcr-1(ok247)/hT2 and dcr-1(pk1351)/hT2 heterozygous embryos or wild-type N2 embryos (Fig. 1B), both dcr-1(ok247) and dcr-1(pk1351) deletion mutations reduced the number of TUNEL signals in cps-6(sm116) embryos to that seen in wild-type embryos (Fig. 1C), suggesting that dcr-1 does function in generating TUNEL-reactive DNA breaks that fail to be resolved in cps-6(sm116) embryos. Similarly, dcr-1(ok247) significantly reduced the number of TUNEL signals in crn-2(tm1177) embryos, which are defective in another apoptotic nuclease CRN-2 (Fig. 1D) (16). dcr-1(ok247) also reduced the number of TUNEL-stained cells in nuc-1(e1392) embryos (Fig. 1D). Taken together, these results indicate that DCR-1 functions in creating TUNEL-reactive DNA breaks in apoptotic cells that are later resolved by other apoptotic nucleases such as CPS-6, CRN-2 and NUC-1 and that dcr-1 likely acts upstream of cps-6, crn-2 and nuc-1 to promote apoptotic DNA degradation.
Fig. 1.

Role of DCR-1 in creating TUNEL-reactive DNA breaks in apoptotic cells. (A–G) TUNEL staining was monitored in the indicated strains (48). In most cases, anti-GFP immunostaining and TUNEL staining were done to quantify the number of TUNEL-stained cells in dcr-1(lf) homozygous embryos (48), which were identified as embryos lacking GFP derived from dcr-1(lf)/hT2(qIs48) mothers. In animals carrying Psur-5sur-5::gfp as a transgenic marker (E, F), dcr-1(lf) homozygous transgenic embryos were identified as embryos with nuclear GFP expression in many cells but lacking pharyngeal GFP expression (qIs48). RNAi experiments were done as described (14). All cps-6(sm116) strains also contain dpy-5(e61). smIs182 is an integrated transgene carrying Pdcr-1DCR-1. smIs196 is an integrated transgene carrying Pdcr-1DCR-1(D1742E). smIs201 is an integrated transgene carrying Pdcr-1tDCR-1. Each numbered array represents an independent transgenic line (F, G). The y axis represents average number of TUNEL-stained cells scored and error bars represent standard error of mean (s.e.m). 15 comma stage embryos were scored for each strain. The significance of differences between different strains was determined by unpaired t test (A) or one-way analysis of variance (ANOVA), followed by Tukey's test (B–G). *, P < 0.001; **, P < 0.05.
dcr-1 promotes apoptosis
We next investigated whether dcr-1 affects programmed cell death in C. elegans. We examined the appearance of apoptotic cell corpses in developing embryos, which is a sensitive assay for detecting weak cell death defects (14, 16, 49). We observed significantly fewer cell corpses in both dcr-1(ok247) and dcr-1(pk1351) mutants at most embryonic stages than in wild-type embryos (Fig. 2A). Thus dcr-1 appears to be a pro-apoptotic factor. The cell death defect displayed by the dcr-1(lf) mutants is stronger than that observed in the cps-6(sm116) mutant or animals deficient in most crn genes, in which cell death in developing embryos is delayed rather than reduced (14, 16). Moreover, both dcr-1(ok247) and dcr-1(pk1351) mutations reduced the number of persistent cell corpses in ced-1(e1735) embryos, in which engulfment of cell corpses is blocked (Fig. 2B)(50), confirming that loss of dcr-1 reduces the number of embryonic cells undergoing cell death.
Fig. 2.

Function of dcr-1 downstream of ced-3 to promote cell death. Embryonic cell corpses were counted in the following animals: (A) N2, dcr-1(ok247), dcr-1(pk1351), and cps-1(sm24) animals; (B) ced-1(e1735), ced-1(e1735); dcr-1(ok247), and ced-1(e1735); dcr-1(pk1351) animals; (C) ced-1(e1735) smIs111; egl-1(n3082) and ced-1(e1735) smIs111; dcr-1(ok247); egl-1(n3082) animals. Stages of embryos examined were: comma, 1.5-fold, 2-fold, 2.5-fold, 3-fold, and 4-fold. The y axis represents average number of cell corpses scored and error bars represent standard deviation (S.D.). 15 embryos were counted for each embryonic stage. The significance of differences between different genetic backgrounds was determined by two-way ANOVA, followed by Bonferroni comparison. *, P < 0.001; **, P < 0.05. All other points had P values > 0.05.
To investigate whether a general reduction in embryonic cell death may indirectly affect apoptotic DNA degradation, we performed the TUNEL assay on the cps-1(sm24) mutant, a CED-3 protease suppressor mutant that exhibits a similar cell death defect to that of the dcr-1(lf) mutants (Fig. 2A)(14). Although cps-1(sm24) causes reduced embryonic cell death at most embryonic stages like the dcr-1(lf) mutations, it did not affect the number of TUNEL-stained cells in the crn-2(tm1177) embryos (Fig. 1D). This result suggests that unlike cps-1 dcr-1 facilitates cell death by promoting apoptotic DNA degradation.
We investigated whether reduction of embryonic cell death caused by dcr-1(lf) mutations might result in survival of cells that normally die and thus yield extra "undead" cells in the anterior pharynx of mutant animals (14). Both dcr-1(ok247) and dcr-1(pk1351) mutants had low numbers of extra cells in the anterior pharynx (Table S1). When combined with other weak cell death mutations, such as the partial loss-of-function mutation (n2438) in the gene encoding the CED-3 caspase, dcr-1(lf) mutations significantly increased the number of extra cells in the anterior pharynx (Table S1). For example, an average of 1.3 extra cells were seen in ced-3(n2438) animals, whereas on average 3.1 extra cells were present in dcr-1(lf); ced-3(n2438) animals (Table S1). Although cps-6(sm116) or crn-2(tm1177) individually also increased the number of extra cells in the ced-3(n2438) mutant background, neither cps-6(sm116) nor crn-2(tm1177) increased the number of extra cells in the dcr-1(ok247); ced-3(n2438) mutant (Table S1), indicating that dcr-1, cps-6 and crn-2 may act in the same pathway to promote apoptosis and DNA degradation.
Because apoptotic DNA degradation occurs in response to caspase activation (5, 7, 14), we performed epistasis analysis to examine whether activation of ced-3 leads to activation of dcr-1, using an integrated transgene (smIs111) expressing an activated form of CED-3 (acCED-3) under the control of the egl-1 promoter (51). When smIs111 was placed in the ced-1(e1735); egl-1(n3082) mutant background, in which almost all naturally occurring somatic cell deaths in C. elegans are blocked by a loss-of-function mutation (n3082) in the cell death initiator egl-1, acCED-3 still induced ectopic cell death and an average of 16.5 and 11 persistent cell corpses in 3-fold and 4-fold late stage embryos, respectively (Fig. 2C). The dcr-1(ok247) mutation significantly reduced the number of ectopic cell deaths induced by acCED-3 in ced-1(e1735) smIs111; egl-1(n3082) embryos, indicating that dcr-1 likely acts downstream of ced-3 to promote apoptosis and DNA degradation.
DCR-1 co-factors in RNA processing do not affect apoptosis
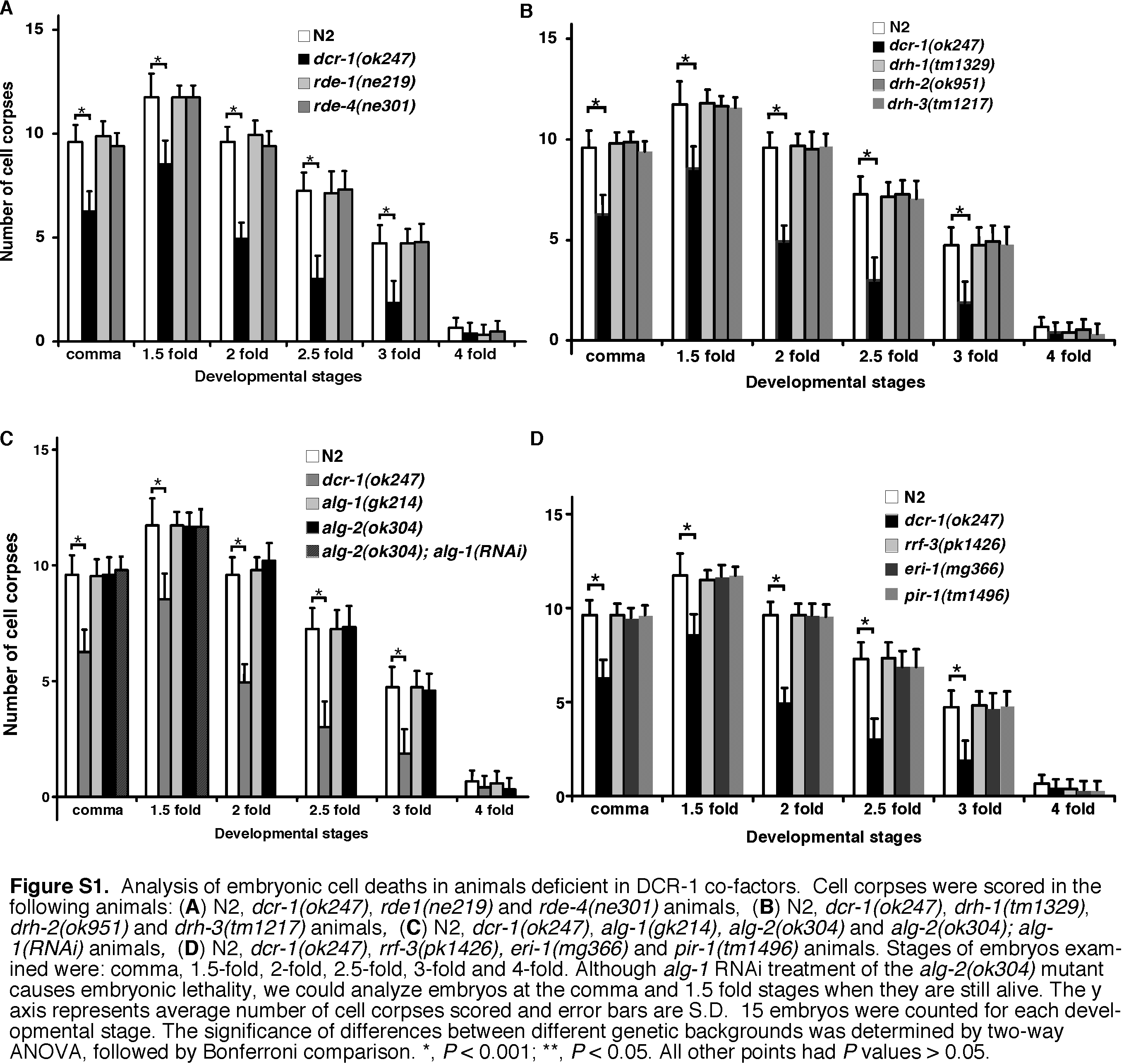
In C. elegans, various co-factors interact with DCR-1 to regulate processing of dsRNA and generate small RNAs, including miRNAs, siRNAs, and siRNAs derived from endogenous triggers (endo-siRNAs) (21, 26, 52). For example, ALG-1 and ALG-2 (Argonaute-Like Gene) are required for miRNA production, RDE-1, RDE-4, DRH-1, and DRH-2 are required for siRNA production, and ERI-1 and RRF-3 are required for endo-siRNA production. We thus examined whether these DCR-1 co-factors also functioned in regulating apoptosis and DNA degradation. Loss of DCR-1 co-factors did not affect the number of TUNEL-stained cells in the cps-6(sm116) mutant or cps-6(RNAi)-treated animals (Table S2). Moreover, unlike dcr-1(lf) mutants, which have reduced number of embryonic cell corpses, the numbers of embryonic cell corpses in animals deficient in DCR-1 co-factors were comparable to those seen in wild-type animals (Fig. S1). These results indicate that defects in the generation of miRNAs, siRNAs, endo-siRNAs, or other small RNAs do not seem to affect apoptosis or apoptotic DNA degradation and that DCR-1 likely mediates apoptotic DNA degradation through a previously uncharacterized mechanism independent of its roles in RNA processing.
DCR-1 is a substrate of the CED-3 caspase
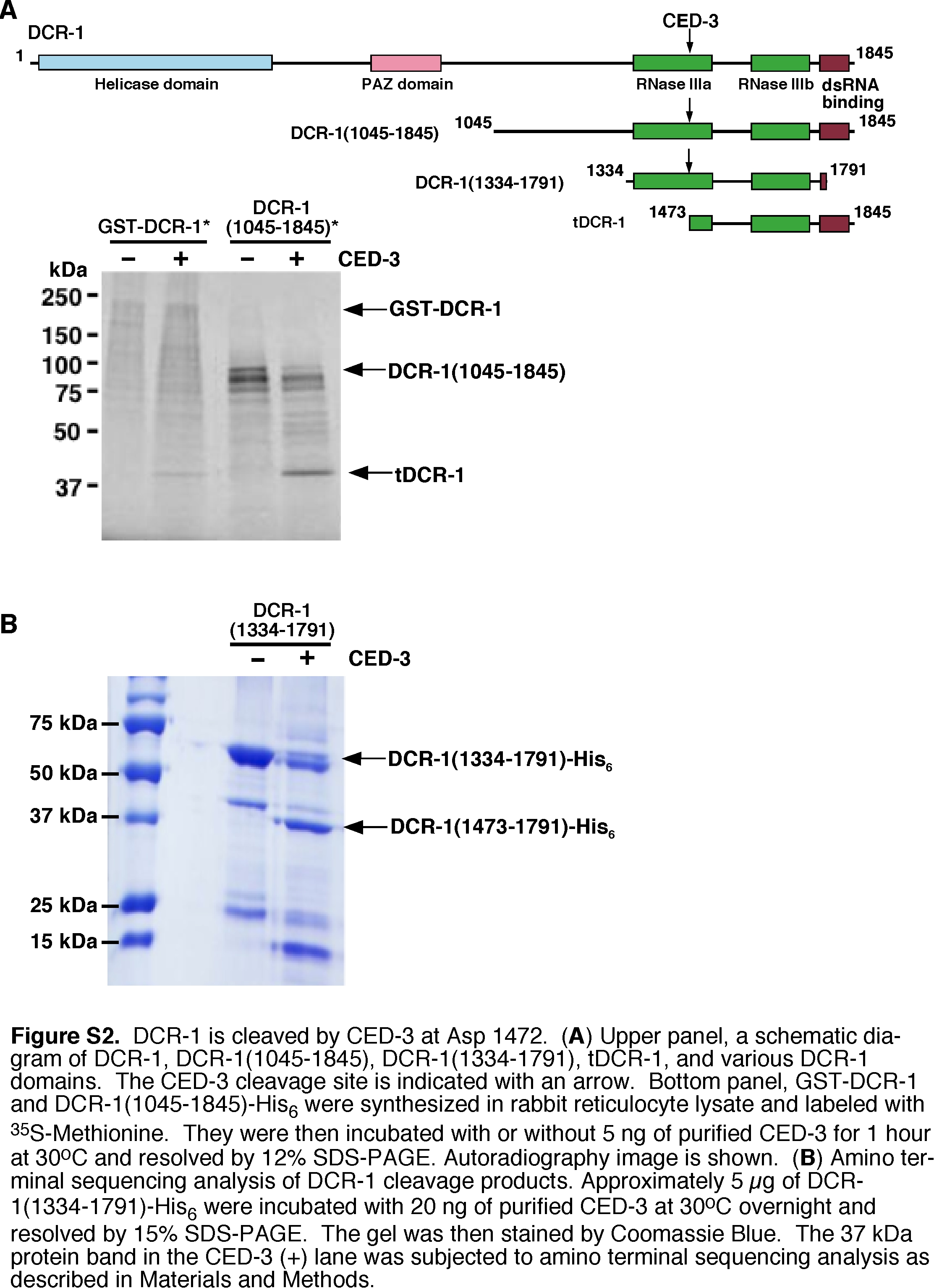
Given that dcr-1 acts downstream of ced-3 to promote apoptosis and DNA degradation, we tested whether DCR-1 itself might be a substrate of the CED-3 protease. A glutathione S transferase DCR-1 fusion (GST-DCR-1) was synthesized and labeled with 35S-Methionine in rabbit reticulocyte lysate and incubated with purified CED-3 protease. 35S-Met-labeled GST-DCR-1 was cleaved by CED-3, yielding two primary cleavage products of approximately 193 kD and 43 kD (Fig. 3A, lanes 1 and 2). A DCR-1 deletion mutant, DCR-1(1045–1845), was also cleaved by CED-3 to yield the same 43-kD cleavage product (Fig. S2A), suggesting that CED-3 cleavage occurs at the carboxyl terminus of DCR-1. To determine the CED-3 cleavage site in DCR-1, we expressed and purified a truncated DCR-1 protein with a six-Histidine tag, DCR-1(1334–1791)-His6, and incubated the purified protein with the CED-3 protease. The ~37-kD C-terminal DCR-1 cleavage product was subjected to amino terminal sequencing analysis (Fig. S2B)(48). Residues 1473 to 1477 of DCR-1 (GIETI) were found to be the N-terminus of this DCR-1 cleavage product, suggesting that DCR-1 was cleaved by CED-3 between Asp 1472 and Gly 1473. When Asp 1472 of DCR-1 was replaced by Glu through site-directed mutagenesis, the resulting mutant protein GST-DCR-1(D1472E) was not cleaved by CED-3 (Fig. 3A, lanes 3 and 4), confirming that Asp 1472 is the CED-3 cleavage site. Thus DCR-1 is a substrate of CED-3 in vitro.
Fig. 3.

Activation of a deoxyribonuclease activity after cleavage of DCR-1 by CED-3. (A) Cleavage of DCR-1 at Asp 1472. Upper panel, a schematic diagram of DCR-1, tDCR-1, tDCR-1(del), and various DCR-1 domains. The CED-3 cleavage site is indicated. Bottom panel, cleavage of DCR-1 by CED-3 at Asp 1472 abolished the dsRNA dicing activity of DCR-1. Lanes 1–4, GST-DCR-1 and GST-DCR-1(D1472E) were synthesized in rabbit reticulocyte lysate and labeled with 35S-Methionine. They were then incubated with or without 5 ng of purified CED-3 for 1 hour and resolved by 12% SDS-PAGE. Lanes 5–8, GST-DCR-1 was synthesized in rabbit reticulocyte lysate and purified by Glutathione Sepharose resins. Purified GST-DCR-1 was first incubated with or without 5 ng purified CED-3 for 2 hours before adding α-32P UTP labeled dsRNA probes. Purified fly Dicer-2 protein was included as a positive control. Samples were resolved by 20% native polyacrylamide gel. Autoradiography image is shown. (B) Activation of DNase activity after cleavage of DCR-1 by CED-3. GST-DCR-1 and GST-DCR-1(D1472E) were synthesized and purified as in (A) in the presence of both Methionine and 35S-Methionine (50:1) and shown in lanes 10 and 11. pUC19 plasmid DNA (200 ng) was incubated with purified GST-DCR-1 or GST-DCR-1(D1472E) or both (lane 6) in the presence or absence of 5 ng purified CED-3 for 1 hour at 37°C (48). Twice amount of GST-DCR-1(D1472E) was added in lane 6. One half of the samples was resolved on 1% agarose gel and then stained with ethidium bromide before a UV image was taken (upper panel). 2 units of TdT, α-32P dCTP, and CoCl2 (250 µM) were added to the remaining reactions and incubated for another 2 hours at 37°C. The samples were resolved on 1% agarose gel, fixed and dried for autoradiography (Bottom panel). In lane 9, only one fourth of the sample was loaded. 1 Kb Plus DNA ladder was used (Invitrogen). (C) DNase activity of tDCR-1. tDCR-1-Flag and tDCR-1(del)-Flag were expressed in C. elegans, purified on an anti-Flag affinity column, eluted by Flag peptides (48), and resolved on 15% SDS-PAGE, before subjected to silver staining (lanes 1 and 2). The nuclease activity assay and TdT labeling were done as in (B) with two different plasmids (pUC19 and pGEX-5X1). The TdT labeling methodology is expected to more efficiently label a linear substrate versus a nicked substrate.
Cleavage of DCR-1 by CED-3 destroys the dsRNA dicing activity but activates a DNase activity
Full-length DCR-1 contains a Helicase domain at its N-terminus, a PAZ domain in the middle, and two RNase III-like domains (indicated as RNase IIIa and RNase IIIb, respectively) and a dsRNA-binding domain at the C-terminus (Fig. 3A). Cleavage of DCR-1 by CED-3 at Asp 1472, which is in the middle of the RNase IIIa domain, presumably would destroy the RNase IIIa domain but leave RNase IIIb unaffected. Consistent with the findings that both RNase III domains of Dicer are required for binding and cleaving dsRNA (29, 41), CED-3 cleavage of GST-DCR-1 abolished the 22 to 23 nucleotide dicing activity of DCR-1 on dsRNA (Fig. 3A, lanes 5–8). However, the C-terminal cleavage product of DCR-1, which we named tDCR-1, still contains one intact RNase III domain (RNase IIIb) that may retain some endonuclease activity. Given that DCR-1 is also involved in mediating apoptotic DNA degradation, we examined whether DCR-1 or its CED-3 cleavage products might possess a deoxyribonuclease activity. GST-DCR-1 and GST-DCR-1(D1472E) synthesized in rabbit reticulocyte lysate were affinity purified using Glutathione Sepharose resins and incubated with pUC19 supercoiled plasmid DNA substrate in the presence or absence of the CED-3 protease (48). Purified CED-3 protease or GST-DCR-1 alone did not cleave or nick plasmid DNA (Fig. 3B, upper panel, lanes 1–3). Interestingly, incubation of CED-3 with GST-DCR-1 resulted in nicking of the plasmid DNA and the mobility shift of the plasmid DNA from the supercoiled form (SC) to the nicked open circle form (Fig. 3B, upper panel, lane 4). This plasmid DNA nicking activity required the presence of Mg2+ and was not observed when GST-DCR-1(D1472E) was incubated with CED-3 (Fig. 3B, upper panel, lanes 5, 7, 8), indicating that CED-3 cleavage is required to activate this new Mg2+-dependent deoxyribonuclease activity of DCR-1. Incubation of GST-DCR-1(D1472E) with GST-DCR-1 and CED-3 did not prevent the activation of this DNase activity by CED-3 (Fig. 3B, upper panel, lane 6). Moreover, when the CED-3 cleavage site in DCR-1 was replaced by a site (ENLYFQG) only recognized by the Tobacco Etch Virus (TEV) protease (Fig. S3A), the resulting DCR-1 mutant, GST-DCR-1(1472TEV), was not activated by CED-3 to generate a DNase activity (Fig. S3B, lanes 1–4). By contrast, purified TEV protease cleaved GST-DCR-1(1472TEV) but not GST-DCR-1 to activate a similar DNase activity (Fig. S3A, Fig. S3B, lanes 5–10). Together, these results indicate that this CED-3-activated DNase activity is derived from cleavage of DCR-1 at Asp 1472 rather than from a protein co-purified with DCR-1 and activated by CED-3 cleavage.
Since the RNase III enzyme is known to generate 3’ OH ends (30, 39) and DCR-1 is implicated in generating 3’ OH DNA breaks in C. elegans apoptotic cells that are labeled by TUNEL (Fig. 1), we tested whether CED-3-activated DCR-1 produced 3’ OH ends in vitro that could be labeled by α-32P-dCTP catalyzed by terminal deoxynucleotidyl transferase (TdT). BamHI, a restriction endonuclease, completely linearized plasmid DNA and generated 3’ OH DNA ends that were strongly labeled by TdT (Fig. 3B, bottom panel, lane 9). GST-DCR-1 but not GST-DCR-1(D1472E), when treated with CED-3, generated both nicked and linearized plasmid DNAs that were labeled by TdT in a Mg2+-dependent manner (Fig. 3B, bottom panel, lanes 3–8). This result indicates that cleavage of DCR-1 by CED-3 activates a deoxyribonuclease activity that can nick DNA and create 3’ OH ends.
tDCR-1 has the DNase activity
We further investigated whether tDCR-1, a CED-3 cleavage product that retains a complete RNase IIIb domain and a dsRNA-binding domain, is responsible for the newly generated DNase activity. tDCR-1 tagged with a Flag epitope was expressed in C. elegans under the control of heat-shock promoters. A mutant tDCR-1, in which one half of the RNase IIIb domain is deleted [tDCR-1(del)], was similarly expressed in C. elegans. After heat-shock induction, tDCR-1-FLAG and tDCR-1(del)-FLAG were purified from worm lysate on an anti-Flag affinity column (Fig. 3C, lanes 1–2)(48) and tested for the DNase activity. tDCR-1 nicked supercoiled plasmid DNA and caused a shift of some of the DNA to the nicked open circle form, whereas tDCR-1(del)-FLAG failed to do so (Fig. 3C, upper panel, lanes 3–5). TdT labeling indicated that tDCR-1 generated both nicked and linearized plasmid DNAs with 3’ OH ends (Fig. 3C, bottom panel, lane 4), whereas tDCR-1(del) did not nick or linearize plasmid DNA. We observed similar results with two other plasmids with different sizes (Fig. 3C, lanes 6–8 and Fig. S3C), indicating that the nicking activity of tDCR-1 may be relatively sequence non-specific. Thus tDCR-1 functions as a DNase to nick DNA and generate 3’ OH ends.
Cleavage of DCR-1 by CED-3 is important for its pro-apoptotic activity
We tested whether cleavage of DCR-1 by CED-3 is required for promoting cell death in C. elegans. Expression of DCR-1 under the control of the endogenous dcr-1 promoter (Pdcr-1DCR-1) fully rescued the reduced-embryonic-cell-corpse phenotype of dcr-1(ok247) animals (Fig. 4A). This construct also rescued the abnormal vulva phenotype (bursting vulva and protruding vulva) of dcr-1(ok247) animals (Table S3), which is due to a defect in biogenesis of miRNAs (21). We were unable to examine rescue of the RNAi defect of dcr-1(ok247) animals, because dcr-1(ok247) homozygous animals derived from dcr-1(ok247)/+ heterozygous mothers show no RNAi defect due to maternal rescue of dcr-1 (21). Pdcr-1DCR-1(D1472E), which drives the expression of the CED-3-resistant DCR-1 mutant, rescued the vulva defects of dcr-1(ok247) animals but failed to rescue the reduced-embryonic-cell-corpse phenotype of dcr-1(ok247) animals (Table S3 and Fig. 4B), indicating that cleavage of DCR-1 by CED-3 is critical for promoting cell death but is not needed for biogenesis of miRNAs. On the other hand, expression of tDCR-1 under the control of the dcr-1 promoter (Pdcr-1tDCR-1) fully rescued the cell death defect of dcr-1(ok247) animals but did not rescue their vulva defects (Fig. 4C and Table S3), confirming that tDCR-1 is sufficient to mediate DCR-1’s pro-apoptotic function.
Fig. 4.

Requirement of DCR-1 cleavage by CED-3 for its pro-apoptotic activity. Cell corpses were scored in the following animals: (A) N2, dcr-1(ok247), dcr-1(ok247); ex[Pdcr-1DCR-1] arrays #1–3, (B) N2, dcr-1(ok247), dcr-1(ok247); ex[Pdcr-1DCR-1(D1472E)] arrays #1–3, (C) N2, dcr-1(ok247), dcr-1(ok247); ex[Pdcr-1tDCR-1] arrays #1–3, (D) N2, dcr-1(ok247), dcr-1(ok247); ex[Pdcr-1DCR-1(E1E2)] arrays #1–3, (E) N2, dcr-1(ok247), dcr-1(ok247); ex[Pdcr-1DCR-1(E1D1E2)] arrays #1–3, (F) ced-1(e1735), ced-1(e1735); ex[PhsptDCR-1] arrays #1–3, (G) ced-1(e1735), ced-1(e1735); ex[PhsptDCR-1(del)] arrays #1–3. Stages of embryos examined were: comma, 1.5-fold, 2-fold, 2.5-fold, 3-fold, and 4-fold. The y axis represents average number of cell corpses scored and error bars represent S.D. 15 embryos were counted for each developmental stage. The significance of differences between different genetic backgrounds was determined by two-way ANOVA, followed by Bonferroni comparison. *, P < 0.001; **, P < 0.05. All other points had P values > 0.05.
We generated three integrated transgenes, smIs182, smIs196 and smIs201, which carry Pdcr-1DCR-1, Pdcr-1DCR-1(D1742E), and Pdcr-1tDCR-1, respectively. These transgenes were crossed into the cps-6(sm116); dcr-1(ok247) mutant and their ability to rescue the DNA degradation defect of the dcr-1(ok247) mutant was assessed. Both smIs182 (Pdcr-1DCR-1) and smIs201 (Pdcr-1tDCR-1) restored the number of TUNEL-stained cells in cps-6(sm116); dcr-1(ok247) embryos to the level seen in cps-6(sm116) embryos alone (Fig. 1E), suggesting that both transgenes rescue the DNA degradation defect of the dcr-1(ok247) mutant. In contrast, cps-6(sm116); dcr-1(ok247); smIs196 embryos had few TUNEL-stained cells (Fig. 1E), indicating that the Pdcr-1DCR-1(D1742E) transgene was unable to rescue the DNA degradation defect of the dcr-1(ok247) mutant. These results further indicate that tDCR-1 is sufficient to replace DCR-1 in promoting DNA degradation and apoptosis, whereas the CED-3-resistant DCR-1 mutant is not.
We also induced global overexpression of tDCR-1 under the control of the heat-shock promoters (PhsptDCR-1) and examined whether tDCR-1 could induce ectopic apoptosis. When expressed in the ced-1(e1735) mutant that is defective in engulfment of apoptotic cells and thus allows easy detection of increased apoptosis, tDCR-1 caused an increase of approximately 45 to 50% in the number of apoptotic cells (Fig. 4F), whereas the DNase-defective tDCR-1(del) mutant had no such activity (Fig. 4G). Interestingly, in the strong ced-3(n2433) mutant that is deficient in cell death, tDCR-1 overexpression failed to induce any apoptosis (Table S4), possibly because tDCR-1 needs to cooperate with other CED-3-activated cell death pathways to induce apoptosis. Yet, overexpression of tDCR-1 but not tDCR-1(del) still created a significant number of TUNEL-stained cells (Fig.1G), indicating that tDCR-1 can initiate 3’ OH nicks in chromosome DNA independent of CED-3. Together, these results indicate that cleavage of DCR-1 by CED-3 during apoptosis is critical for its role in promoting apoptosis and produces a cleavage product, tDCR-1, that initiates apoptotic DNA degradation through its DNase activity.
Conserved acidic residues are important for both DCR-1 cleavage of RNA and tDCR-1 cleavage of DNA
Because DCR-1 is a dsRNA processing enzyme and the yeast RNA III, Rnt1p, can cleave the DNA strand of DNA-RNA hybrids in vitro (53), tDCR-1 may cleave DNA through the same catalytic mechanism used for dsRNA cleavage. We thus examined whether catalytic residues that are critical for Dicer’s ribonuclease activity are also important for tDCR-1’s DNase activity (29, 41). Structural analysis of Giardia Dicer reveals four highly conserved acidic residues that are involved in binding and positioning of the two catalytic divalent metal cations in each RNase III domain (Fig. 5A; they are named E1, D1, D2 and E2 for simplicity)(41). The D1 and E2 residues in both RNase III domains have been shown to be important for the dicing activity of human dicer (29). We generated two DCR-1 mutants with altered acidic residues in the RNase IIIb domain, the E1E2 double mutant (E1617A, E1729A) and the E1D1E2 triple mutant (E1617A, D1621A, E1729A). We then assayed their dsRNA dicing activity and CED-3-activated DNase activity. The DCR-1(E1E2) mutant showed significantly reduced dsRNA dicing activity, which was dependent on the presence of Mg2+ (Fig. 5B, lanes 1–4). The CED-3-activated DNase activity of DCR-1(E1E2) was also impaired (Fig. 5C, lanes 1–4). In comparison, no detectable dsRNA dicing activity or CED-3-activated DNase activity was observed with the DCR-1(E1D1E2) triple mutant (Fig. 5B, lane 5 and Fig. 5C, lanes 5–6). These results and the observations that the dsRNA dicing activity and the CED-3-activated DNase activity of DCR-1 were both dependent on Mg2+ indicate that the catalytic mechanism of DNA cleavage by tDCR-1 likely is similar to that for dsRNA cleavage by DCR-1. They also provide further evidence that the CED-3-activated DNase activity comes from Dicer.
Fig. 5.

Catalytic residues important for DCR-1’s dsRNA dicing activity and tDCR-1’s DNase activity. (A) Alignment of conserved regions of Aquifex aeolicus (Aa) RNase III and RNase IIIa and RNase IIIb domains of Giardia intestinalis (Gi), Homo sapiens (Hu), and C. elegans (Ce) dicer nucleases. Highly conserved catalytic acidic residues are highlighted in red. (B) Catalytic residues important for DCR-1’s dsRNA dicing activity. The dsRNA dicing assay was done as in Fig. 3A. Wild-type (WT) and mutant GST-DCR-1 proteins (E1E2 and E1D1E2), in which the indicated acidic residues were replaced by Alanine, were synthesized and purified as in Fig. 3B. Purified GST-DCR-1 proteins were shown in the right panel. (C) Catalytic residues important for tDCR-1’s DNase activity. The DNase activity assay on the pUC19 plasmid and TdT labeling were done as in Fig. 3B.
We also tested whether these catalytic residues are important for DCR-1’s functions in vivo. Consistent with its reduced RNase and DNase activities in vitro, DCR-1(E1E2) expressed under the control of the dcr-1 promoter [Pdcr-1DCR-1(E1E2)] partially rescued the vulva defects, the reduced-embryonic-cell-corpse defect, and the DNA degradation defect of dcr-1(ok247) animals (Table S3, Fig. 1F, and Fig. 4D). On the other hand, expression of the DCR-1(E1D1E2) mutant [Pdcr-1DCR-1(E1D1E2)], which had no detectable RNase or DNase activity in vitro (Fig. 5B, C), failed to rescue all three defects of dcr-1(ok247) animals that we assayed (Table S3, Fig. 1F, and Fig. 4E). Thus, the in vitro dsRNA dicing activity and the CED-3-activated DNase activity of two DCR-1 mutants correlate with their abilities to promote small RNA processing and apoptosis in C. elegans.
Discussion
The Dicer ribonuclease plays a critical role in processing dsRNA substrates into short dsRNA species such as siRNAs and miRNAs with diverse regulatory functions (30, 39, 40). Our findings that C. elegans DCR-1 is involved in generating TUNEL-reactive DNA breaks in apoptotic cells that are later resolved by downstream apoptotic nucleases such as CPS-6 and NUC-1 and that DCR-1 promotes, and even is required for, apoptosis in sensitized genetic backgrounds reveal an unexpected role of DCR-1 in apoptosis and in initiating apoptotic DNA degradation. The observations that inactivation of crucial DCR-1 co-factors in generating siRNAs, miRNAs, endo-siRNAs, and other small RNAs did not affect either apoptotic DNA degradation or apoptosis indicate that DCR-1 likely promotes apoptosis through a distinct mechanism that is not related to dsRNA processing and gene silencing.
Dicer is a member of the RNase III family that recognizes and cleaves dsRNA substrates to various lengths and is not known to contain a deoxyribonuclease activity (39, 40). All RNase III family enzymes contain at least one characteristic RNase III domain and most also have a dsRNA-binding domain (dsRBD). Some, like Dicer, contain two RNase III domains and several additional domains, including the PAZ domain and the DExH helicase domain. Biochemical and structural analyses of the Dicer enzymes indicate that two RNase III domains form an intramolecular dimer, which constitutes a single processing center, with each domain contributing to the hydrolysis of one RNA strand of the duplex substrate through several highly conserved acidic residues that bind and position two catalytic Mg2+ cations (29, 41). Interestingly, CED-3 cleaves C. elegans DCR-1 in the middle of its first RNase III domain, yielding a C-terminal cleavage product (tDCR-1) with only one complete RNase III domain, and presumably, a disabled dsRNA processing center. Indeed, cleavage of DCR-1 by CED-3 abolishes the dsRNA dicing activity of DCR-1. However, cleavage of DCR-1 by CED-3 appears to also activate a deoxyribonuclease activity that can nick and linearize DNA to generate 3’ OH DNA ends through the same highly conserved catalytic residues and Mg2+ cations and this DNase activity is not observed in full-length DCR-1. The observations that tDCR-1 similarly nicked and linearized DNA in vitro and fully rescued the cell death defects of the dcr-1(lf) mutant when expressed under the control of the dcr-1 promoter and the findings that expression of DCR-1(D1472E), the CED-3-resistant mutant, fully rescued the developmental defects of the dcr-1(lf) mutant but failed to rescue the cell death defects of the dcr-1(lf) mutant indicate that cleavage of the DCR-1 ribonuclease by CED-3 activates a DNase activity that is necessary and sufficient to promote DNA degradation and apoptosis and that the pro-apoptotic function of DCR-1 is separable from DCR-1’s developmental functions. The proteolytic mechanism we discovered, through which a ribonuclease is disabled and converted into a DNase, may have more general implications on regulation of RNases and DNases, RNA and DNA binding proteins, and their associated cellular functions.
In mammals, the apoptotic nuclease DFF40 is kept in check in normal cells by its cognate inhibitor DFF45 (5–8). During apoptosis, executor caspases such as caspase-3 and caspase-7 are activated to cleave a plethora of caspase targets, which lead to rapid cell disassembly. One of these targets is DFF45, which is cleaved and inactivated by caspases to unleash DFF40 and to initiate the chromosome fragmentation process. Intriguingly, no DFF40 or DFF45 homologues are found in C. elegans, in which other key cell death components are highly conserved, including apoptotic nucleases endoG and DNase II (4). We propose that tDCR-1 is a functional analog of DFF40 and that a conserved, caspase-mediated mechanism activates the apoptotic DNA degradation process in both C. elegans and mammals.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank T. Blumenthal, N. Pace, M. Yarus, L. He, X.D. Liu, and S. Marquez for comments, Y. Kohara for dcr-1 cDNA, Q.H. Liu for purified fly Dicer-2 protein, K. Morita for alg-1 RNAi construct, and members of the Xue lab for helpful discussions. This work was supported by a Burroughs Wellcome Fund Award (D.X.), a grant from MEXT of Japan (S.M.), and NIH grants R01 GM59083 and R01 GM79097 (D.X.).
Footnotes
Supporting Online Material
Materials and Methods
Figs. S1 to S3
Tables S1 to S4
References and Notes
References and Notes
- 1.Wyllie AH. Nature. 1980;284:555. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 2.Widlak P, Garrard WT. J. Cell Biochem. 2005;94:1078. doi: 10.1002/jcb.20409. [DOI] [PubMed] [Google Scholar]
- 3.Samejima K, Earnshaw WC. Nat. Rev. Mol. Cell Biol. 2005;6:677. doi: 10.1038/nrm1715. [DOI] [PubMed] [Google Scholar]
- 4.Parrish JZ, Xue D. Chromosoma. 2006;115:89. doi: 10.1007/s00412-005-0038-0. [DOI] [PubMed] [Google Scholar]
- 5.Liu X, Zou H, Slaughter C, Wang X. Cell. 1997;89:175. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, et al. Proc. Natl. Acad. Sci. U. S. A. 1998;95:8461. doi: 10.1073/pnas.95.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enari M, et al. Nature. 1998;391:43. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 8.Sakahira H, Enari M, Nagata S. Nature. 1998;391:96. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 9.Widlak P, Li P, Wang X, Garrard WT. J. Biol. Chem. 2000;275:8226. doi: 10.1074/jbc.275.11.8226. [DOI] [PubMed] [Google Scholar]
- 10.Durrieu F, et al. Curr. Biol. 2000;10:923. doi: 10.1016/s0960-9822(00)00620-5. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, et al. Proc. Natl. Acad. Sci. U. S. A. 1998;95:12480. doi: 10.1073/pnas.95.21.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawane K, et al. Nat. Immunol. 2003;4:138. doi: 10.1038/ni881. [DOI] [PubMed] [Google Scholar]
- 13.Li LY, Luo X, Wang X. Nature. 2001;412:95. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 14.Parrish J, et al. Nature. 2001;412:90. doi: 10.1038/35083608. [DOI] [PubMed] [Google Scholar]
- 15.Buttner S, et al. Mol. Cell. 2007;25:233. doi: 10.1016/j.molcel.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 16.Parrish JZ, Xue D. Mol. Cell. 2003;11:987. doi: 10.1016/s1097-2765(03)00095-9. [DOI] [PubMed] [Google Scholar]
- 17.Parrish JZ, Yang C, Shen B, Xue D. EMBO J. 2003;22:3451. doi: 10.1093/emboj/cdg320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu YC, Stanfield GM, Horvitz HR. Genes Dev. 2000;14:536. [PMC free article] [PubMed] [Google Scholar]
- 19.Hammond SM, Bernstein E, Beach D, Hannon GJ. Nature. 2000;404:293. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 20.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Nature. 2001;409:363. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 21.Grishok A, et al. Cell. 2001;106:23. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 22.Hutvagner G, et al. Science. 2001;293:834. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 23.Knight SW, Bass BL. Science. 2001;293:2269. doi: 10.1126/science.1062039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pham JW, Pellino JL, Lee YS, Carthew RW, Sontheimer EJ. Cell. 2004;117:83. doi: 10.1016/s0092-8674(04)00258-2. [DOI] [PubMed] [Google Scholar]
- 25.Lee YS, et al. Cell. 2004;117:69. doi: 10.1016/s0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- 26.Duchaine TF, et al. Cell. 2006;124:343. doi: 10.1016/j.cell.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 27.Elbashir SM, Lendeckel W, Tuschl T. Genes Dev. 2001;15:188. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W. EMBO J. 2002;21:5875. doi: 10.1093/emboj/cdf582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Cell. 2004;118:57. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 30.Carmell MA, Hannon GJ. Nat. Struct. Mol. Biol. 2004;11:214. doi: 10.1038/nsmb729. [DOI] [PubMed] [Google Scholar]
- 31.Liu Q, et al. Science. 2003;301:1921. doi: 10.1126/science.1088710. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, et al. Science. 2004;305:1437. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 33.Rivas FV, et al. Nat. Struct. Mol. Biol. 2005;12:340. doi: 10.1038/nsmb918. [DOI] [PubMed] [Google Scholar]
- 34.Carthew RW, Sontheimer EJ. Cell. 2009;136:642. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tabara H, Yigit E, Siomi H, Mello CC. Cell. 2002;109:861. doi: 10.1016/s0092-8674(02)00793-6. [DOI] [PubMed] [Google Scholar]
- 36.Tabara H, et al. Cell. 1999;99:123. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- 37.Simmer F, et al. Curr. Biol. 2002;12:1317. doi: 10.1016/s0960-9822(02)01041-2. [DOI] [PubMed] [Google Scholar]
- 38.Kennedy S, Wang D, Ruvkun G. Nature. 2004;427:645. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- 39.MacRae IJ, Doudna JA. Curr. Opin. Struct. Biol. 2007;17:138. doi: 10.1016/j.sbi.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Jaskiewicz L, Filipowicz W. Curr. Top. Microbiol. Immunol. 2008;320:77. doi: 10.1007/978-3-540-75157-1_4. [DOI] [PubMed] [Google Scholar]
- 41.Macrae IJ, et al. Science. 2006;311:195. doi: 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- 42.Mochizuki K, Fine NA, Fujisawa T, Gorovsky MA. Cell. 2002;110:689. doi: 10.1016/s0092-8674(02)00909-1. [DOI] [PubMed] [Google Scholar]
- 43.Volpe TA, et al. Science. 2002;297:1833. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 44.Bernstein E, et al. Nat. Genet. 2003;35:215. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 45.Baulcombe D. Nature. 2004;431:356. doi: 10.1038/nature02874. [DOI] [PubMed] [Google Scholar]
- 46.Hatfield SD, et al. Nature. 2005;435:974. doi: 10.1038/nature03816. [DOI] [PubMed] [Google Scholar]
- 47.Forstemann K, et al. PLoS Biol. 2005;3:e236. doi: 10.1371/journal.pbio.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Materials and Methods are available on Science Online.
- 49.Stanfield GM, Horvitz HR. Mol. Cell. 2000;5:423. doi: 10.1016/s1097-2765(00)80437-2. [DOI] [PubMed] [Google Scholar]
- 50.Ellis RE, Jacobson DM, Horvitz HR. Genetics. 1991;129:79. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kokel D, Li Y, Qin J, Xue D. Nat. Chem. Biol. 2006;2:338. doi: 10.1038/nchembio791. [DOI] [PubMed] [Google Scholar]
- 52.Pavelec DM, Lachowiec J, Duchaine TF, Smith HE, Kennedy S. Genetics. 2009;183:1283. doi: 10.1534/genetics.109.108134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lamontagne B, Hannoush RN, Damha MJ, Abou Elela S. J. Mol. Biol. 2004;338:401. doi: 10.1016/j.jmb.2004.02.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.