

Abstract
Preeclampsia is a systemic disease that results from placental defects and occurs in about 5–8% of pregnancies worldwide. Preeclampsia is a disease of many theories, wherein investigators put forward their favorite mechanistic ideas, each with a causal appeal for the pathogenesis of preeclampsia. In reality, the patho-mechanism of preeclampsia remains largely unknown. Preeclampsia, as diagnosed in patients today, is likely a heterogeneous collection of disease entities that share some common features but also exhibit important differences. Therefore, one single mechanism may never be found to explain all the variants of preeclampsia. Current research must focus on evaluating such diverse mechanisms, as well as the possible common effectors pathways. Here we provide a discussion of several possible mechanisms and putative theories proposed for preeclampsia, with particular emphasis on the recent discovery of a new genetic mouse model offering new opportunities to explore experimental therapies.
Introduction
Preeclampsia is a devastating pregnancy-associated disorder characterized by the onset of hypertension, proteinuria and edema. Despite intensive investigation, our current understanding of the pathophysiology is limited. Emergent delivery of the baby alleviates the maternal symptoms of preeclampsia, but also leads to increased risks of morbidity for the baby due to iatrogenic prematurity. It is estimated that about 15% of preterm births are due to preeclampsia. In screening for this disease, hypertension associated with pregnancy is a useful clinical feature, however, it is not a specific finding and is often confused with gestational hypertension. Preeclampsia affects about 5–8% of all pregnant women. Surprisingly, the incidence of preeclampsia has increased in recent years [1] and could be much higher in developing countries.
Recent speculations on the pathogenesis of preeclampsia are focused mainly on the maternal symptoms of preeclampsia. However, such attempts have failed to consider an important feature of this disease, except special cases (such as postpartum preeclampsia), preeclampsia is a pregnancy-induced disease that originates in the ‘hypoxic placenta’.
History of preeclampsia
Eclampsia has been recognized clinically since the time of Hippocrates. Two thousand years ago, Celsus described pregnancy-associated seizures that disappeared after delivery of the baby. Because these symptoms emerged without any warning signs, the condition was named ‘eclampsia’, the Greek word for ‘lightning’. In the mid 19th century, Rayer and Lever described the association of proteinuria with eclampsia [2, 3]. In 1884, Schedoff and Porockjakoff first observed the link between hypertension and eclampsia. Based on these early observations, physicians and scientists in 20th century began to observe that proteinuria and hypertension were strong predictive indicators for the onset of eclampsia. This prequel of eclampsia was termed pre-eclampsia [4].
Basic Pathology and Physiology of Preeclampsia
Hypertension
Hypertension in preeclampsia can lead to serious complications in both maternal and neonatal health. However, the etiology of hypertension in preeclampsia remains unclear. In normal human pregnancy, there is increased cardiac output with expanded circulatory volume along with a decrease in peripheral vascular resistance (Figure 1) [5, 6]. During normal human gestation, blood pressure is slightly decreased (with minimal changes in systolic pressure but with evident diastolic blood pressure drop) because of the dilation of maternal vessels (Figure 1) [6]. Such vessel dilation allows for fluid expansion in the mother and helps protect against placental hypoperfusion (Figure 1) [7]. However, in preeclamptic pregnancy, plasma volume is significantly decreased despite the presence of massive edema [5]. As a result, there is reduced systemic perfusion, which can lead to potential damage to the maternal organs and to the baby [8] (Figure 1).
Figure l. Patho-physiology of Hypertension in Preeclampsia.

When compared to normal pregnancy, preeclampsia is associated with constricted, high resistance vessels, lower plasma volume, high sensitivity to vasoactive substances, presence of auto-antibodies against angiotenein type I (AT1) receptor, and low plasma level of 2-ME. PRA, plasma rennin activity; 2-ME, 2-methoxyestradiol.
In preeclamptic women, plasma renin activity (PRA) is lower when compared to that of normal pregnant women [9] (Figure 1). Renin, a key enzyme in the renin-angiotensin system, acts as a volume sensor, and lower PRA has been associated with expansion of circulatory volume [10]. Does PRA suppression in preeclampsia simply suggest that preeclampsia is associated with volume-dependent hypertension? The answer is not clear at this point and more studies are required. In preeclampsia, increased vascular sensitivity for vasoactive substances, such as angiotensin II, is reported [11] (Figure 1). In addition, increasing number of studies suggest the presence of agonistic auto-antibodies to angiotensin receptor type I (AT(1)-AAs) in the sera of women with preeclampsia [12] (Figure 1). The injection of such AT(1)-AAs from preeclamptic women induces key features of preeclampsia such as hypertension, proteinuria, glomerular endotheliosis, placental abnormalities and embryonic defects in pregnant mice. Such symptoms in AT(1)-AAs-injected pregnant mice are attenuated with losartan, an AT1 receptor antagonist, or when neutralizing peptide against AT(1)-AAs is administered. This evidence demonstrates that despite the suppression of PRA in preeclampsia, activation of the angiotensin receptor might be key to understanding the mechanism/s of hypertension in preeclampsia. The underlying mechanisms that drive the production of AT(1)-AAs in preeclampsia are still unknown. In the 1980’s, several reports demonstrated efficacy of angiotensin-converting enzyme-inhibitor (captopril) in preeclamptic women, revealing significant improvement of hypertension [13, 14]. These findings suggest that preeclampsia-associated hypertension may result from an overactive angiotensin receptor signaling, or a vasoactive substance-induced vasoconstriction (Figure 1). Unfortunately, with respect to the angiotensin receptor-mediated vasoconstriction and hypertension in preeclampsia, AT1 receptor antagonists and ACE-inhibitors cannot be used in the clinic due to their serious teratogenic effects.
Remodeling of Spiral Artery/Acute Atherosis in the Placenta
In human placental development, cytotrophoblasts (CTB) differentiate into two different types of invasive trophoblasts: multinuclear syncytiotrophoblasts or extravillous trophoblasts (EVT) [15, 16]. Such differentiation plays a pivotal role in the establishment of utero-placental circulation, which occurs at around 12–13 weeks of gestation [17]. EVT subsequently invade into the uterine vasculature (endovascular invasion), and make direct contact with maternal blood [16]. It is speculated that such vascular remodeling via the invasion of trophoblasts, including replacement of smooth muscle layer of spiral arteries by trophoblast, results in vessels that are resistant to vasoactive-substances, thereby making such vessels independent from the control of maternal blood pressure regulation. Consequently, normal placental circulation is characterized by dilated vessels with low resistance [18]. In preeclampsia, however, such trophoblast invasion is shallow and the spiral arteries are not remodeled appropriately [19]. As a result, the placental circulation does not carry sufficient blood supply to meet the embryonic demand due to these high resistance/non-dilated vessels.
Acute atherosis is another prominent vascular alteration that is often observed in preeclampsia, and also in idiopathic intrauterine growth retardation [19, 20]. Such vasculopathy of the spiral arteries is defined by fibrinoid necrosis of the vessel wall, accumulation of lipid-laden macrophages, and a mononuclear perivascular infiltrate [21]. Interestingly, similar vascular lesions have been observed in the vessels of patients with autoimmune diseases such as lupus vasculopathy [22], and in renal, cardiac and hepatic transplant-graft rejection [21]. Immunofluorescence analysis reveals extensive vascular deposition of non-specific IgM and complement in these lesions [23]. Acute atherosis may contribute to the impairment of feto-placental circulation and efficient nutrient/gas exchange [24]. It is not clear whether such acute atherosis is the consequence of incomplete remodeling of vessels due to shallow invasion of trophoblasts.
Kidney alterations and proteinuria
Increased proteinuria during pregnancy is associated with poor outcome in preeclampsia. The kidney defect in preeclampsia is characterized by a distinct glomerular lesion known as ‘glomerular endotheliosis’, defined by an enlarged glomerular volume with swelling of endothelial cells and occlusion of capillary lumens. Mesangial cells of the glomeruli may also exhibit swelling. Such changes in the endothelium are unique to preeclamptic glomeruli and are not found in endothelium associated with other organs of preeclamptic women. Despite the presence of proteinuria, glomerular podocytes appear relatively normal. Furthermore, glomerular endotheliosis is not observed in glomeruli of patients with other hypertensive disorders. Glomerular endotheliosis may contribute to the increased proteinuria and decreased glomerular filtration rate observed in preeclampsia [25, 26].
Recent reports have suggested that circulating angiogenic factors in the blood of preeclamptic women are responsible for the emergence of glomerular endotheliosis. Among such molecules, the increased level of soluble-vascular endothelial growth factor (VEGF) type1 receptor (also known as soluble Fms-like tyrosine kinase-1, sFlt1) is believed to contribute to the onset of glomerular endotheliosis. sFlt1 (~100 kDa), a transcriptional variant of full-length Flt1, is a secreted protein without the transmembrane/cytoplasmic domains and serves as an endogenous inhibitor of angiogenesis. Interestingly, the injection of sFlt1 protein [25] and adenoviral-delivery of sFlt1 [26] induces glomerular endotheliosis-like lesion in mice and rats. Such lesions are also found in mice treated with VEGF-neutralizing antibody (VEGF-Ab) [25]. In addition, mice injected with VEGF-Ab and sFlt1 exhibit proteinuria and a decrease in podocyte expression of nephrin [25]. These findings suggest that depletion of VEGF in the glomeruli may be responsible for the glomerular endotheliosis and disruption of glomerular microvascular homeostasis [25]. In this regard, a recent report demonstrates that a human-specific splicing variant of VEGF type1 receptor (designated sFlt1-14) that is qualitatively different from the previously described soluble receptor (sFlt1) and functioning as a potent VEGF inhibitor [27]. sFlt1-14 is generated in a cell type-specific fashion, primarily in nonendothelial cells such as vascular smooth muscle cells. The expression of sFlt1-14 is elevated in the placenta of women with preeclampsia, specifically induced in abnormal clusters of degenerative syncytiotrophoblasts known as syncytial knots. More studies are required to know the implication of sFlt1-14 in preeclampsia. Nevertheless, it likely that the non-specific antibody detection methods to assay sFlt1 may have detected this novel human specific isoform (sFlt1-14) instead of sFlt1, in the many of the previously reported studies [28].
Pathogenesis of Preeclampsia
Placental hypoxia
Decreased blood circulation in the uteroplacental unit (possibly as a result of abnormal placentation) may partially explain the pathogenesis of preeclampsia [29, 30]. In 1939, Page demonstrated that relative ischemia in the placenta was associated with eclampsia. In fact, Lunell et al. described a 50% decrease in uteroplacental circulation in preeclamptic women. In support of this observation, in 1940, Ogden et al. described that a 50% reduction of uteroplacental circulation in dogs (by clamping descending aorta) resulted in about 25mm/Hg increase in blood pressure. Subsequently, several animal studies have validated the association between preeclampsia-like symptoms and reduced placental perfusion [31]. These studies indicate that the reduction in placental circulation is likely a fundamental factor in the onset of preeclampsia. It has been hypothesized that abnormal implantation in the first trimester of pregnancy [32] and/or defects in vascular remodeling [19] are major factors contributing to the reduction of placental circulation. However, the precise mechanisms leading to placental hypoxia in preeclampsia still remain unclear.
Placental Hypoxia in the Onset of Maternal Syndrome of Preeclampsia
How does placental hypoxia cause the maternal symptoms of preeclampsia? This important question is still an open debate. One theory is that reduced placental perfusion results in the production of numerous placenta-derived circulatory agents, which cause the maternal symptoms of preeclampsia.
Among these factors, sFlt1 is speculated to be an important candidate molecule associated with the pathogenesis of preeclampsia [33–36]. The underlying theory of how sFlt1 is involved in the pathogenesis of preeclampsia stems from the evidence from a research group, Vuorela and colleagues from Finland, that VEGF is bound to a circulating protein in the amniotic fluid and maternal serum [33]. In 2000, the same research group reported that sFlt1 is significantly elevated in the amniotic fluid of preeclamptic women [34]. This seminal work by Vuorela and colleagues was the first report implicating sFlt1 in preeclampsia. Subsequently, Susan Fisher and colleagues demonstrated that cytotrophoblasts from preeclamptic placentae exhibit higher levels of sFlt1 in vitro when compared to control cells from normal placentae [36]. In 2003, Sugimoto and colleagues demonstrated that the neutralization of VEGF with sFlt1 or anti-VEGF antibodies in mice leads to proteinuria [25]. Later in 2003, Maynard et al. reported that the concentration of sFlt1 in maternal serum of preeclamptic women was increased when compared to normal pregnant women, and that adenoviral delivery of sFlt1 in rats caused endotheliosis and hypertension in males, non-pregnant females and also pregnant female rats [26].
It is speculated that excess sFlt1 neutralizes both free-VEGF and free–placental growth factor (PlGF) in maternal circulation, resulting in endothelial damage and the onset of the clinical syndrome [25, 26]. Although the placenta is believed to be responsible for the excess sFlt1 production in preeclampsia, this has not been explicitly demonstrated due to the pan-specificity of the antibodies to Flt1 and sFlt1. Clinical studies have demonstrated that sFlt1 is elevated in blood of women with preeclampsia [28] and that PlGF concentration is suppressed in urine of woman with preeclampsia [28, 37, 38]. Interestingly, preeclampsia does not develop in all women with high sFlt1 or low PlGF levels, and furthermore can occur in some women with low sFlt1 and high PlGF levels [28]. Therefore, while the involvement of placenta-derived sFlt1 in the pathogenesis of preeclampsia is still being explored, the utility of sFlt1 and PlGF levels as diagnostic biomarkers for predicting preeclampsia is independently being evaluated. In the AT(1)-AA-induced preeclampsia-like disease in mice, elevation of sFlt1 is also found and speculated to contribute to the kidney glomerular endothelial damage, however, a connection to hypertension was not found [12]. Therefore, additional in-depth mechanistic studies are required to elucidate the contribution of sFlt1 and PlGF to the pathogenesis of preeclampsia.
Soluble endoglin (sEndoglin), which is also increased in maternal serum in preeclampsia, is another circulating factor which causes a preeclampsia-like phenotype in rodents [39]. It is speculated that adenoviral expression of sEndoglin induces a preeclampsia-like phenotype in rats via inhibition of angiogenesis and endothelial nitric oxide synthase (eNOS) activation. Such effects of adenoviral-delivered sEndoglin can be enhanced by the co-infection with sFlt1 expressing adenovirus [39], suggesting that sEndoglin may augment the effects of sFlt1. While such investigations may shed light on the pathogenesis of preeclampsia, it is important to consider that using an adenovirus system to deliver factors relies on its capacity to infect many organs throughout the body, including the liver. Adenoviral delivery leads to concentration of sFlt1 and sEndoglin that is substantially higher in these animal models when compared to physiological levels observed in preeclampsia [28, 40]. These data in the rats were analyzed using an enzyme-linked immunosorbent assay system specific for mouse sFlt1 and human sEndoglin in rats infected with adenovirus carrying cDNA for mouse sFlt1 and human sEndoglin. There was no evaluation of endogenous rat specific sFlt1 or sEndoglin in these animals. Additionally, increased viral infections may cause liver dysfunction in a non-specific manner and may contribute to the vascular dysfunction. Furthermore, the duration and change rate of exposure to these molecules may be different from physiological condition. Therefore these models need further characterization to determine their physiological relevance to human preeclampsia [26, 39].
Role of Hypoxia-Inducible Factors in Preeclampsia
Hypoxia inducible factors (HIFs) play a pivotal role in the regulation of tissue oxygen tension and gene expression. In particular, HIF-1α is the master regulator of oxygen homeostasis. The accumulation and increased activity of HIFs have been shown in the placentae of preeclamptic women [41]. HIFs may play important roles in the onset of preeclampsia via alteration in the expression of hypoxia target-genes, such as VEGF and sFlt1. In addition, Caniggia et al. have shown that accumulation of HIF-1α induces the elevation of transforming growth factor (TGF)-β3, and that the resultant expression of TGF-β3 suppresses trans-differentiation of trophoblasts [42]. Trophoblast invasion is a critical step for the remodeling of spiral arteries, which allows the placenta to supply enough blood for efficient nutrient/gas exchanges with the fetus. Together, these studies indicate that suppression of HIF-1α may serve as a possible therapeutic strategy for the treatment of preeclampsia.
Th1 Immunity and Natural Killer Cells in Preeclampsia
The maternal immune response against the fetus and placenta has also been suggested to play an important role in the pathogenesis of preeclampsia. In the maternal decidua, invading trophoblasts encounter maternal immune cells, mainly natural killer (NK) cells. NK cells are normally cleared by full term. However, in preeclampsia, NK cells remain active in the maternal decidua [43]. Such activation of NK cells might be responsible for Th1-predominant inflammatory response profiles observed in preeclampsia, such as increased interferon-γ and tumor necrosis factor-α levels. Such NK cell-derived Th1 cytokines may therefore play a role in the pathogenesis of preeclampsia, perhaps by inhibiting trophoblast invasion locally, and/or also by the induction of endothelial damage and inflammation systemically [43]. However, these potential mechanisms require further investigation.
Deficiency of Catechol-O-Methyltransferase/2-Methoxyestradiol in Preeclampsia
Catechol-O-methyltransferase (COMT), a well-studied candidate gene in psychiatric disorders such as schizophrenia, is a catabolic enzyme involved in the degradation of a number of bioactive molecules such as catecholamines and catecholestrogens. Estradiol is metabolized by cytochrome p450 and the resultant 17-hydroxyestradiol (one of the catecholestrogens) is a substrate for COMT. COMT converts 17-hydroxyestradiol into 2-methoxyestradiol (2-ME), as a rate-limiting step in estrogen breakdown (Figure 2). 2-ME inhibits HIF-1α by possibly destabilizing microtubules in trophoblast [44], and can act in some cases as an anti-angiogenic molecule. 2-ME is currently being evaluated as a new therapeutic agent in the treatment of cancer, and clinical trials with oral administration of 2-ME are underway. Interestingly, during pregnancy, the concentration of maternal circulatory 2-ME immediately increases [44] and peaks at term (18–96.21 nM between 37–40th week of pregnancy) [45]. In preeclampsia, the plasma level of 2-ME is suppressed [44].
Figure 2. The Role of COMT)/2-methoxyestradiol (2-ME in Pregnancy.
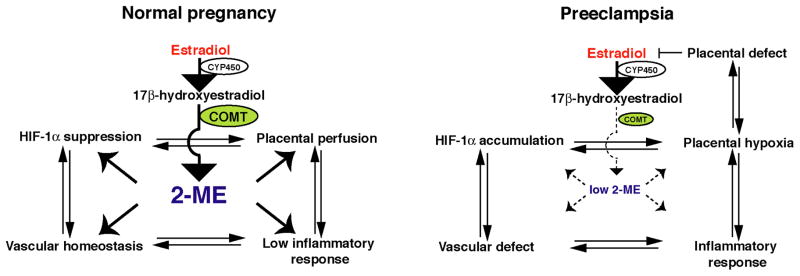
In normal pregnancy, 2-ME may play a role in regulating hypoxia-inducible factor (HIF)-1α in diverse ways. In preeclampsia, low COMT/2-ME may induce accumulation of HIF-1α, vascular defect, placental hypoxia and inflammatory responses in the placenta. Such response may induce placental defects and result in suppression of placental-derived estradiol and further reduction in 2-ME levels. CYP450, cytochrome 450.
Suppression of placental COMT in preeclampsia was first described in 1988 [46], however, the significance of this phenomenon was not examined until recently. It has been reported that COMT deficiency in mice is associated with placental hypoxia and preeclampsia-like symptoms [44]. COMT deficient mice (COMT−/−) display a preeclampsia-like phenotype, including pregnancy-induced hypertension with proteinuria and increased fetal wastage as a result of the absence in 2-ME. Interestingly, administration of exogenous 2-ME ameliorates hypertension, proteinuria, placental defects, fetal wastage, acute atherosis, and glomerular and placental endothelial damage in pregnant COMT−/− mice (Figure 2).
How does deficiency in COMT and 2-ME lead to the preeclampsia-like phenotype in mice? This may be linked to HIF-1α accumulation in the placenta of COMT−/− mice. When COMT is present, 2-ME acts to suppress HIF-1α accumulation and sFlt1 induction. In the COMT−/− mice, however, HIF-1α accumulation is associated with an increased inflammatory response and endothelial damage. In this regard, COMT−/− mice-treated with 2-ME showed a decrease in NK cells recruitment, interferon-γ production and endothelial damage (Figure 2). In addition, 2-ME can induce trophoblast invasion specifically under hypoxic conditions via suppression of HIF-1α–mediated TGF-β3 upregulation (unpublished data), suggesting that 2-ME may play an important role in maintaining placental homeostasis. This result is consistent with HIF-1α/TGF-β3 theory proposed by Caniggia et al [42]. Furthermore, 2-ME may directly function as a vasodilator in pregnant women [47, 48] (Figure 1).
The question remains how COMT might be suppressed in human preeclampsia. The activity of the COMT enzyme displays a tri-modal frequency distribution in human populations because of the presence of a functional polymorphism in the coding sequence [49]. This polymorphism (rs4680: G-A nucleotide substitution) results in a Val to Met amino acid substitution at position amino acid residue 158 [49]. The human COMTMet158 variant has a lower stability and exhibits a lower enzymatic activity at physiologic temperature [49]. The allelic frequency of this polymorphism is found to be about 25–30% of human population. This functional COMT polymorphism is associated with fetal growth restriction and abnormalities [50]. Furthermore, Nackley et al. reported that the combination of multiple single nucleotide polymorphisms (SNPs) in the COMT gene results in a significant decrease in COMT mRNA stability [51]. These findings suggest that the emergence of preeclampsia could be associated with genomic alterations in the COMT gene. However these theories have to be tested and many diverse SNP analyses should be conducted in multiple families in different population cohorts. Additionally, COMT deficiency may not explain all variants of the preeclampsia phenotype in humans; thus if one set of patient groups is found not to exhibit relevant COMT polymorphisms, it does not mean that all patients will follow this trend. Preeclampsia most likely emerges due to diverse patho-mechanisms and COMT may be relevant in only a select patient population. Additionally, COMT deficiency may not be due to SNPs alone but also may result from other transcriptional/translational control mechanisms leading to decreased protein levels.
With regard to COMT suppression, drugs currently used in the clinic for the treatment of preeclampsia should be evaluated carefully. Hydralazine is a well-known vasodilator and is a widely accepted drug for the treatment of preeclampsia. However, hydralazine has also been shown to inhibit placental COMT activity [52]. Importantly, some reports indicate that the administration of hydralazine is associated with placental abruption [53], which is a complication of the last half of pregnancy frequently associated with preeclampsia and often resulting in severe maternal and fetal morbidity and mortality. In light of recent findings regarding COMT/2-ME deficiency in preeclampsia, the COMT-suppressing activity of hydralazine needs careful reassessement. COMT suppression is also observed in placental explants following alpha-methyldopa administration, another antihypertensive drug that is used in the preeclamptic women [52]. Therefore, suppression of COMT/2-ME needs to be carefully evaluated for its connection with possible drug-exacerbated preeclampsia.
Perspective
Although decreased placental perfusion via poor placentation are likely key factors in the onset of preeclampsia, they are unlikely to be the only factors which lead to preeclampsia. For example, perfusion defects can also lead to fetal growth restriction even in the absence of preeclampsia. Furthermore, only one-third of babies born to preeclamptic women exhibit growth restriction even in the presence of placental defects [54]. Placental perfusion defects (vide supra) are often a consequence of abnormal implantation and shallow invasion of trophoblasts; however these findings are also found in the placentae of pregnant women with intrauterine growth restriction and preterm babies of non-preeclamptic women [55]. Preeclamptic placentae are sometimes larger than in normal pregnancy, and large babies in women with obesity and gestational diabetes have been associated with increased risk of preeclampsia [56]. Therefore, while decreased perfusion of the placenta is a key feature of preeclampsia, it is likely not sufficient to explain all symptoms associated with preeclampisa.
An interesting alternate posibility is the consideration of fetal/placental weight ratio (FP ratio) as an important determinant for the onset of preeclampsia. In fact, FP ratio is lower in fetal distress but significantly increased in preeclampsia when compared to normal pregnancy [57]. Experimental models suggested that an increased FP ratio is associated with a decreased placental blood supply and normal/increased demand of the embryo [58, 59]; consequently an imbalance emerges between embryonic demand and placental blood supply. Therefore, the efficiency of placental blood supply (not the size) may be important in the pathogenesis of preeclampsia. In this regard, pregnant COMT−/− mice exhibit increased FP ratio associated with placental hypoxia [44], similar to that observed in preeclamptic women.
In conclusion, more work is required to obtain new clinically useful biomarkers and to better understand the patho-mechanisms of this disease and the discovery of sFlt1-14 may shed novel insights into the pathogenesis of preeclampsia (Figure 3). It should be emphasized that the field of preeclampsia research should be cautious in not using biomarkers assessment in patients and preliminary rodent studies to derive conclusion regarding the pathogenesis of this disease.
Figure 3. Biology of preeclampsia.

Many different mechanisms have been reported for preeclampsia and they are listed here. NK cell, natural killer cell; 2-ME, 2-methoxyestradiol; HIFs, hypoxia-inducible factors; AT(1)-AAs, auto-antibodies for angiotensin receptor type I; VEGF, vascular endothelial growth factor; sEndoglin, soluble endoglin; sFlt1, soluble Fms-like tyrosine kinase-1.
Acknowledgments
The work in the authors’ laboratory is funded by grants from the NIH Grants (DK 55001, DK 62987, DK 13193, DK 61688) and research fund of the Division of Matrix Biology at the Beth Israel Deaconess Medical Center.
Footnotes
Disclosure
No conflict of interest is reported at the current time.
References
- 1.Martin J, Hamilton B, Sutton P, et al. Births: Final Data for 2006. National Vital Statistics Reports. 2009 [PubMed] [Google Scholar]
- 2.Lever JCW. Cases of puerperal convulsions, with remarks. Guy’s Hosp Reports. 1843:495–517. [Google Scholar]
- 3.Rayer P. Traité des Maladies des Reins et des Altérations Sécrétion Urinaire. Vol. 3. Paris: J.B. Baillière; pp. 1839–1841. [Google Scholar]
- 4.Cook H, Briggs J. Clinical observations on blood pressure. Johns Hopkins Hosp Rep. 1903;11:451–455. [Google Scholar]
- 5.Cope I. Plasma and blood volume changes in pregnancies complicated by pre-eclampsia. Journal of Obstetrics and Gynaecology of the British Commonwealth. 1961;68:413–416. doi: 10.1111/j.1471-0528.1961.tb02746.x. [DOI] [PubMed] [Google Scholar]
- 6.MacGillivray I, Rose GA, Rowe B. Blood pressure survey in pregnancy. Clin Sci. 1969;37:395–407. [PubMed] [Google Scholar]
- 7.Visser W, Wallenburg HC. Maternal and perinatal outcome of temporizing management in 254 consecutive patients with severe pre-eclampsia remote from term. Eur J Obstet Gynecol Reprod Biol. 1995;63:147–154. doi: 10.1016/0301-2115(95)02260-0. [DOI] [PubMed] [Google Scholar]
- 8.Redman CW. Maternal plasma volume and disorders of pregnancy. Br Med J (Clin Res Ed) 1984;288:955–956. doi: 10.1136/bmj.288.6422.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown MA, Zammit VC, Mitar DA, et al. Renin-aldosterone relationships in pregnancy-induced hypertension. Am J Hypertens. 1992;5:366–371. doi: 10.1093/ajh/5.6.366. [DOI] [PubMed] [Google Scholar]
- 10.Blumenfeld JD, Laragh JH. Management of hypertensive crises: the scientific basis for treatment decisions. Am J Hypertens. 2001;14:1154–1167. doi: 10.1016/s0895-7061(01)02245-2. [DOI] [PubMed] [Google Scholar]
- 11.Gant NF, Daley GL, Chand S, et al. A study of angiotensin II pressor response throughout primigravid pregnancy. J Clin Invest. 1973;52:2682–2689. doi: 10.1172/JCI107462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou CC, Zhang Y, Irani RA, et al. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med. 2008;14:855–862. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hurault de Ligny BH, Ryckelynck JP, Mintz P, et al. Captopril therapy in preeclampsia. Nephron. 1987;46:329–330. doi: 10.1159/000184380. [DOI] [PubMed] [Google Scholar]
- 14.Coen G, Cugini P, Gerlini G, et al. Successful treatment of long-lasting severe hypertension with captopril during a twin pregnancy. Nephron. 1985;40:498–500. doi: 10.1159/000183532. [DOI] [PubMed] [Google Scholar]
- 15.Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science. 1994;266:1508–1518. doi: 10.1126/science.7985020. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Y, Fisher SJ, Janatpour M, et al. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest. 1997;99:2139–2151. doi: 10.1172/JCI119387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jauniaux E, Hempstock J, Greenwold N, et al. Trophoblastic oxidative stress in relation to temporal and regional differences in maternal placental blood flow in normal and abnormal early pregnancies. Am J Pathol. 2003;162:115–125. doi: 10.1016/S0002-9440(10)63803-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karimu AL, Burton GJ. The effects of maternal vascular pressure on the dimensions of the placental capillaries. Br J Obstet Gynaecol. 1994;101:57–63. doi: 10.1111/j.1471-0528.1994.tb13011.x. [DOI] [PubMed] [Google Scholar]
- 19.Meekins JW, Pijnenborg R, Hanssens M, et al. A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies. Br J Obstet Gynaecol. 1994;101:669–674. doi: 10.1111/j.1471-0528.1994.tb13182.x. [DOI] [PubMed] [Google Scholar]
- 20.Zeek PM, Assali NS. Vascular changes in the decidua associated with eclamptogenic toxemia of pregnancy. Am J Clin Pathol. 1950;20:1099–1109. doi: 10.1093/ajcp/20.12.1099. [DOI] [PubMed] [Google Scholar]
- 21.Labarrere CA. Acute atherosis. A histopathological hallmark of immune aggression? Placenta. 1988;9:95–108. doi: 10.1016/0143-4004(88)90076-8. [DOI] [PubMed] [Google Scholar]
- 22.Sugimoto T, Kanasaki K, Morita Y, et al. Lupus vasculopathy combined with renal infarction: unusual manifestation of lupus nephritis. Intern Med. 2005;44:1185–1190. doi: 10.2169/internalmedicine.44.1185. [DOI] [PubMed] [Google Scholar]
- 23.Hustin J, Foidart JM, Lambotte R. Maternal vascular lesions in pre-eclampsia and intrauterine growth retardation: light microscopy and immunofluorescence. Placenta. 1983;4(Spec No):489–498. [PubMed] [Google Scholar]
- 24.Sebire NJ, Sepulveda W. Correlation of placental pathology with prenatal ultrasound findings. J Clin Pathol. 2008;61:1276–1284. doi: 10.1136/jcp.2008.055251. [DOI] [PubMed] [Google Scholar]
- 25.Sugimoto H, Hamano Y, Charytan D, et al. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt1) induces proteinuria. J Biol Chem. 2003;278:12605–12608. doi: 10.1074/jbc.C300012200. [DOI] [PubMed] [Google Scholar]
- 26.Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sela S, Itin A, Natanson-Yaron S, et al. A novel human-specific soluble vascular endothelial growth factor receptor 1: cell-type-specific splicing and implications to vascular endothelial growth factor homeostasis and preeclampsia. Circ Res. 2008;102:1566–1574. doi: 10.1161/CIRCRESAHA.108.171504. [DOI] [PubMed] [Google Scholar]
- 28.Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–683. doi: 10.1056/NEJMoa031884. Epub 2004 Feb 2005. [DOI] [PubMed] [Google Scholar]
- 29.Lunell NO, Nylund LE, Lewander R, et al. Uteroplacental blood flow in pre-eclampsia measurements with indium-113m and a computer-linked gamma camera. Clin Exp Hypertens B. 1982;1:105–117. doi: 10.3109/10641958209037184. [DOI] [PubMed] [Google Scholar]
- 30.Genbacev O, Joslin R, Damsky CH, et al. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J Clin Invest. 1996;97:540–550. doi: 10.1172/JCI118447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberts JM, Lain KY. Recent Insights into the pathogenesis of pre-eclampsia. Placenta. 2002;23:359–372. doi: 10.1053/plac.2002.0819. [DOI] [PubMed] [Google Scholar]
- 32.Redline RW, Patterson P. Pre-eclampsia is associated with an excess of proliferative immature intermediate trophoblast. Hum Pathol. 1995;26:594–600. doi: 10.1016/0046-8177(95)90162-0. [DOI] [PubMed] [Google Scholar]
- 33.Vuorela-Vepsalainen P, Alfthan H, Orpana A, et al. Vascular endothelial growth factor is bound in amniotic fluid and maternal serum. Hum Reprod. 1999;14:1346–1351. doi: 10.1093/humrep/14.5.1346. [DOI] [PubMed] [Google Scholar]
- 34.Vuorela P, Helske S, Hornig C, et al. Amniotic fluid--soluble vascular endothelial growth factor receptor-1 in preeclampsia. Obstet Gynecol. 2000;95:353–357. doi: 10.1016/s0029-7844(99)00565-7. [DOI] [PubMed] [Google Scholar]
- 35.Helske S, Vuorela P, Carpen O, et al. Expression of vascular endothelial growth factor receptors 1, 2 and 3 in placentas from normal and complicated pregnancies. Mol Hum Reprod. 2001;7:205–210. doi: 10.1093/molehr/7.2.205. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Y, McMaster M, Woo K, et al. Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am J Pathol. 2002;160:1405–1423. doi: 10.1016/S0002-9440(10)62567-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krauss T, Pauer HU, Augustin HG. Prospective analysis of placenta growth factor (PlGF) concentrations in the plasma of women with normal pregnancy and pregnancies complicated by preeclampsia. Hypertens Pregnancy. 2004;23:101–111. doi: 10.1081/PRG-120028286. [DOI] [PubMed] [Google Scholar]
- 38.Levine RJ, Thadhani R, Qian C, et al. Urinary placental growth factor and risk of preeclampsia. Jama. 2005;293:77–85. doi: 10.1001/jama.293.1.77. [DOI] [PubMed] [Google Scholar]
- 39.Venkatesha S, Toporsian M, Lam C, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 40.Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992–1005. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 41.Rajakumar A, Brandon HM, Daftary A, et al. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta. 2004;25:763–769. doi: 10.1016/j.placenta.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 42.Caniggia I, Mostachfi H, Winter J, et al. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3) J Clin Invest. 2000;105:577–587. doi: 10.1172/JCI8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308:1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 44.Kanasaki K, Palmsten K, Sugimoto H, et al. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature. 2008;453:1117–1121. doi: 10.1038/nature06951. [DOI] [PubMed] [Google Scholar]
- 45.Berg D, Sonsalla R, Kuss E. Concentrations of 2-methoxyoestrogens in human serum measured by a heterologous immunoassay with an 125I-labelled ligand. Acta Endocrinol (Copenh) 1983;103:282–288. doi: 10.1530/acta.0.1030282. [DOI] [PubMed] [Google Scholar]
- 46.Barnea ER, MacLusky NJ, DeCherney AH, et al. Catechol-o-methyl transferase activity in the human term placenta. Am J Perinatol. 1988;5:121–127. doi: 10.1055/s-2007-999669. [DOI] [PubMed] [Google Scholar]
- 47.Yan J, Chen C, Lei J, et al. 2-methoxyestradiol reduces cerebral vasospasm after 48 hours of experimental subarachnoid hemorrhage in rats. Exp Neurol. 2006;202:348–356. doi: 10.1016/j.expneurol.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 48.Gui Y, Zheng XL, Zheng J, et al. Inhibition of rat aortic smooth muscle contraction by 2-methoxyestradiol. Am J Physiol Heart Circ Physiol. 2008;295:H1935–1942. doi: 10.1152/ajpheart.00723.2008. [DOI] [PubMed] [Google Scholar]
- 49.Tunbridge EM, Harrison PJ, Weinberger DR. Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry. 2006;60:141–151. doi: 10.1016/j.biopsych.2005.10.024. Epub 2006 Feb 2014. [DOI] [PubMed] [Google Scholar]
- 50.Sata F, Yamada H, Suzuki K, et al. Functional maternal catechol-O-methyltransferase polymorphism and fetal growth restriction. Pharmacogenet Genomics. 2006;16:775–781. doi: 10.1097/01.fpc.0000230116.49452.c0. [DOI] [PubMed] [Google Scholar]
- 51.Nackley AG, Shabalina SA, Tchivileva IE, et al. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–1933. doi: 10.1126/science.1131262. [DOI] [PubMed] [Google Scholar]
- 52.Barnea ER, Fakih H, Oelsner G, et al. Effect of antihypertensive drugs on catechol-O-methyltransferase and monoamine oxidase activity in human term placental explants. Gynecol Obstet Invest. 1986;21:124–130. doi: 10.1159/000298941. [DOI] [PubMed] [Google Scholar]
- 53.Magee LA, Cham C, Waterman EJ, et al. Hydralazine for treatment of severe hypertension in pregnancy: meta-analysis. Bmj. 2003;327:955–960. doi: 10.1136/bmj.327.7421.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eskenazi B, Fenster L, Sidney S, et al. Fetal growth retardation in infants of multiparous and nulliparous women with preeclampsia. Am J Obstet Gynecol. 1993;169:1112–1118. doi: 10.1016/0002-9378(93)90265-k. [DOI] [PubMed] [Google Scholar]
- 55.Khong TY, De Wolf F, Robertson WB, et al. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br J Obstet Gynaecol. 1986;93:1049–1059. doi: 10.1111/j.1471-0528.1986.tb07830.x. [DOI] [PubMed] [Google Scholar]
- 56.Rosenberg TJ, Garbers S, Lipkind H, et al. Maternal obesity and diabetes as risk factors for adverse pregnancy outcomes: differences among 4 racial/ethnic groups. Am J Public Health. 2005;95:1545–1551. doi: 10.2105/AJPH.2005.065680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riss P, Bartl W. Placental function, fetal distress, and the fetal/placental weight ratio in normal and gestotic pregnancies. Int J Biol Res Pregnancy. 1982;3:10–13. [PubMed] [Google Scholar]
- 58.Constancia M, Hemberger M, Hughes J, et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417:945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- 59.Angiolini E, Fowden A, Coan P, et al. Regulation of placental efficiency for nutrient transport by imprinted genes. Placenta. 2006;27 (Suppl A):S98–102. doi: 10.1016/j.placenta.2005.12.008. [DOI] [PubMed] [Google Scholar]