

Abstract
The aza-Diels–Alder cycloaddition of 1,2,4-triazines with alkynes offers a rapid and convenient method for the synthesis of highly substituted pyridines, but often requires harsh conditions and long reaction times. The present study offers a solution to these limitations by use of a temporary tether established by a Lewis acid–base complexation of in situ generated alkynylboranes and triazines bearing a Lewis basic donor. The cycloaddition reactions take place within 20 min at 40 °C and provide direct access to a broad range of pyridines with complete and predictable regiocontrol. The carbon—boron bond can be further functionalised by cross-coupling allowing further functionality to be introduced after cycloaddition.
Keywords: boranes, cycloadditions, pyridines, regioselectivity, triazines
Introduction
Pyridines are a fundamentally important class of aromatic molecules.[1] They are present in many bioactive compounds and they play a key role in a number of biological processes. From a synthetic viewpoint, the ready quaternisation of the basic pyridine ring limits the functionalisation of this aromatic system by electrophilic substitution processes. Ring substitution is, therefore, often dictated by the availability of a halide substituent, or related group that allows elaboration by substitution or transition-metal-catalysed coupling. An alternative approach to pyridines is by means of ring synthesis and a number of approaches are now established.[2] In this regard, the inverse electron demand aza-Diels–Alder reaction of triazines constitutes a useful and much studied method, however this process has largely focused on the use of enamine dienophiles as alkyne surrogates because alkynes themselves only participate in [4+2] cycloadditions with triazines under very harsh conditions. Moreover, such processes are often poorly regioselective and are relatively low yielding.[3]
With regard to inverse electron demand aza-Diels–Alder reactions, we have recently become interested in the use of directed cycloadditions for the mild and regiocontrolled synthesis of aromatic and heteroaromatic compounds.[4] Central to our design was the use of an alkyne bearing a Lewis acid acceptor that would promote pre-association with a diene bearing a complementary Lewis base (Scheme 1). The resulting complex would provide a platform for rate enhancements in the ensuing cycloaddition, and this rate enhancement was exemplified by the reaction of tetrazines with in situ generated alkynyldifluoroboranes at ambient temperatures.
Scheme 1.

Directed cycloaddition reactions.
In considering an appropriate alkyne-substituted Lewis acid, boron-based acceptors are of particular interest as they deliver organoboron products of potential value for further organic synthesis.[5] We report herein the employment of this concept in a mild and versatile route to pyridine boronic acid derivatives by means of directed triazine cycloadditions.
Results and Discussion
To establish a typical reactivity profile for non-activated triazines and alkynes, we opted to explore the cycloaddition reactions of readily available alkynes and triazine. Indeed, we found that triazine 1 a was particularly reluctant to undergo efficient reaction with phenylacetylene, providing the corresponding product in low yield after prolonged heating, albeit with high regiocontrol.[6] Moreover, we attempted a similar reaction with an alkynylboronate and found that this approach generated the corresponding pyridine boronic acid derivative, again in very low yield, but with high regioselectivity (Scheme 2).[7]
Scheme 2.
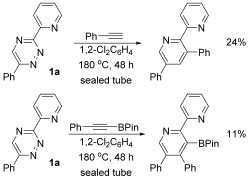
Triazine cycloaddition reactions of alkynes. Pin=pinacol.
The poor reactivity of diene 1 a with alkynes made it an ideal choice for evaluating the potential of our proposed directed cycloaddition, and we set out to explore the reaction of this compound with alkynytrifluoroborate 2 a, our results are depicted in Table 1. Fluorophilic Lewis acids are known to transform alkynyltrifluoroborate salts into the corresponding difluoroboranes,[8], [9] and so we employed BF3⋅OEt2 to promote formation of our BF2-appended alkyne in situ. Remarkably, simply stirring this Lewis acid and substrate combination in CH2Cl2 at room temperature provided the desired cycloadduct (entry 1). The yield could be improved by increasing the temperature and the concentration of alkynydifluoroborane (entries 2 and 3). Finally, TMSCl was also found to be a competent fluorophile, albeit slightly less effective than BF3⋅OEt2 in this case (entry 4). Confirmation of the Lewis acid–base interaction between the pyridyl and BF2 substituents in the product, as well as the regioselectivity, was confirmed by X-ray crystallography. Figure 1 shows the expected tetrahedral geometry around the B atom.
Table 1.
Directed cycloaddition of 1 and alkynyltrifluoroborates.[a]
 | ||||
|---|---|---|---|---|
| Entry | Lewis acid[a] [(equiv)] | T [°C] | t [min] | Yield3[%] |
| 1 | BF3⋅OEt2 (2) | 25 | 16 h | 29 |
| 2 | BF3⋅OEt2 (2) | 40 | 10 | 43 |
| 3 | BF3⋅OEt2 (3) | 40 | 10 | 84 |
| 4 | Me3SiCl (3) | 40 | 10 | 73 |
A 1:1 stoichiometry of Lewis acid and alkyne was used in all cases.
Figure 1.

X-ray crystal structure representation of 3, H atoms omitted for clarity.
A minor side product observed in the cycloadditions of 1 a and 2 a was the product of direct acetylide addition at the heteroaromatic ring. This compound was isolated in 12 % yield under the optimal conditions (Table 1, entry 3), and its structure was also verified by X-ray crystallography (Figure 2).[10]
Figure 2.

X-ray crystal structure representation of 4.
Notwithstanding the propensity for competing direct addition processes, the optimal conditions of the cycloaddition were found to be quite general across a small selection of alkynes, allowing the corresponding pyridines 5–7 to be generated in moderate to high yield (Figure 3).
Figure 3.

Pyridine products from the directed cycloaddition of 1 a. Conditions: 1 a (1 equiv), alkyne (3 equiv) and BF3⋅OEt2 (3 equiv) heated at 40 °C in CH2Cl2 for 10 min.
Having established reaction conditions for the mild cycloaddition of triazine 1 a with alkynyltrifluoroborates, we set out to explore the scope of this chemistry for the preparation of bipyridyldifluoroboranes, our results are shown in Table 2. We began by employing an isomer of triazine 1 a and were pleased to find that pyridines 8 and 9 were formed in high yield (entries 1 and 2). Expanding to more heavily substituted triazines provided the opportunity to access fully substituted pyridines under mild conditions (entries 3–6). This approach is completely regioselective because of the nature of the directed reaction; therefore, this approach represents a powerful method for assembling highly functionalised products with entirely predictable regiocontrol. Finally, less heavily substituted pyridines can also be accessed by this strategy, compounds 14 and 15 were both prepared from triazine 1 e in good yield.
Table 2.
Directed cycloaddition of triazines and alkynyltrifluoroborates.
 | |||||
|---|---|---|---|---|---|
| Entry | Triazine | Alkyne (R2) | Product | Lewis acid[a] [(equiv)] | Yield3 a[%] |
| 1 |  |
Ph; 2 a |  |
Me3SiCl (3) | R=Ph; 75 (8) |
| 2 |  |
Me3SiCl (3) | R=C6H9; 83 (9) | ||
| 3 |  |
Ph; 2 a | 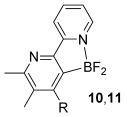 |
BF3⋅OEt2 (3) | R=Ph; 72 (10) |
| 4 | |
BF3⋅OEt2 (3) | R=C6H9; 76 (11) | ||
| 5 |  |
Ph; 2 a |  |
BF3⋅OEt2 (3) | R=Ph; 82 (12) |
| 6 | Bu; 2 c | BF3⋅OEt2 (3) | R=Bu; 62 (13) | ||
| 7 |  |
Ph; 2 a |  |
BF3⋅OEt2 (3) | R=Ph; 50 (14) |
| 8 | Bu; 2 c | Me3SiCl (3) | R=Bu; 62 (15) | ||
Having had broad success with pyridyl directing groups, we decided to establish whether other Lewis bases could direct the cycloaddition reaction. Indeed, we were pleased to find that amides also functioned as competent directing groups, providing access to pyridines 16–20 in good overall yield (Scheme 3). Interestingly, the less substituted triazine substrate 1 h was significantly less efficient, providing poor yields of the corresponding pyridines even when the reaction was conducted at low temperature. In this case, the crude mixtures were relatively complex, but the major side product in each case, 24, appeared to result from alkyne addition to the ring.[11]
Scheme 3.

Alternative directing groups. [a] The reaction was conducted at 40 °C for 20 min.
A further issue that we wished to clarify was the importance of the positioning of the directing group. In principle, the Lewis basic donor could also be incorporated at the 6-position of the triazine giving rise to isomeric pyridine products. As shown in Scheme 4, the cycloaddition of 25 was found to proceed in good yield, although the reaction required a longer time period and returned a small amount of starting triazine 25 (≈10 %). We also prepared 27 to probe the effect of having two competing directing groups on reaction regiochemistry. Interestingly, the reaction proceeded with high selectivity to provide 28 a, albeit in modest yield,[12] and <5 % of regioisomer 28 b (as judged by LC-MS analysis). This preliminary data suggest that substrates bearing a directing group at the 3-position are optimal, but that the inclusion of directing groups at C6 are viable. Further studies aimed at understanding the scope of directing-group positioning are currently being pursued.
Scheme 4.

Incorporation of a directing group at C6.
Although the main objective of this study was to demonstrate that the directed cycloaddition could deliver faster reactions than the traditional aza-Diels–Alder process, we recognised the potential value of the products that are armed with a carbon—boron bond. We decided to explore the Pd-catalysed cross coupling of two representative difluoroboranes, 9 and 17, which contain multiple functionality and a hindered borane unit. In the event, both reactions required some optimisation, but delivered the corresponding biaryl products in acceptable yields (Scheme 5).
Scheme 5.

Reactions of cycloadducts. RuPHOS=2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl, DME=1,2-dimethoxyethane.
Conclusion
We have developed a mild and regiocontrolled method for the synthesis of highly substituted pyridines by means of a Lewis base directed cycloaddition of triazines and in situ generated alkynylboranes. This method proceeds with a range of alkynes and triazines, although it appears to be advantageous to have the Lewis base directing group at C3 of the diene cycloaddition partner. As well as providing a convenient means for generating bipyridines, this method is compatible with amide directing groups and the presence of the carbon—boron bond allows further functionalisation to take place through cross-coupling reactions.
Experimental Section
General procedure for the cycloaddition of alkynyltrifluoroborates and triazines
Synthesis of 3: A solution of 6-phenyl-3-(2-pyridyl)-1,2,4-triazine 1 a (50 mg, 0.21 mmol) and potassium (phenylethynyl)trifluoroborate 2 a (132 mg, 0.64 mmol) in CH2Cl2 (2 mL) was treated with BF3⋅OEt2 (55 μL, 0.64 mmol). The reaction was stirred for 10 min and then quenched with brine (10 mL). The mixture was extracted with CH2Cl2 (3×15 mL) and the extract dried over MgSO4, filtered and the solvent evaporated. The residue was purified chromatographically over silica gel (gradient; starting with petroleum ether, ending with ethyl acetate) to afford 3-(difluoroboryl)-4,5-diphenyl-2,2′-bipyridine 3 (63 mg, 84 %) as a colourless solid. M.p 225–226 °C. 1H NMR (400 MHz, CDCl3): δ=7.18–7.22 (2 H, m), 7.25–7.33 (6 H, m), 7.35–7.40 (2 H, m), 7.61–7.67 (1 H, m), 8.26 (1 H, td, J=7.5, 1.5 Hz), 8.40 (1 H, d, J=8.0), 8.59 (1 H, d, J=5.5 Hz), 8.67 ppm (1 H, s); 13C NMR (100.6 MHz, CDCl3): δ=118.9, 125.0, 127.4, 127.7, 127.8, 127.9, 128.2, 129.8, 129.9, 138.2, 138.5, 141.4, 144.1, 151.7, 151.9, 154.6, 154.9 ppm; 19F NMR (235.1 MHz, CDCl3): δ=−156.4 ppm; FTIR:  =3058 (w), 2925 (w), 1626 (s), 1578 (m), 1555 (m), 1489 (s), 1452 (m), 1433 (s), 1158 (m), 1131 (s), 1100 (s), 1007 (m), 910 (m) cm−1. HRMS: (ESI) m/z calcd for C22H1511BF2N2Na: 379.1194 [M+Na+], found 379.1204.
=3058 (w), 2925 (w), 1626 (s), 1578 (m), 1555 (m), 1489 (s), 1452 (m), 1433 (s), 1158 (m), 1131 (s), 1100 (s), 1007 (m), 910 (m) cm−1. HRMS: (ESI) m/z calcd for C22H1511BF2N2Na: 379.1194 [M+Na+], found 379.1204.
Acknowledgments
This work was supported by The University of Sheffield, the EPSRC and the FP7 Marie Curie Actions of the European Commission via the ITN ECHONET Network (MCITN-2012–316379).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201403916.
References
- [1].Jones G. In: Comprehensive Heterocyclic Chemistry II Vol. 5. Katritzky A, Rees CW, Scriven EV, editors. Oxford: Pergamon; 1996. p. 167. (Eds: [Google Scholar]
- [2].p. 3787.
- [2a].Varela JA, Saá C. Chem. Rev. 103 doi: 10.1021/cr030677f. ; [DOI] [PubMed] [Google Scholar]
- [2b].Henry GD. Tetrahedron. 2003;60 ; [Google Scholar]
- [2c].Hill MD. Chem. Eur. J. 2004;16 [Google Scholar]
- [3].p. 2869.
- [3a].Boger DL. Tetrahedron. 39 ; [Google Scholar]
- [3b].Raw SA, Taylor RJK. Adv. Heterocycl. Chem. 1983;100 ; [Google Scholar]
- [3c].Foster RAA, Willis MC. Chem. Soc. Rev. 2010;42 doi: 10.1039/c2cs35316d. [DOI] [PubMed] [Google Scholar]
- [4].p. 160.
- [4a].Vivat JF, Adams H, Harrity JPA. Org. Lett. 12 ; [Google Scholar]
- [4b].Kirkham JD, Butlin RJ, Harrity JPA. Angew. Chem. 2010;124 doi: 10.1002/anie.201200917. ; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51 ; [Google Scholar]
- [4c].Crépin DF, Harrity JPA. Org. Lett. 2012;15 doi: 10.1021/ol401952k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hall DG. Boronic Acids. Weinheim: Wiley-VCH; 2005. [Google Scholar]
- [6].The high selectivity observed in the reaction of 1 a. Diring S, Retailleau P, Ziessel R. J. Org. Chem. 2007;72:10181. doi: 10.1021/jo7019866. with phenylacetylene mirrors that observed by Ziessel and co-workers: [DOI] [PubMed] [Google Scholar]
- [7].For related [4+2] cycloaddition reactions of alkynylboronates, see: p. 2901.
- [7a].Hilt G, Smolko KI. Angew. Chem. 115 ; [Google Scholar]; Angew. Chem. Int. Ed. 2003;42 ; [Google Scholar]
- [7b].Helm MD, Moore JE, Plant A, Harrity JPA. Angew. Chem. 2003;117 ; [Google Scholar]; Angew. Chem. Int. Ed. 2005;44 ; [Google Scholar]
- [7c].Moore JE, York M, Harrity JPA. Synlett. 2005 ; [Google Scholar]
- [7d].Delaney PM, Moore JE, Harrity JPA. Chem. Commun doi: 10.1039/b607322k. ; [DOI] [PubMed] [Google Scholar]
- [7e].Browne DL, Vivat JF, Plant A, Gomez-Bengoa E, Harrity JPA. J. Am. Chem. Soc. 2006;131 doi: 10.1021/ja902460n. [DOI] [PubMed] [Google Scholar]
- [8].Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J. Org. Chem. 1995;60:3020. [Google Scholar]
- [9].Alkynyl difluoroboranes are subject to rapid and reversible disproportionation to dialkynyl fluoroboranes and trialkynylboranes, which can all undergo cycloaddition. Nonetheless, these products can all converge to the corresponding difluoroboranes products following further disproportionation. For a discussion, see: D. F. P. Crépin, J. P. A. Harrity, J. Jiang, A. J. H. M. Meijer, A.-C. M. A. Nassoy, P. Raubo, J. Am. Chem. Soc2014136. , 8642.
- [10].For related nucleophilic addition reactions to triazines, see: p. 1378.
- [10a].Konno S, Ohba S, Sagi M, Yamanka M. Chem. Pharm. Bull. 35 ; [Google Scholar]
- [10b].Ye L, Haddadin MJ, Lodewyk MW, Ferreira AJ, Fettinger JC, Tantillo DJ, Kurth MJ. Org. Lett. 1987;12 [Google Scholar]
- [11].Compound 241131. R=Ph was characterised by H, C NMR and HRMS analysis, whereas R=Bu was tentatively characterised by H NMR and HRMS analysis only.
- [12].CCDC-1005645 (417328 a. ), 1005646 ( ), 1005647 ( ) and 1005648 ( ) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.