

Abstract
Toll is a cell surface receptor with well described roles in the developmental patterning of invertebrates and innate immunity in adult Drosophila. Mammalian toll-like receptors represent a family of Toll orthologs that function in innate immunity by recognizing molecular motifs unique to pathogens or injured tissue. One member in this family of pattern recognition receptors, toll-like receptor 3 (TLR3), recognizes viral double-stranded RNA and host mRNA. We examined the expression and function of TLRs in the nervous system and found that TLR3 is expressed in the mouse central and peripheral nervous systems and is concentrated in the growth cones of neurons. Activation of TLR3 by the synthetic ligand polyinosine:polycytidylic acid (poly I:C) or by mRNA rapidly causes growth cone collapse and irreversibly inhibits neurite extension independent of nuclear factor κB. Mice lacking functional TLR3 were resistant to the neurodegenerative effects of poly I:C. Neonatal mice injected with poly I:C were found to have fewer axons exiting dorsal root ganglia and displayed related sensorimotor deficits. No effect of poly I:C was observed in mice lacking functional TLR3. Together, these findings provide evidence that an innate immune pattern recognition receptor functions autonomously in neurons to regulate axonal growth and advances a novel hypothesis that this class of receptors may contribute to injury and limited CNS regeneration.
Keywords: Toll-like receptor-3, axon, polyinosine:polycytidylic acid, poly I:C, RNA, CNS, danger theory
Introduction
Neurons in the adult CNS are able to grow new axons in permissive environments, but have a limited ability to regenerate axons in the context of CNS injury (Ramon y Cajal, 1928; Richardson et al., 1980; David and Aguayo, 1981, 1985). The regenerative failure of CNS axons can primarily be attributed to an inhibitory environment composed of glial-derived cues (Caroni and Schwab, 1988; Schwab and Caroni, 1988; Mukhopadhyay et al., 1994; Filbin, 1995; Schwab, 1996; Tang et al., 1997; Chen et al., 2000; GrandPre et al., 2000). In the intact CNS, these known inhibitory molecules appear to function principally as organizing and stabilizing factors for proper neuronal architecture. The fact that they impede axonal regeneration after injury raises the issue as to whether neurons can recognize signals specific to a damaged CNS landscape. That is, can cues found only in the context of active injury or cell death also discourage new axonal growth? Framed in this sense, the questions we ask regarding CNS regenerative failure are analogous to the questions raised in “danger theory” of immunity.
Danger theory suggests that immunity primarily functions to identify tissue injury or destruction rather than functioning to distinguish self from nonself (Matzinger, 1998). For example, danger theory is conceptually congruent with the absence of an immune response to commensal intestinal bacteria, but a vigorous response to pathogenic bacteria, whereas self–nonself theories struggle to explain such phenomena. Danger signals can be pathogen derived or host derived, sharing the common feature of locally produced molecules that activate resident innate immunity within tissues (Colaco, 1998; Heeg et al., 1998; Krieg et al., 1998; Matzinger, 1998; Stockwin et al., 2000; Smyth et al., 2001; Brown and Lillicrap, 2002; Vabulas et al., 2002; Wallin et al., 2002; Paterson et al., 2003; Bergmann-Leitner and Leitner, 2004). Toll-like receptors (TLRs) likely serve the major function of transducing danger signals. TLRs are a family of innate immune receptors that recognize a spectrum of protein, lipid-based, or nucleic acid motifs and in turn initiate inflammatory responses (Medzhitov and Janeway, 2000; Janeway and Medzhitov, 2002; Takeda and Akira, 2004; Beutler, 2004). The intense investigation of mammalian TLRs followed a discovery in Drosophila where loss-of-function of Toll in adult flies led to a defect in innate immunity and overwhelming fungal infection (Lemaitre et al., 1996). Yet, the original discovery of Toll was based on its function in establishing normal dorsal-ventral patterning of the Drosophila embryo (Anderson et al., 1985a,b). Later studies provided evidence that Toll expressed by muscle functions in axonal guidance of the RP3 and other motor neurons in Drosophila (Halfon et al., 1995; Rose et al., 1997; Rose and Chiba, 1999).
We thus considered the possibility that TLRs function in the mammalian nervous system to regulate axonal growth either during development or in the context of injury. We used a simple screening assay of axonal growth from DRG explants and found the TLR3 agonist poly I:C was a potent negative regulator of axonal growth. We also show mammalian mRNA, shown previously to activate immune cells through TLR3 (Kariko et al., 2004), similarly functions to inhibit axonal growth. The effect of poly I:C is dependent on the presence of functional TLR3 in neurons, and appears to be active at the growth cone because it causes profound growth cone collapse, but does not elicit cell death in sensory neurons. In wild-type but not TLR3−/− neonatal mice, administration of poly I:C into the CNS causes defects in achieving relevant sensory motor reflexes, and, corresponding loss of axons in dorsal roots. Localization and function for TLRs in microglia, the resident innate immune cell of the CNS, has been described previously (Laflamme et al., 2001; Nguyen et al., 2001; Dalpke et al., 2002; Lehnardt et al., 2002, 2003, 2006; Olson and Miller, 2004; Ebert et al., 2005; Jack et al., 2005; Jung et al., 2005; Kielian et al., 2005; Lotz et al., 2005; Su et al., 2005; Zhang et al., 2005; Andaloussi et al., 2006; Esen and Kielian, 2006; Glezer et al., 2006; Town et al., 2006). Previous reports have shown the presence and potential function of TLR3 in a human neuroblastoma cell line (Prehaud et al., 2005; Nessa et al., 2006), in primary mouse cortical neurons (Tang et al., 2007) and in human histopathologic tissue sections of the brain (Jackson et al., 2006). This is the first report of the functional significance of TLR3 in primary neurons in vitro and in vivo, as well as the subcellular localization of TLR3 in growth cones. Because injury to the nervous system can occur in the context of infection or following insults such as trauma or infarction, termed sterile injury, recognition of both infectious and sterile injury by neurons through a single receptor thus provides a novel mechanism whereby neurons “sense” danger.
Materials and Methods
Mice.
Swiss Webster and C57BL/6 mice were obtained from Taconic Farms (Germantown, NY) and Charles River Laboratories (Wilmington, MA), respectively. These mice were maintained and bred in the Harvard Institutes of Medicine animal housing facility under pathogen-free conditions. MyD88−/− mice, a kind gift from Dr. S. Akira (Osaka University, Osaka, Japan) via Dr. H. Plough (Harvard Medical School, Boston, MA) and Dr. D. Golenbock (University of Massachusetts Medical School, Worcester, MA) were used after five generations of back-crossing to the C57BL/6 background, with C57BL/6 mice serving as controls. TLR3−/− mice, a kind gift from Dr. R. Flavell (Yale University School of Medicine, New Haven, CT) were used after more than six generations of back-crossing to the C57BL/6 background, with C57BL/6 mice serving as controls (Alexopoulou et al., 2001).
Primary cell culture preparation.
DRG neurons were isolated from embryonic day 14 (E14) mouse embryos or E9 White Leghorn chick embryos, as indicated in the text. Ganglia were dissociated with trypsin (Invitrogen, Eugene, OR) for 20 min at 37°C, followed by inactivation with horse serum (Invitrogen) and trituration with a fire-polished pipette. Cells were plated on poly-d-lysine and laminin-coated culture slides (BD Biosciences, Franklin Lakes, NJ). For explant cultures, whole DRGs were plated directly on culture slides. Cells were grown in DMEM alone for acute studies of neurite outgrowth or DMEM supplemented with 2% heat-inactivated horse serum (Invitrogen), 1% penicillin/streptomycin (Invitrogen), 10 ng/ml nerve growth factor (NGF; R&D Systems, Minneapolis, MN), and N2 supplement (Invitrogen) for overnight cultures. Cortical and hippocampal mouse neurons were purified and cultured as described previously (Lehnardt et al., 2002, 2003). Cultures were free of macrophages, as determined by F4/80 immunofluorescence (data not shown). Cells were allowed to adhere for 30 min, after which media containing PBS, poly I:C (Amersham Biosciences, Arlington Heights, IL), poly dI:dC (Amersham Biosciences), or lipopolysaccharide (LPS; Escherichia coli) (Sigma, St. Louis, MO) was added. All reagents were purchased from Sigma unless noted.
Growth cone collapse.
Explant cultures were generated as described above from E9 chick embryos. After 24 h, culture media containing PBS or poly I:C was added to explants for 30 min. Explants were then immediately fixed and stained for neurofilament (Millipore, Temecula, CA) and f-actin (Invitrogen) as described below. To quantify the collapsing effects of poly I:C, the growth cones of all neurites extending outward from each explant, free of underlying glial cells, were scored morphologically as either collapsed or not collapsed. A collapsed growth cone was defined has having a tapered axonal terminal without spread lamellipodia (Cox et al., 1990), as visualized by neurofilament and f-actin staining. Data presented is the percentage of collapsed growth cones relative to the total number of growth cones scored.
Immunofluorescence microscopy.
Cells were fixed in 4% paraformaldehyde (PFA) in PBS or a solution of 4% PFA and 20% sucrose to preserve growth cone morphology. Embryonic immunohistochemical sections were obtained by drop-fixing whole E14 mouse embryos in 4% PFA overnight. Embryos were then cryoprotected in 30% sucrose and sectioned at 20 μm. For neonate histology, postnatal day 11 (P11) mouse pups were perfused intracardially with 4% PFA after anesthetization with Avertin. Brains and spinal cords from these animals were then cryoprotected in 30% sucrose overnight and sectioned at 20 μm. The following primary antibodies were used: mouse neurofilament mAb (1:300; Millipore), goat TLR3 polyclonal antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), mouse TLR3 monoclonal antibody (1:1000; Apotech, Epalinges, Switzerland), mouse NeuN mAB (1:100; Millipore), rabbit nuclear factor-κB (NF-κB) p65 polyclonal antibody (1:100; Santa Cruz Biotechnology), Isolectin IB4 (1:100; Invitrogen), and Alexa 488 or rhodamine phalloidin (1:20; Invitrogen). Relevant cy-3- (Jackson ImmunoResearch, West Grove, PA) or Alexa-488- (Invitrogen) conjugated secondary antibodies were used for visualization. 4,6,diamidino-2-phenylindole (DAPI) stain (Invitrogen) served to visualize nuclei. Immunofluorescent images were obtained using a Nikon (Tokyo, Japan) Eclipse 660 microscope with a Spot cooled CCD camera using Spot image software (version 3.2.4 for Macintosh). Confocal images were obtained using a Zeiss (Oberkochen, Germany) LSM 510 UV upright confocal with UV laser, three visible wavelength lasers, and META spectral emission detectors. For each well the total number of neurons with neurites was counted and normalized to the control well for each day of plating. A neurite was defined as a process extending from the cell body by at least one cell diameter plating density and cell survival was assayed by DAPI staining. Four fields were randomly chosen for each well and the average number of DAPI+ nuclei was determined.
TUNEL staining.
Terminal transferase dUTP nick-end labeling (TUNEL) was used to assay fragmented DNA in DRG neurons. TUNEL staining was performed using the manufacturer's protocol for in situ cell death detection (Roche, Welwyn Garden City, UK). As a positive control, some wells were treated with 1 U/ml DNase I for 30 min before fixation. In these wells, ∼100% of neurons were TUNEL positive (data not shown).
RT-PCR.
Total RNA was isolated from E14 mouse DRGs or brain with Trizol reagent (Invitrogen) and then reverse transcribed using SuperScript One-Step reverse transcription (RT)-PCR with Platinum Taq (Invitrogen) in a 50 μl reaction volume. After an initial incubation for 30 min at 50°C, 35 cycles were performed (95°C for 30 s, 60°C for 30 s, and 72°C for 30 s), followed by a final extension for 5 min at 72°C. In all experiments, amplification of β-actin was run in parallel as a positive control for RNA quality and quantity. Primers used were as follows: TLR3 (control DRG), sense TTGTCTTCTGCACGAACCTG, antisense GAGAAAGTGCTCTCGCTGGT; TLR3 (control and TLR3−/− brain), sense CAGGGATTGCACCCATAATC, antisense CCCGTTCCCAACTTTGTAGA; MyD88 (control and MyD88−/− brain), sense TCCCTGGACTCCTTCATGTT, antisense TGTCCTAGGGGGTCATCAAG.
Neonate injections and behavioral testing.
P4 neonatal mice were injected intrathecally with 3 μl of either PBS or poly I:C (2 mg/ml) at a final concentration of 3 μg/ml. Behavioral testing was performed at P5, P7, P9, and P11. In righting reflex studies, animals were positioned on their backs and the amount of time needed for each pup to return to an upright position on all paws was recorded in seconds. In negative geotaxis studies, animals were placed with their heads facing downward on an inclined plane with a 45° slope and the time needed for the pup to turn around with its head facing upward on the inclined plane was recorded. The maximum amount of time allowed for these tests was 30 s. Pups that did not complete the tasks were assigned the maximum score of 30 s.
Dorsal root quantification.
Dorsal roots were isolated at the lumbar 1–5 segments (L1–L5), put in separate vials, washed with PBS, and then postfixed with 1% osmium tetroxide. After postfixation, samples were washed in PBS, dehydrated in graded alcohol series and embedded in Epon. Sections were cut at 1 μm thickness and stained with alkaline toluidine blue. The sections were then observed under light microscope (Nikon Eclipse E600) and images were acquired using a digital color camera (Nikon Digital Sight DS-5M). These images were used to count the myelinated fibers in each root.
Results
In the present study, we used host- and pathogen-associated molecular motifs recognized by TLRs to investigate whether CNS axonal growth can be inhibited by cues specific to an injured CNS. Initially, we screened agonists for TLRs 2–9 by treatment of DRGs isolated from E9 chicks, which suggested that the TLR3 agonist, poly I:C, (Alexopoulou et al., 2001), dramatically inhibited axonal growth (Fig. 1 A). Poly I:C is a synthetic double-stranded RNA molecule used to simulate the signature of viral infection. In immune cells, poly I:C functions as a specific ligand for TLR3 at concentrations ranging from 25 to 100 μg/ml (Alexopoulou et al., 2001; Doyle et al., 2002, 2003; Jiang et al., 2003; Wietek et al., 2003; Yamamoto et al., 2003; Sarkar et al., 2004; Schmidt et al., 2004). We found that neurons were exceptionally sensitive to poly I:C at concentrations 1.0–20 μg/ml, which are lower than those typically used to activate immune cells. Overnight treatment with poly I:C led to an almost complete inhibition of neurite outgrowth in E9 chick DRG explants, as compared with treatment with PBS or poly dI:dC (Fig. 1 A). Similarly, application of 20 μg/ml of poly I:C for 4 h to dissociated DRG neurons isolated from E14 mouse embryos caused a marked decrease in the total number of neurons extending neurites in culture (Fig. 1 B,C, black bars). In addition, hippocampal neurons exposed to 20 μg/ml poly I:C for 12 or 24 h showed limited neurite outgrowth compared with vehicle controls (Fig. 1 G). Data indicate neurites ceased growing because there was no evidence of neurite retraction (Fig. 1 C,G). Neither the double-stranded DNA control, poly dI:dC, PBS alone, nor the TLR4 agonist LPS had any significant effect on neurite outgrowth of mouse DRGs (Fig. 1 B,C, black bars). To assess whether the observed effect was caused by cell death, we evaluated cell number and TUNEL positivity in these cultures. No change was observed in the number of DRG neurons in any treatment group as determined by DAPI staining (Fig. 1 C, gray bars). Furthermore, there was no increase in the number of TUNEL+ neurons after poly I:C exposure (data not shown). The inhibitory effect of poly I:C on neurite outgrowth was significant at a concentration of 1 μg/ml, and nearly maximal at 10 μg/ml (Fig. 1 F). These initial findings suggest that poly I:C inhibits neurite outgrowth, but has no effect on neuronal viability. This data also suggests that the potency of poly I:C in this functional bioassay of neurite extension is relatively greater than the potency of poly I:C observed in immunologic systems.
Figure 1.
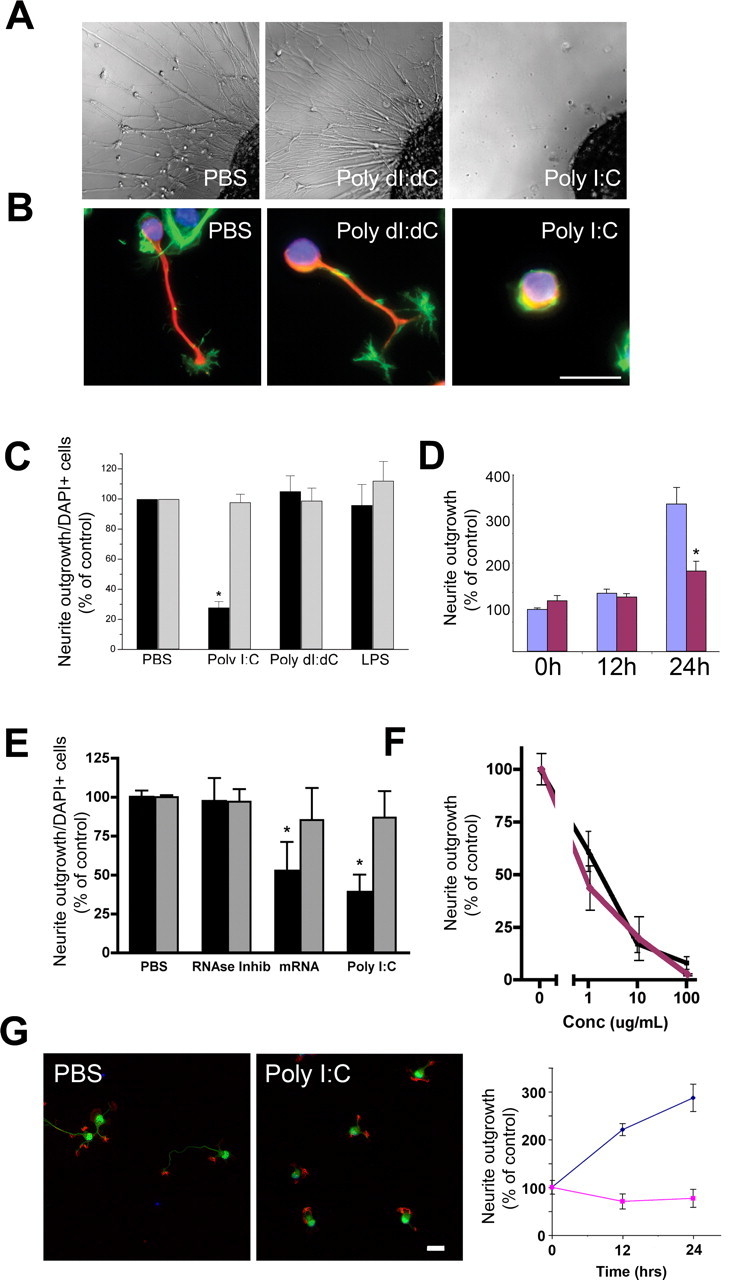
Poly I:C is a negative regulator of neurite outgrowth. A, Differential interference contrast images of E9 chick DRG explants cultured overnight in the presence of the TLR3 agonist poly I:C (20 μg/ml) or relevant controls, poly dI:dC (20 μg/ml), or PBS. B, Neurofilament (red) and f-actin (green) staining of E14 mouse sensory neurons cultured for 4 h in the presence of poly I:C, poly dI:dC, or PBS. Scale bar, 25 μm. C, Quantification of neurite outgrowth (black bars) and DAPI staining (gray bars) from B in percent for both measures. D, Quantification (percent) of neurite outgrowth of mouse DRGs exposed to poly I:C (20 μg/ml) (red bars) or vehicle control (purple bars) for 0, 12, or 24 h. Cells were grown for 1 d before exposure. E, Quantification of neurite outgrowth (black bars) and DAPI staining (gray bars) from mouse sensory neurons cultured for 4 h in the presence of poly I:C, poly(A)+ mRNA, RNase inhibitor, or PBS. F, Dose–response curve for poly I:C (black line) and RNA (purple line) effects on neurite outgrowth of mouse sensory neurons. G, Neurofilament (green) and f-actin (red) staining of E18 mouse hippocampal neurons cultured for 24 h in the presence of poly I:C (20 μg/ml) or PBS control. Scale bar, 25 μm. Graph at right represents time course of neurite length of hippocampal neurons exposed to poly I:C (purple line) or PBS (blue line). Error bars in this and subsequent figures represent SEM. *p < 0.01, Student's t test.
It has been reported previously that duplex structures within mammalian derived mRNA function as TLR3 agonists (Kariko et al., 2004). To determine whether this potential endogenous, host derived ligand for TLR3 acted as an inhibitor of axonal growth we assessed the activity of brain-derived mRNA in a neurite outgrowth assay. Mouse brain-derived poly(A)+ mRNA, like poly I:C, caused a significant reduction in neurite growth. DRG neurons exposed to mouse brain mRNA in the presence of an RNase inhibitor showed a 50% reduction in the number of neurons extending neurites, without a negative impact on cell survival (Fig. 1 E). RNase inhibitor alone had no effect on neurite outgrowth. In terms of neurite outgrowth, a similar dose-effect profile was seen for RNA compared with poly I:C (Fig. 1 F). The effects of mRNA cannot be attributed to endotoxin functioning through TLR4 (Poltorak et al., 1998; Qureshi et al., 1999) because neurons do not express TLR4 (Lehnardt et al., 2002, 2003), the preparation of mRNA had undetectable endotoxin, and because LPS alone has no effect on neurite outgrowth (Lehnardt et al., 2003). In addition, RNase degraded mRNA failed to inhibit neurite outgrowth (data not shown), thus indicating that primary or secondary structures within mRNA are necessary for neurite inhibiting activity. Together, these findings demonstrate that the TLR3 agonist poly I:C, as well as mRNA, inhibit neurite outgrowth in DRG neurons without impacting neuronal viability.
Negative regulators of axonal outgrowth can induce rapid changes in the morphology and motility of growth cones leading to axonal deflection, arrest, or growth cone collapse and neurite retraction (Filbin, 2003). We asked whether poly I:C functioned to induce growth cone collapse by examining growth cone morphology of sensory neurons after acute exposure to poly I:C. E9 chick DRG explants were plated on culture slides precoated with poly-d-lysine and laminin and allowed to extend neurites overnight. Poly I:C or PBS was then added to culture media for 30 min, at which time cells were fixed and stained. Growth-cone morphology was visualized by F-actin staining using rhodamine phalloidin. Acute exposure to poly I:C caused a fourfold increase in growth cone collapse compared with controls (Fig. 2 A,B). The rapidity of this response (<5 min) suggests that poly I:C induced inhibition of growth cone motility and neurite extension occurs independent of transcriptional regulation.
Figure 2.
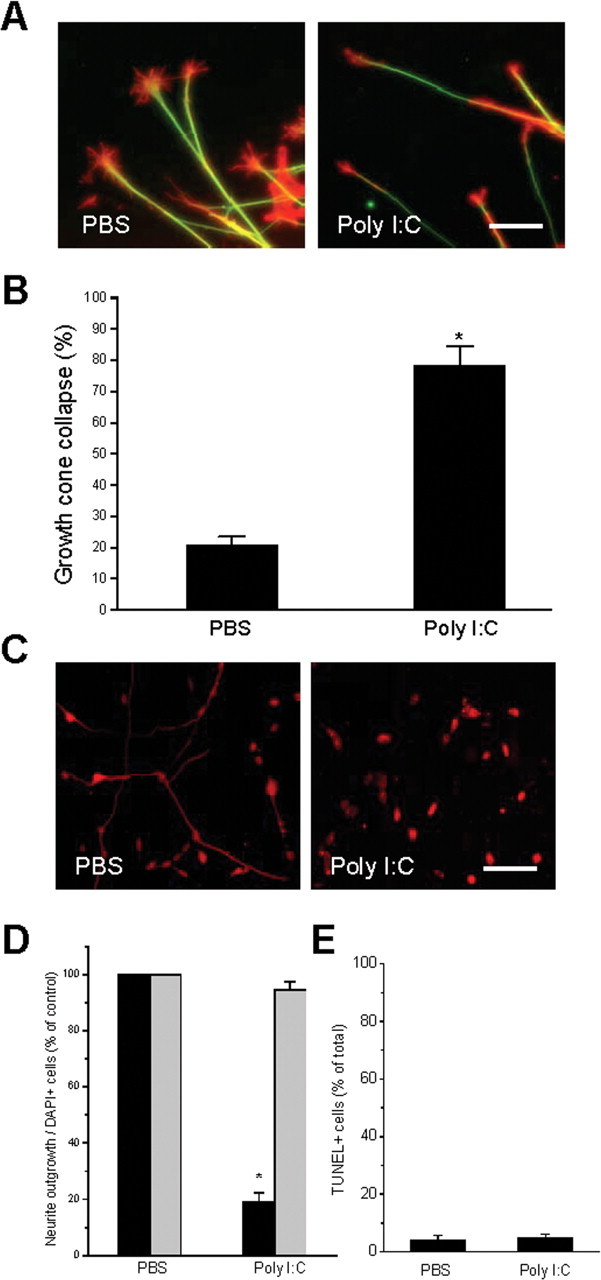
Poly I:C causes growth cone collapse of existing neurites and inhibition of neurite outgrowth is not reversible within 24 h. Explants of E9 chick DRGs were plated overnight and exposed to either PBS or poly I:C (20 μg/ml) for 30 min. A, Immunofluorescent staining of f-actin (red) and neurofilament (green) in DRG explants shows growth-cone collapse as a result of exposure to poly I:C. Scale bar, 25 μm. B, Quantification of explants assayed for growth-cone collapse. Exposure to poly I:C for 30 min caused a significant increase in the total number of collapsed growth cones in DRG explants. C, Neurofilament staining of a representative field of chick sensory neurons treated acutely with PBS or poly I:C (20 μg/ml) for 4 h, washed, then grown overnight in DMEM plus NGF. Scale bar, 100 μm. D, Quantification of the effects of poly I:C on neurite outgrowth (black bars) and cell survival (gray bars) 24 h after 4 h treatment. E, Quantification of TUNEL+ neurons from D. *p < 0.01, Student's t test.
We investigated whether neurite inhibition induced by poly I:C was reversible by culturing DRG neurons for 4 h in the presence of poly I:C or PBS, then rinsing and replacing the media with DMEM supplemented by NGF. Washout of poly I:C did not result in recovery of neurite extension rates to those of PBS-treated cells (Fig. 2 C,D, black bars). There were no significant differences between PBS and poly I:C exposure in the number of cells detected by DAPI staining (Fig. 2 D, gray bars) or in the number of TUNEL+ cells 24 h after the exposures (Fig. 2 E). Together, these results demonstrate that poly I:C causes growth cone collapse and inhibits neurite outgrowth in DRG neurons, implicating its associated receptor, TLR3, as a negative regulator of axonal growth.
The above experiments demonstrate that the TLR3 agonist poly I:C causes inhibition of neurite outgrowth and growth cone collapse in sensory neurons. We then determined whether TLR3 was expressed by neurons and its localization. TLR3 transcripts can be detected by RT-PCR in E14 mouse DRGs (Fig. 3 A). Relative to β-actin, TLR3 mRNA is expressed in greater quantity in DRGs than it is in spleen. Histologic staining for TLR3 in sections from mouse DRGs demonstrates that the protein is found in neurons in vivo (Fig. 3 B). As a control for the specificity of the commercial antibody used, DRGs from TLR3−/− mice failed to show immunoreactivity (Fig. 3 B). In cultured sensory neurons from E14 DRGs, immunoreactivity is localized to soma, neurites and is concentrated in filopodial structures along the leading edge of the growth cone (Fig. 3 C,D). Interestingly, the pattern of staining mirrors that of actin filaments within the growth cone (Fig. 3 D). Using confocal microscopy, we found TLR3 is at least partly colocalized with EAA1, a marker of endosomes, indicating at least some TLR3 is intracellular (Fig. 4 A). We also observed TLR3 staining in cortical neurons isolated from embryonic mouse brain (Fig. 4 B) as well as neurons isolated from embryonic mouse spinal cord (data not shown). TLR3 protein was not found in cultured sensory neurons isolated from TLR3−/− mice (Fig. 3 B,C) again confirming the specificity of the α-TLR3 antibody. These results indicate that TLR3 is present in neurons and is suitably localized to modulate growth cone morphology and motility.
Figure 3.
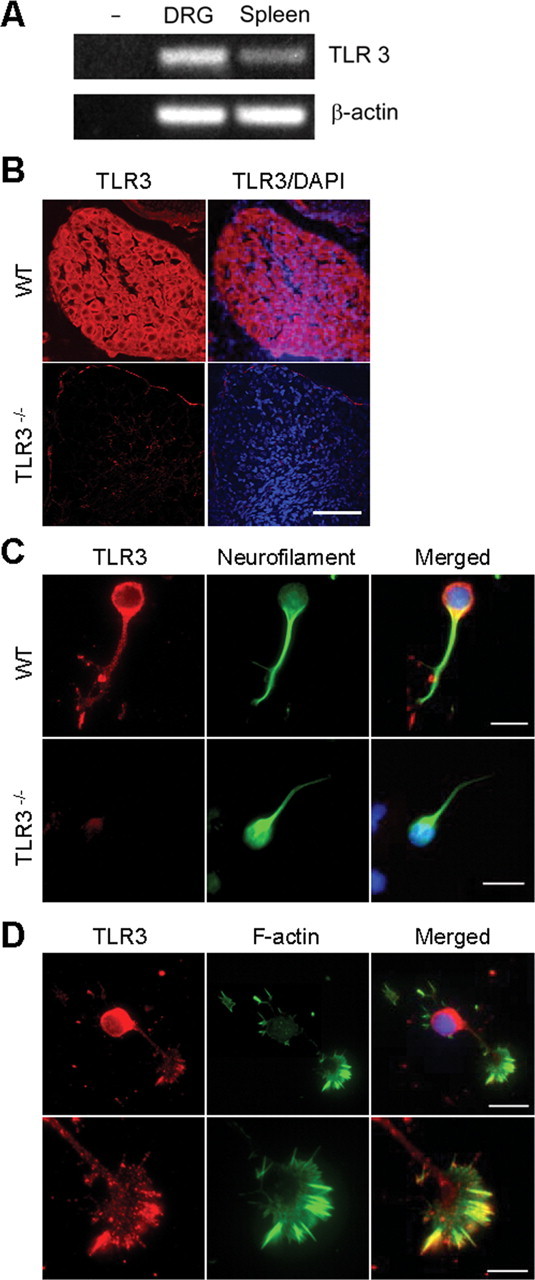
TLR3 transcripts and protein are expressed in mouse DRG neurons and colocalize within the growth cone. A, Total RNA was extracted from E14 mouse DRG or adult spleen. TLR3 and actin transcripts were identified by RT-PCR. TLR3 transcripts were detected in DRG and spleen extracts, but not in the absence of RNA. B, Confocal images of immunofluorescent staining for TLR3 (red) and DAPI (blue) in wild-type and TLR3−/− mouse DRGs in situ. Scale bar, 40 μm. C, TLR3 (red) and neurofilament (green) staining of dissociated DRG neurons isolated from E14 wild-type and TLR3−/− embryos. Scale bars: 25 μm. D, TLR3 (red) and f-actin (green) staining of dissociated DRG neurons and growth cones. Scale bars: top, 25 μm; bottom, 10 μm.
Figure 4.

TLR3 is partly endosomal and found in multiple neuronal subtypes. A, Confocal imaging of a mouse DRG growth cone containing TLR3 (red) and the endosomal marker, EAA1 (green). B, E17 cortical neurons at high magnification (top) and low magnification (bottom) stained with TLR3 (red) and neurofilament-M (NFM) (green). Scale bar, 25 μm.
We have shown above that poly I:C inhibits neurite outgrowth and that TLR3 is expressed by sensory neurons. To prove that neuronal TLR3 mediates the inhibitory effects of poly I:C on neurite outgrowth, we examined the effects of poly I:C in neurons lacking functional TLR3. Sensory neurons were isolated from mice bearing a targeted loss-of-function mutation in the TLR3 gene (Alexopoulou et al., 2001) or strain matched controls. TLR3 genotypes were confirmed by RT-PCR (data not shown). E14 DRG neurons isolated from wild-type (C57BL/6) or TLR3−/− mouse embryos were plated on poly-d-lysine/laminin coverslips for 30 min and treated with poly I:C or poly dI:dC for 4 h before fixation and staining. In contrast to wild-type neurons, poly I:C did not significantly diminish neurite outgrowth in TLR3−/− cultured neurons (Fig. 5 A). No significant differences in neurite outgrowth were detected between wild-type or TLR3−/− neurons grown in the presence of PBS or poly dI:dC. The TIR (Toll/interleukin-1 receptor) domain containing adaptor protein MyD88 serves a central signaling role for most TLRs except for TLR3, which signals independent of MyD88. As an additional control to exclude potential signaling of poly I:C through other TLRs, we studied the effects of poly I:C on neurons bearing a loss-of-function mutation in MyD88. As anticipated, neurite outgrowth in MyD88−/− neurons, like wild-type neurons, was significantly inhibited by poly I:C. In other words, loss of MyD88 function had no impact (Fig. 5 B). In immune cells, most of the downstream signaling events induced by TLR3 occur through the transcriptional regulator NF-κB and the ISRE (Fitzgerald et al., 2003a,b; Wietek et al., 2003; Yamamoto et al., 2003; Wang et al., 2004; Zhang et al., 2004; Schoenemeyer et al., 2005). Although TLR3-mediated induction of interferon-β synthesis is well described (Prehaud et al., 2005), interferon-β itself has no effect on neurite outgrowth and thus the effects of TLR3 activation in neurons cannot be attributed to secondary synthesis of interferon-β (data not shown). Although the swiftness of the effects of poly I:C on growth cone morphology (30 min) suggest that it likely functions independent of transcriptional regulation, we nonetheless examined neurons to determine whether NF-kB activation occurs by assaying for nuclear translocation of its p65 subunit. Nuclear translocation of the p65 subunit of NF-κB was not observed in sensory neurons treated with poly I:C (Fig. 5 C). In contrast the resident innate immune cell in the CNS, microglia, (identified by the microglial marker isolectin IB4) showed robust translocation of the NF-κB p65 subunit after stimulation with poly I:C (Fig. 5 C). Furthermore, the inhibitor of NF-κB activity, SN50, had no effect on the ability of poly I:C to inhibit neurite outgrowth in sensory neurons (Fig. 5 D). Thus, the inhibitory effects of poly I:C on neurite outgrowth are dependent on functional TLR3 and appear to be independent of NF-κB signaling, suggesting that the effects do not rely on the canonical transcriptional regulatory pathway activated by TLR3 in immune cells.
Figure 5.

Poly I:C inhibits neurite outgrowth through a TLR3-dependent NFκB- and MyD88-independent pathway. A, Quantification of neurite outgrowth from wild-type and TLR3−/− mouse sensory neurons cultured for 4 h in the presence of poly I:C (20 μg/ml) or poly dI:dC (20 μg/ml), as a negative control. No differences in plating density or cell survival were observed by DAPI staining. B, MyD88 loss-of-function has no impact on poly I:C-mediated inhibition of neurite outgrowth. Quantification of neurite outgrowth from wild-type and MyD88−/− sensory neurons cultured for 4 h in the presence of poly I:C (20 μg/ml) or PBS, as a negative control is shown. C, Nuclear translocation of the p65 subunit of NF-κB occurs in microglia but not neurons after treatment with poly I:C. Immunofluorescent staining of NF-κB localization in microglia and neurons after 1 h exposure to poly I:C (20 μg/ml) or PBS. D, Quantification of neurite outgrowth from sensory neurons cultured for 4 h in the presence of poly I:C (20 μg/ml) or PBS with or without the addition of SN50 (50 μg/ml), an inhibitor of NFκB nuclear translocation. *p < 0.01, Student's t test.
To determine whether our in vitro findings would be consistent in a more physiologically relevant environment in vivo, we injected wild-type (C57BL/6) and TLR3−/− mice intrathecally with poly I:C or saline vehicle at P4. We observed the performance of the injected pups on two well described developmental tasks of sensory–motor function, the righting reflex and negative geotaxis. Beginning at P9, wild-type poly I:C-injected pups were significantly slower in righting themselves than TLR3−/− poly I:C-injected pups (Fig. 6 A). Poly I:C-injected wild-type pups were also significantly slower at performing a negative geotaxis task, compared with saline-injected pups, beginning at P9 (Fig. 6 B). No differences were seen between poly I:C or saline-injected groups in TLR3−/− mice at any time point (Fig. 6 A,B). These results support an in vivo effect of poly I:C on inhibiting sensory-motor development that is dependent on functional TLR3.
Figure 6.

A, B, Effects of poly I:C (3 mg/kg) or PBS intrathecal injection on righting reflex (A) and negative geotaxis (B) in wild-type and TLR3−/− neonatal mice. P4 neonatal mice were injected intrathecally with 3 μl of either PBS or poly I:C (2 mg/ml) at a final concentration of 3 μg/ml. Behavioral testing was performed at P5, P7, P9, and P11. Righting reflex and negative geotaxis tasks were conducted and scored as described in Materials and Methods. *p < 0.01.
To determine whether behavioral deficits observed in poly I:C-injected pups were associated with impaired axonal development, we quantified the number of axons in cross sections of L1–L5 dorsal roots in injected animals. These analyses revealed a marked decrease in the number of axons in wild-type animals injected with poly I:C, but no changes in fiber number were observed in TLR3−/− animals receiving either PBS or poly I:C injections (Fig. 7 A,B). Histochemical observation revealed analogous deficits in fiber outgrowth in sensory ganglia. Neurofilament staining in DRGs from pups injected with poly I:C was greatly reduced in wild-type animals, but no effect was seen in TLR3−/− mice (Fig. 7 C). In wild-type poly I:C-injected animals, neuronal fibers were more sparse, as suggested by punctate neurofilament staining, in contrast to the elaborate web of processes seen in the DRG outflow tracts in the other three groups (Fig. 7 C,arrows). We found no gross neuroanatomic defects in TLR3−/− mice compared with controls (data not shown). These findings support the hypothesis that TLR3 can function as a negative regulator of axonal growth in vivo when stimulated with double stranded RNA.
Figure 7.
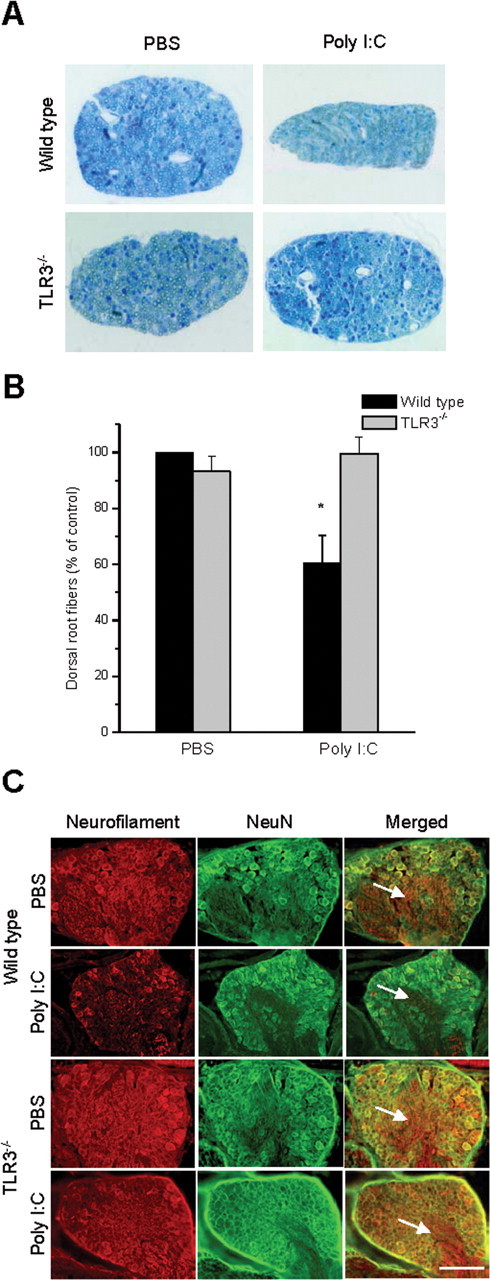
Intrathecal injection of poly I:C inhibits the normal development of sensory neurons in vivo via TLR3. A, Cross-sections of dorsal roots isolated from wild-type and TLR3−/− mice injected intrathecally with poly I:C (3 mg/kg) or PBS. Representative images are from the L3 dorsal root of each animal. B, Quantification of the number of myelinated fibers in dorsal root cross sections for L1–L5. C, Neurofilament (red) and NeuN (green) immunofluorescent images of DRGs from animals in A. Note the smaller ganglion size and lack of neurofilament intensity in ganglion roots (arrows) in wild-type animals injected with poly I:C, whereas there is no change in size or neurofilament staining in the other groups, as expected. Scale bar, 50 μm.
Discussion
In this report, we have defined a novel function for TLR3 in the mammalian nervous system. Activation of TLR3 abolishes neurite outgrowth and causes growth cone collapse. TLR3 is localized in filopodia and lamellipodia at the leading edge of axonal growth cones in a pattern overlying f-actin. Its localization raises the possibility that TLR3 interacts proximally with actin cytoskeletal components within the growth cone, a notion supported by the finding that neuronal TLR3 signaling is independent of NF-κB nuclear translocation.
We demonstrate that mice injected with poly I:C display early sensory–motor deficits and have relevant associated neuroanatomic defects with fewer axons present in dorsal roots. Because resident monocytes in the CNS and PNS express TLR3, we cannot exclude an immune contribution to the axonal injury observed in vivo. Our in vitro findings show that TLR3 can function as a signal transducing receptor in neurons to inhibit axonal outgrowth. It is thus likely that TLR3 agonists cause axonal injury in vivo both through direct effects on neuronal TLR3 and indirect effects mediated through resident innate immune cells.
In light of its known ligands, double stranded RNA characteristic of viral infection as well as duplex structures within mRNA (Alexopoulou et al., 2001; Kariko et al., 2004), TLR3 is potentially relevant to stunted axonal growth and regenerative failure in the context of both pathogen-mediated and sterile CNS injury. A neuronal receptor capable of recognizing features specific to a damaged CNS, where myelin inhibitory cues may be absent, has not been described previously and, thus, adds significant new insight into regenerative failure in the CNS. It stands to reason that such a receptor could confer a protective advantage to individual neurons by discouraging growth into regions of active infection or ongoing cell death. Secondarily, TLR3 may limit axonal growth and regeneration in a number of neurodegenerative states where markers of cell death, such as extracellular RNA, may be misinterpreted. We speculate, for instance, whether neuronal TLR3 recognition of mRNA within Alzheimer's disease plaques (Ginsberg et al., 1999; Marcinkiewicz, 2002) could potentially contribute to the incomplete and dystrophic neurite growth associated with these plaques. The presence of TLR3 on neurons and its effects in limiting axonal outgrowth in response to host- and pathogen-associated ligands provides a new mechanism whereby axonal regeneration in the CNS can be defeated and suggests a novel category of injury-associated cues that could potentially limit CNS regeneration and repair strategies.
Footnotes
This work was supported in part by National Institutes of Health Grants PO1NS38475 and R21 NS056085 (T.V.) and National Multiple Sclerosis Society Grant RG3426A2. J.S.C. and J.A.S. were funded as postdoctoral fellows of the National Multiple Sclerosis Society. R.A.F. is an Investigator of the Howard Hughes Medical Institute. We thank S. Akira, H. Plough, and D. Golenbock for kindly providing reagents, and M. Siegel for technical assistance.
References
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Andaloussi AE, Sonabend AM, Han Y, Lesniak MS. Stimulation of TLR9 with CpG ODN enhances apoptosis of glioma and prolongs the survival of mice with experimental brain tumors. Glia. 2006;54:526–535. doi: 10.1002/glia.20401. [DOI] [PubMed] [Google Scholar]
- Anderson KV, Jurgens G, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: genetic studies on the role of the Toll gene product. Cell. 1985a;42:779–789. doi: 10.1016/0092-8674(85)90274-0. [DOI] [PubMed] [Google Scholar]
- Anderson KV, Bokla L, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell. 1985b;42:791–798. doi: 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- Bergmann-Leitner ES, Leitner WW. Danger, death and DNA vaccines. Microbes Infect. 2004;6:319–327. doi: 10.1016/j.micinf.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- Brown BD, Lillicrap D. Dangerous liaisons: the role of “danger” signals in the immune response to gene therapy. Blood. 2002;100:1133–1140. doi: 10.1182/blood-2001-11-0067. [DOI] [PubMed] [Google Scholar]
- Caroni P, Schwab ME. Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol. 1988;106:1281–1288. doi: 10.1083/jcb.106.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–439. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- Colaco CA. Towards a unified theory of immunity: dendritic cells, stress proteins and antigen capture. Cell Mol Biol (Noisy-le-grand) 1998;44:883–890. [PubMed] [Google Scholar]
- Cox EC, Muller B, Bonhoeffer F. Axonal guidance in the chick visual system: posterior tectal membranes induce collapse of growth cones from the temporal retina. Neuron. 1990;4:31–37. doi: 10.1016/0896-6273(90)90441-h. [DOI] [PubMed] [Google Scholar]
- Dalpke AH, Schafer MK, Frey M, Zimmermann S, Tebbe J, Weihe E, Heeg K. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. 2002;168:4854–4863. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- David S, Aguayo AJ. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;214:931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- David S, Aguayo AJ. Axonal regeneration after crush injury of rat central nervous system fibres innervating peripheral nerve grafts. J Neurocytol. 1985;14:1–12. doi: 10.1007/BF01150259. [DOI] [PubMed] [Google Scholar]
- Doyle S, Vaidya S, O'Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- Doyle SE, O'Connell R, Vaidya SA, Chow EK, Yee K, Cheng G. Toll-like receptor 3 mediates a more potent antiviral response than Toll-like receptor 4. J Immunol. 2003;170:3565–3571. doi: 10.4049/jimmunol.170.7.3565. [DOI] [PubMed] [Google Scholar]
- Ebert S, Gerber J, Bader S, Muhlhauser F, Brechtel K, Mitchell TJ, Nau R. Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J Neuroimmunol. 2005;159:87–96. doi: 10.1016/j.jneuroim.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J Immunol. 2006;176:6802–6811. doi: 10.4049/jimmunol.176.11.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated glycoprotein: a role in myelination and in the inhibition of axonal regeneration? Curr Opin Neurobiol. 1995;5:588–595. doi: 10.1016/0959-4388(95)80063-8. [DOI] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J Exp Med. 2003a;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003b;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Crino PB, Hemby SE, Weingarten JA, Lee VM, Eberwine JH, Trojanowski JQ. Predominance of neuronal mRNAs in individual Alzheimer's disease senile plaques. Ann Neurol. 1999;45:174–181. [PubMed] [Google Scholar]
- Glezer I, Lapointe A, Rivest S. Innate immunity triggers oligodendrocyte progenitor reactivity and confines damages to brain injuries. FASEB J. 2006;20:750–752. doi: 10.1096/fj.05-5234fje. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–444. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- Halfon MS, Hashimoto C, Keshishian H. The Drosophila toll gene functions zygotically and is necessary for proper motoneuron and muscle development. Dev Biol. 1995;169:151–167. doi: 10.1006/dbio.1995.1134. [DOI] [PubMed] [Google Scholar]
- Heeg K, Sparwasser T, Lipford GB, Hacker H, Zimmermann S, Wagner H. Bacterial DNA as an evolutionary conserved ligand signalling danger of infection to immune cells. Eur J Clin Microbiol Infect Dis. 1998;17:464–469. doi: 10.1007/BF01691128. [DOI] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Jackson AC, Rossiter JP, Lafon M. Expression of Toll-like receptor 3 in the human cerebellar cortex in rabies, herpes simplex encephalitis, and other neurological diseases. J Neurovirol. 2006;12:229–234. doi: 10.1080/13550280600848399. [DOI] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BR, Li X. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFκB and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem. 2003;278:16713–16719. doi: 10.1074/jbc.M300562200. [DOI] [PubMed] [Google Scholar]
- Jung DY, Lee H, Jung BY, Ock J, Lee MS, Lee WH, Suk K. TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia: a critical role of IFN-beta as a decision maker. J Immunol. 2005;174:6467–6476. doi: 10.4049/jimmunol.174.10.6467. [DOI] [PubMed] [Google Scholar]
- Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg AM, Love-Homan L, Yi AK, Harty JT. CpG DNA induces sustained IL-12 expression in vivo and resistance to Listeria monocytogenes challenge. J Immunol. 1998;161:2428–2434. [PubMed] [Google Scholar]
- Laflamme N, Soucy G, Rivest S. Circulating cell wall components derived from gram-negative, not gram-positive, bacteria cause a profound induction of the gene-encoding Toll-like receptor 2 in the CNS. J Neurochem. 2001;79:648–657. doi: 10.1046/j.1471-4159.2001.00603.x. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, Wiazewicz N, Wiltfang J, Gerber J, Nau R. Amyloid beta peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem. 2005;94:289–298. doi: 10.1111/j.1471-4159.2005.03188.x. [DOI] [PubMed] [Google Scholar]
- Marcinkiewicz M. BetaAPP and furin mRNA concentrates in immature senile plaques in the brain of Alzheimer patients. J Neuropathol Exp Neurol. 2002;61:815–829. doi: 10.1093/jnen/61.9.915. [DOI] [PubMed] [Google Scholar]
- Matzinger P. An innate sense of danger. Semin Immunol. 1998;10:399–415. doi: 10.1006/smim.1998.0143. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway C., Jr The Toll receptor family and microbial recognition. Trends Microbiol. 2000;8:452–456. doi: 10.1016/s0966-842x(00)01845-x. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- Nessa BN, Tanaka T, Kamino K, Sadik G, Ansar AB, Kimura R, Tanii H, Okochi M, Morihara T, Tagami S, Kudo T, Takeda M. Toll-like receptor 3 mediated hyperphosphorylation of tau in human SH-SY5Y neuroblastoma cells. Psychiatry Clin Neurosci. 2006;60(Suppl 1):S27–33. [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol. 2001;50:630–639. doi: 10.1002/ana.1256. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Paterson HM, Murphy TJ, Purcell EJ, Shelley O, Kriynovich SJ, Lien E, Mannick JA, Lederer JA. Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J Immunol. 2003;171:1473–1483. doi: 10.4049/jimmunol.171.3.1473. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol. 2005;79:12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon y Cajal S. Degeneration and regeneration of the nervous system. London: Hafner; 1928. [Google Scholar]
- Richardson PM, McGuinness UM, Aguayo AJ. Axons from CNS neurons regenerate into PNS grafts. Nature. 1980;284:264–265. doi: 10.1038/284264a0. [DOI] [PubMed] [Google Scholar]
- Rose D, Chiba A. A single growth cone is capable of integrating simultaneously presented and functionally distinct molecular cues during target recognition. J Neurosci. 1999;19:4899–4906. doi: 10.1523/JNEUROSCI.19-12-04899.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose D, Zhu X, Kose H, Hoang B, Cho J, Chiba A. Toll, a muscle cell surface molecule, locally inhibits synaptic initiation of the RP3 motoneuron growth cone in Drosophila . Development. 1997;124:1561–1571. doi: 10.1242/dev.124.8.1561. [DOI] [PubMed] [Google Scholar]
- Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, Sen GC. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol. 2004;11:1060–1067. doi: 10.1038/nsmb847. [DOI] [PubMed] [Google Scholar]
- Schmidt KN, Leung B, Kwong M, Zarember KA, Satyal S, Navas TA, Wang F, Godowski PJ. APC-independent activation of NK cells by the Toll-like receptor 3 agonist double-stranded RNA. J Immunol. 2004;172:138–143. doi: 10.4049/jimmunol.172.1.138. [DOI] [PubMed] [Google Scholar]
- Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, Fitzgerald KA, Golenbock DT. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem. 2005;280:17005–17012. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- Schwab ME. Molecules inhibiting neurite growth: a minireview. Neurochem Res. 1996;21:755–761. doi: 10.1007/BF02532297. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Caroni P. Oligodendrocytes and CNS myelin are nonpermissive substrates for neurite growth and fibroblast spreading in vitro . J Neurosci. 1988;8:2381–2393. doi: 10.1523/JNEUROSCI.08-07-02381.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001;2:293–299. doi: 10.1038/86297. [DOI] [PubMed] [Google Scholar]
- Stockwin LH, McGonagle D, Martin IG, Blair GE. Dendritic cells: immunological sentinels with a central role in health and disease. Immunol Cell Biol. 2000;78:91–102. doi: 10.1046/j.1440-1711.2000.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Zhang Z, Trautmann K, Xu S, Schluesener HJ. TLR and NOD2 ligands induce cell proliferation in the rat intact spinal cord. J Neuropathol Exp Neurol. 2005;64:991–997. doi: 10.1097/01.jnen.0000187051.74265.56. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Tang S, Woodhall RW, Shen YJ, deBellard ME, Saffell JL, Doherty P, Walsh FS, Filbin MT. Soluble myelin-associated glycoprotein (MAG) found in vivo inhibits axonal regeneration. Mol Cell Neurosci. 1997;9:333–346. doi: 10.1006/mcne.1997.0633. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. Microglia recognize double-stranded RNA via TLR3. J Immunol. 2006;176:3804–3812. doi: 10.4049/jimmunol.176.6.3804. [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- Wallin RP, Lundqvist A, More SH, von Bonin A, Kiessling R, Ljunggren HG. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002;23:130–135. doi: 10.1016/s1471-4906(01)02168-8. [DOI] [PubMed] [Google Scholar]
- Wang YY, Li L, Han KJ, Zhai Z, Shu HB. A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-kappaB and ISRE and IFN-beta promoter. FEBS Lett. 2004;576:86–90. doi: 10.1016/j.febslet.2004.08.071. [DOI] [PubMed] [Google Scholar]
- Wietek C, Miggin SM, Jefferies CA, O'Neill LA. Interferon regulatory factor-3-mediated activation of the interferon-sensitive response element by Toll-like receptor (TLR) 4 but not TLR3 requires the p65 subunit of NF-kappa. J Biol Chem. 2003;278:50923–50931. doi: 10.1074/jbc.M308135200. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xu LG, Han KJ, Wei X, Shu HB. PIASy represses TRIF-induced ISRE and NF-kappaB activation but not apoptosis. FEBS Lett. 2004;570:97–101. doi: 10.1016/j.febslet.2004.05.081. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Trautmann K, Schluesener HJ. Microglia activation in rat spinal cord by systemic injection of TLR3 and TLR7/8 agonists. J Neuroimmunol. 2005;164:154–160. doi: 10.1016/j.jneuroim.2005.03.014. [DOI] [PubMed] [Google Scholar]