

Abstract
The rat amyloid-β (Aβ) intracerebroventricular infusion can model aspects of Alzheimer’s disease (AD) and has predicted efficacy of therapies such as ibuprofen and curcumin in transgenic mouse models. High density lipoprotein (HDL), a normal plasma carrier of Aβ, is used to attenuate Aβ aggregation within the pump, causing Aβ-dependent toxicity and cognitive deficits within 3 months. Our goal was to identify factors that might accelerate onset of Aβ-dependent deficits to improve efficiency and cost-effectiveness of model. We focused on: 1) optimizing HDL-Aβ preparation for maximal toxicity; 2) evaluating the role of copper, a factor typically in water that can impact oligomer stability; and 3) determining impact of insulin resistance (type II diabetes), a risk factor for AD. In vitro studies were performed to determine doses of copper and methods of Aβ-HDL preparation that maximized toxicity. These preparations when infused resulted in earlier onset of cognitive deficits within 6 weeks post-infusion. Induction of insulin resistance did not exacerbate Aβ-dependent cognitive deficits, but did exacerbate synaptic protein loss. In summary, the newly described in vivo infusion model may be useful cost-effective method for screening for new therapeutic drugs for AD.
Keywords: Alzheimer’s disease, amyloid-β, cognitive deficit, copper, high density lipoprotein, insulin resistance
INTRODUCTION
Despite the advances in our understanding of the molecular, genetic and environmental causes of Alzheimer’s disease (AD), the emotional and economic burdens associated with this disease continue to grow, due to the growing elderly population and the dearth of effective, disease-modifying medications. New animal models of AD have been developed to examine promising therapeutic prospects. Although each of these models exhibits important aspects of AD pathogenesis, no single model successfully mimics all aspects of the disease process, and no single model appears to adequately predict efficacy in clinical trials.
Transgenic mouse models
Several transgenic mouse models of increased amyloid deposition have been developed which overexpress mutations in human amyloid-β protein precursor (AβPP) and/or presenilin 1 (PS1) that result in early-onset familial AD. Such transgenic mice have become the favored preclinical models to investigate potential disease-modifying therapies [22,27,50,57]. These animals exhibit increased human amyloid-β (Aβ) deposition and some degree of neuroinflammation and neurodegeneration, but lack the neuron loss in selectively vulnerable regions evident with incipient human AD. The absence of neuron loss may be related to the trophic effects of AβPP, which can stimulate neurite and synapse growth and enhance memory function when over-expressed [54,60]. Although some chronic inflammatory changes are seen in AβPP mice, they are less extensive than those seen in autopsy studies of human AD patients [56], which may reflect or even explain the lack of neuron loss in these transgenic models. In contrast, neuron loss has been achieved with overexpression of mutant tau genes that are associated with frontotemporal dementia (FTD) [55], suggesting the absence of extensive tau pathology in the AβPP transgenics may be a factor limiting neuron loss. Interestingly, this inducible tau model is the only model reflecting robust neuron loss and severe brain atrophy similar to AD. The problem is the unclear relevance to AD, since its mutation (P301L) is not found in AD, but in FTD, and P301L has to be dramatically overexpressed to cause this phenomenon.
Triple transgenic mice that over-express [AβPP(Swe)], presenilin-1 [PS1(M146V)], and tau (P301L) exhibit human tau pathology but lack robust neuron loss, and neuron loss in AD is not quantitatively explained by tangles. Therefore the connections between Aβ,tau accumulation and neuron loss in AD remain unresolved. AβPP and/or PS1 transgenic models lack the ability to control amyloid dosing, have high costs of colony maintenance and are associated with patent rights.
Canine models
Another important model is the aging dog, which exhibits neuron loss, age-dependent accumulation of diffuse Aβ deposits, and vascular amyloid deposition, but little or no accumulation of neuritic amyloid deposits in the neuropil and no neurofibrillary tangles [25,63]. This model incorporates many of the neuropathological aspects of AD and is particularly compelling because the pattern of neuronal loss correlates with the extent of amyloid accumulation. The main disadvantages of this model are the high costs of maintaining dogs in the laboratory and their long lifespan.
Rabbit models
Aged rabbits maintained on a high cholesterol diet or supplemented with trace levels of copper in their drinking water, exhibit significant tau and amyloid pathology and neuron loss; they have been useful for evaluating the efficacy of drugs for AD such as galantamine [58, 71]. The advantages of this model are moderate neuron loss, tau pathology, and Aβ deposition. It has similarities to the canine model. The disadvantages are that it is less well-characterized for cognitive effects (perhaps because of neuron loss in cerebellum) and that its animal costs are higher than costs for smaller rodents.
Rodent amyloid injection/infusion models
Aβ injections
We and Yankner/Kowalls group performed the first acute injection models, investigating the impact of synthetic Aβ [34] and amyloid plaque cores isolated from human AD patients [17] on acute neurodegeneration and on inflammatory clearance of amyloid plaques [16]. Initial reports of the effects of amyloid peptide injections on neurotoxicity were difficult to interpret, since the vehicle (acetonitrile) appeared to impact toxicity when injected alone, and acute low-dose injections of PBS-soluble Aβ42 had relatively minimal effects on toxicity [66] or cognition [70]. Subsequent studies determined that acute injections of some forms of Aβ42 could induce behavioral deficits [29,43] and neuroinflammatory changes [62,69]. Notably, acute injections of Aβ42 oligomers purified from cells transfected with human AβPP have demonstrated that specific oligomeric species are responsible for deficits in object recognition observed with acute injection models [6, 53,64,68].
Chronic Aβ40 infusion
The fact that single acute injections of purified Aβ do not adequately model the pathophysiology of AD triggered attempts to chronically infuse Aβ into the ventricular system. The use of Aβ40 is particularly advantageous for chronic infusion studies because it is more soluble than Aβ42, and therefore is less prone to aggregation in the pump. The potential utility of Aβ40 infusions for inducing AD pathology is supported by evidence suggesting that the main genetic risk factor for sporadic AD, the presence of an apolipoprotein E4 (ApoE4) allele, increases parenchymal Aβ40 accumulation [23,28,40,44,67]. Nevertheless its role is complex, since Aβ40 limits Aβ42 aggregation and may even have protective effects [37]. Chronic infusions of Aβ40 typically result in behavioral deficits, but the reported extent and duration of these effects vary across studies [5,48]. A longitudinal study of Aβ40 infusion has suggested mild and reversible cognitive impairments [65]. Infusion of Aβ40 alone does not produce Thioflavin S-positive amyloid deposits, but Aβ-laden phagocytes are detectable in periventricular regions, presumably clearing the infused Aβ [18]. Injection of transforming growth factor β1 (TGFβ1), an immunomodulator, in conjunction with Aβ40 infusion facilitated the deposition of ThioflavinS positive cotton wool plaques frequently associated with the vasculature, demonstrating the important of inflammatory clearance mechanisms in attenuating deposition [18]. Cotton wool plaques (CWP) were first described in a Finnish kindred with a presenilin-1 Δ9 mutation and are large, ball-like plaques lacking dense amyloid cores associated with dystrophic neurites and microgliosis [35]. Further attempts to replicate this model of Aβ40 infusion-induced plaques using recombinant peptide failed, possibly since plaque induction may also require the presence of an unknown contaminant of the purified TGFβ used in the original study (TGFβ 1.2, R&D, a product that was discontinued in 1993). In a transgenic animal model of a TGFβ1 and AβPP/PDAβPP overexpression, TGFβ1 has been hypothesized to play a role in vascular but not neuropil amyloid deposition [72]. Lack of success in reproducibility with recombinant TGFβ1/Aβ40 infusion model to induce cotton wool plaques remains unexplained and may relate to a Aβ40-specific effect or that there was an unknown contaminant in the original TGFβ1 preparation that was necessary for induction of amyloid plaques. One possibility, copper, is discussed below.
Injection or infusion of Aβ (25–35 fragment)
Acute injections [42] or infusions of an Aβ fragment (25–35), which is thought to mediate Aβ-related toxicity has been shown to induce deficits in cognition and LTP [13,14,20,21,24,32,61]. The wide use of Aβ25-35 in many laboratories suggests that these effects are fairly reproducible. However, this synthetic fragment of Aβ is not found in the brain and does not represent all aspects of native Aβ. In particular, it does not include domains that interact with other molecules, such as those that are co-deposited in neuritic plaques (ApoE, α1-antichymotrypsin, ACT, heparan sulfate proteoglycans). Nevertheless, like Aβ, Aβ25-35 aggregates rapidly to form toxic species, but this is a disadvantage with chronic infusion since aggregation may occur within the infusion pump itself, resulting in precipitation and reduction in delivery. More significantly, it remains unclear whether Aβ25-35 can model the pathophysiological processes and protein-protein or protein-lipid interactions in AD.
Chronic infusion of Aβ42
Many researchers believe that Aβ42 is thought to play a more prominent role in AD pathophysiology than Aβ40 [78]. Chronic infusions with Aβ42 alone induce progressive cognitive deficits that correlate with increasing infusion duration [47]. Indeed, the Aβ42 model system has been used as a preclinical model to test various therapeutic interventions for AD [73–75]. Much like Aβ25-35, Aβ42 demonstrates an increased propensity of aggregation, particularly within the infusion pump when it is used in chronic infusion paradigms.
Problems with the Aβ42 chronic infusion models relate to lack of characterization of the pump infusate over time and lack of control for aggregation during infusion period. Another disadvantage is that although infused animals exhibit some Aβ42 toxicity, they do not form parenchymal amyloid plaques. Nevertheless, animal models that demonstrate oligomeric toxicity in the absence of plaque deposition may be useful since soluble oligomers concentrations may be better predictors of memory dysfunction than plaque density [38], and may play a causal role in AD [31,36].
Chronic co-infusion using human HDL with either Aβ42 or mixed Aβ42 and Aβ40
One approach to optimizing the utility of chronic Aβ42 infusion models is to limit the aggregation of Aβ42 within the infusion pump by co-infusing it with human HDL. HDL is a normal carrier of Aβ in the serum [33] and appears to inhibit Aβ42 aggregation [51]. Aβ40 also limits aggregation [37]. In our laboratory, we have co-infused HDL with Aβ40 and Aβ42, which results in increased oxidative damage, synaptic dysfunction, inflammatory changes, plaque deposition, and cognitive impairment compared to infusion of vehicle (HDL) [19]. HDL contains apolipoproteins such as apolipoprotein J (ApoJ) and ApoE, which bind Aβ, and although, HDL is carrier for Aβ in plasma, it is not a normal carrier in the brain. Nevertheless Aβ is found bound to HDL-like particles in cerebrospinal fluid. The Aβ-HDL infusion model has been useful for preclinical assessments of the efficacy of potential therapeutic interventions for AD [8–11,19,39,45]. However, different preparations of HDL and Aβ42 have not consistently induced cognitive deficits, suggesting that variations between batches of HDL and/or Aβ or additional unknown factors may be affecting aggregation within the infusion pump or toxicity in the brain parenchyma.
The purpose of this study was to improve the Aβ42/HDL infusion model in order to produce more consistent results. Our goals included developing in vitro methods for pre-screening preparations of Aβ oligomers for toxicity in order to predict cognitive deficits in our in vivo infusion models of AD. We wanted to avoid infusion of preparations that, for unforeseen reasons, do not yield stable toxic oligomers. We also focused on optimizing the ratio of Aβ/HDL concentrations in our preparation and the impact of two environmental risk factors that might confer vulnerability to Aβ infusion in vivo: copper exposure and insulin resistance (Type II diabetes).
Copper levels in drinking water can vary dramatically between different regions and may therefore be a crucial determinant of the reproducibility of Aβ infusion models. Epidemiological studies have suggested that higher levels of dietary copper intake are associated with an increased risk for cognitive decline [46]. Rabbits ingesting high levels of copper in drinking water who are on a high cholesterol diet show increased neuronal accumulation of Aβ [58]. Copper may also modulate Aβ aggregation [1] and vulnerability to oxidative damage [4]. Therefore, we investigated impact of doses of copper on Aβ toxicity and aggregation in vitro and then infused preparations with similar ratios in vivo to determine impact on cognitive performance.
Insulin resistance (Type II diabetes) is another major environmental risk factor for AD (see reviews [7, 12]), and may modulate the effects of Aβ infusion on cognitive deficits and synaptic loss. We induced insulin resistance in infused rats by maintaining them on a high fructose diet, a commonly used model for hyperinsulinemia, hypertriglyceridemia, and hypercholesterolemia [52].
MATERIALS AND METHODS
In vivo studies
Animals
Retired breeder Sprague-Dawley rats were used in this study. Surgical and animal care procedures were carried out following guidelines set out in the NIH Guide for the Care and Use of Laboratory Animals. Animals were maintained on a 12-hour light-dark cycle and provided with food and water ad libitum. Rats were either fed a control diet or a high fructose diet to induce insulin resistance [79] which is a well-accepted model [52]. Insulin resistance was induced by feeding rats 60% High Fructose Diet (Harlan Teklad; TD.89247, Madison WI) for 8 months prior to surgery.
Mini-osmotic pumps
Three in vivo studies were performed using mini-osmotic pumps that release their contents at a rate of 0.25 μl/hour in each lateral ventricle. All animals received continuous infusions for at least one month. In the first study, Aβ pumps were prepared as previously described, combining Aβ42 (25 μg/mL), Aβ40 (100 μg/mL), and HDL (750 μg/mL) using 10 rats per group [19]. In the second study, Aβ42 (25 μg/mL) was co-infused with different concentrations of HDL using 5 rats per group. In the third in vivo study, 6 to 7 rats per group were put on a high fructose diet to induce insulin resistance or a normal diet and infused with a toxic oligomeric preparation of Aβ42, HDL and copper sulfate at pump concentrations of copper sulfate (2 μM), HDL (210 μg/mL), and Aβ42 (120 μg/mL), which enhanced Aβ toxicity in in vitro studies. Pumps were replaced every 28 days to ensure continuous Aβ infusion.
Surgery
Animals received isoflurane anesthesia during the intracerebro-ventricular pump implantation procedure. A 10 mm incision was made in the skin overlying the skull. A double stainless steel catheter system with 3 mm spacing and 4 mm depth (Plastic One, 3280PD, Roanoke, VA) was stereotaxically implanted into the lateral ventricles at coordinates −0.6 mm AP, −1.4 mm ML to Bregma and fixed with medical grade acrylic cement. A 10 mm length of polyethylene tubing was used to connect each catheter to a mini-osmotic pumps (Alzet 1004, 100 μl volume, 0.25 μl/hr release rate) containing the infusion preparations, which were inserted through the cranial incision and implanted subcutaneously on the back. The cranial incision was closed with sterile stainless steel staples.
Morris water maze
Rats were tested on the Morris water maze task using a 2-meter diameter tank as previously described [19]. Water temperature was maintained at 21°C. Distal stationary cues were placed around the walls of room. No proximal or mobile cues were present. Swim latency and path length were determined with a video tracking system (HVS Image Ltd.; Buckingham, UK). Rats were first trained on a visible Morris water maze task for 3 consecutive days, with 4 trials per day. In this version of the task, the platform protrudes 1 cm above the surface of the water and is moved to a different location for each day of testing. They were subsequently tested on the hidden Morris water maze task, where the platform is submerged 2 cm below the surface of the water and is no longer visible to the swimming rat. Prior to the initial trial of the hidden platform task, rats were placed on platform for 30 seconds for spatial orientation. They were then trained for 5 consecutive days with 4 trials per day divided across two blocks. Rats were introduced to the pool facing the tank wall from one of 8 possible start points in a pseudo-randomized fashion, and allowed to search for the hidden platform for a maximum of 60 seconds. If the rat failed to locate the platform, it was guided to the platform by the experimenter. Rats were allowed to remain on the platform for 30 seconds before the start of the next trial.
Perfusion and tissue collection
Prior to euthanasia, rats were terminally anesthetized with pentobarbital and perfused with a non-fixative protease inhibitor buffer [19]. The brain was removed, and one hemisphere was immersion-fixed in 4% paraformaldehyde and paraffin-embedded, and the other hemisphere was dissected and snap frozen for biochemical analysis as previously described [19].
Western blots
Post-synaptic density protein-95 (PSD-95), NMDA-receptor 2B (NR2B), and actin were assayed in the lysis buffer fractions. Samples (30 μg) were loaded and electrophoresed onto a 7.5–15% Tris-glycine gel, and which was transferred to a PVDF membrane and blocked for 1 hour at room temperature in 5% milk with 0.05% Tween20 in PBS. The membrane was incubated overnight at 4°C with the following primary antibodies: mouse anti-PSD95 antibody (1:1,000; Upstate, Lake Placid, NY), mouse anti-NR2B (1:500; Biosource International, Camarillo, CA) and mouse anti-actin (1:2000; Chemicon International, Temecula, CA). Membranes were then incubated for 6 minutes at room temperature with horseradish peroxidase-conjugated anti-mouse (1:25,000) or anti-rabbit (1:100,000) secondary antibodies and developed with Super Femto chemiluminescence kits (Pierce, Rockford, IL).
Immunostaining and image analysis
8 μm paraffin sections were baked for 40 minutes at 55°C and cleared with Citrisolvent. Sections were then quenched in 0.3% hydrogen peroxide and methanol for 30 minutes at room temperature, and washed 3 times with TTBS buffer solution (pH 7.4). Sections for Aβ staining were pre-treated with 50% formic acid for 20 minutes at room temperature. Sections for Ne-uN were steamed for 1 hour using a citrate buffer. Sections for Iba-1 were treated with 0.3% Triton in TBS buffer for 10 minutes at room temperature. All sections were subsequently blocked using 5% normal horse serum (NHS) or normal goat serum (NGS) containing 3% BSA/TTBS for 1 hour at 37°C. Primary antibody was incubated for 1 hour at 37°C and 24 hours at 4°C. Aβ deposits were labeled with 10G4 (Aβ5-13) (1:500 [76]). Microglia were labeled with anti-Iba-1 rabbit polyclonal antibody (1:200; Wako, Richmond VA). Neuron loss was determined through staining with anti-NeuN antibody (1:5,000; Chemicon, Temecula, CA). Sections were then blocked with 1.5% NHS or NGS containing 3% BSA/TTBS for 1 hour, incubated with secondary antibody (Vector biotinylated horse anti-mouse or goat anti-rabbit antibody; 1:1200) for 1 hour, then treated with ABC reagent (Vector Labs, Burlingame, CA) followed by metal peroxidase di-aminobenzidine (DAB; Pierce, Rockford, IL) for single labeling. Microscopic images were acquired and analyzed using NIH Image software to quantify amyloid plaque, microglia, and neuron counts, size, and density in the cortical and hippocampal layers.
In vitro studies
Aβ oligomer preparation
Aβ42 oligomer was prepared as previously described [77]. Briefly, double-lyophilized synthetic Aβ1-42 peptide (Charles Glabe, UCI) was monomerized by solubilization in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP). Fibril formation was minimized during evaporation by the addition of sterilized water to achieve a concentration of 315 μg/ml and subsequently by adding HDL (EMD Biosciences, La Jolla, CA; stock H solution 525 μg/ml) to achieve desired Aβ:HDL ratios. For the pre-aggregated Aβ preparations, HDL (EMD Biosciences, cat. 437641) and copper sulfate (Sigma, St. Louis, MO) were added to 1 mg/ml of Aβ42 (after HFIP evaporation). This solution was continuously stirred at 600 rpm for 48 hours at 25°C.
Aβ oligomer characterization by Western blot
Fresh preparations of co-incubated and pre-aggregated Aβ oligomers were incubated at 37°C for 2 days. 500 ng samples of each Aβ oligomer preparation were electrophoresed at 100 V on a 10–20% Tris-Tricine SDS gel for 45 minutes to 1 hour, transferred to a PDVF membrane and blocked for 1 hour at 37°C with 10% nonfat milk in PBS and 0.1% gelatin. The blots were then incubated probed with 6E10 (1:10,000) and A11 (1:4,000) overnight at 4°C, followed by goat anti-mouse (1:10,000) or anti-rabbit (1:50,000) horseradish peroxidase conjugated secondary antibodies for 1 hour at 25°C, and then developed with Supersignal (Pierce, 1:80).
Cell culture
To evaluate the impact of HDL and copper-induced Aβ oligomer toxicity, Aβ oligomer preparations with or without HDL and copper were added to human APPsw transfected N2A cells (gift of Dr. S. Sisodia) cultured in 50% Dulbecco’s modified Eagle’s medium, 50% Opti-MEM (Invitrogen, Carlsbad, CA), 5% fetal bovine serum, 200 μg/ml Glutamax. Cells were plated at equal densities (8000 cells/well) with 1.5% bovine calf serum and maintained at 37°C in an atmosphere of 5% CO2. The medium was removed before treatment and replaced with 70% Dulbecco’s modified Eagle’s medium and 30% Opti-MEM with 0.1% BSA. After 48 hours of incubation, lactate dehydrogenase (LDH) levels leaking into media were measured as an index of toxicity, using a CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, Madison, WI), with absorption read at 490 nm. Maximal LDH release was determined by measuring LDH release after lysing cells with 1% Triton-X. Each data point was determined in triplicate, and the S.D. values did not exceed 5%.
Statistical analysis
Assays comparing Aβ oligomer preparations with or without HDL and copper were analyzed by one-way ANOVAs, followed by post-hoc Bonferroni/Dunn tests (SPSS 14.0 for Windows; SPSS Inc., Chicago IL). Western blot densitometry was analyzed by one-way ANOVA analysis (Graph Pad InStat v.3.05, San Diego, CA). Coefficient of correlation and significance of the degree of linear relationship between parameters were determined with a simple regression model using StatView (SAS Institute Inc, Cary, NC). All experiments were performed in triplicate or quadruplicate (n = 3 – 4). Performance on the hidden platform version of the Morris water maze was analyzed using repeated-measures ANOVAs and independent-samples t-tests (SPSS 14.0 for Windows; SPSS Inc., Chicago IL).
RESULTS
Our previous work indicated that co-infusion of Aβ40 and Aβ42 with HDL caused cognitive and/or synaptic deficits [19], but the relationship between HDL dose, toxicity and amyloid deposition when only Aβ42, the more toxic form of Aβ, was infused, remained unclear. Therefore, we co-infused a continuous dose of Aβ42 (25μg/200 μl) with different doses of HDL and examined amyloid deposition and neurotoxicity, as defined by loss of NeuN in the entorhinal cortex (Fig. 1). HDL co-infusion caused a dose-dependent decrease in Aβ deposition measured by two monoclonal antibodies to Aβ, but a dose-dependent increase in toxicity. HDL alone was not toxic even at the highest doses used (data not shown).
Fig. 1. Impact of HDL dose on Aβ deposition and neurotoxicity.
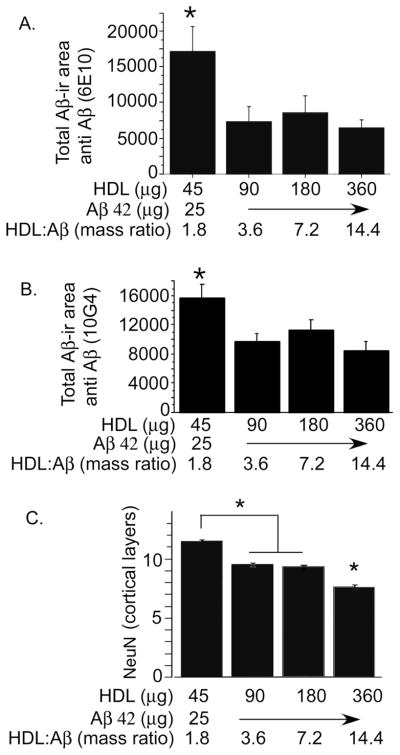
Aβ42 was icv infused at 25 μg/200 μl (0.2% DMSO) with different mass ratios of HDL. Rats were sacrificed at 4 weeks post-infusion and evaluated for Aβ deposits using monoclonal antibodies 6E10 (A) or 10G4 (B). Neuron nuclei staining in layer 2 of the entorhinal cortex was evaluated using NeuN (C). Values are shown as the mean ± SD. * p < 0.05 represent a significant difference between vehicle (HDL) and treatment (HDL + Aβ).
Since high molecular weight Aβ oligomers appear to play a critical role in AD pathogenesis [3,30,36], we investigated the impact of HDL on neurotoxicity induced by pre-aggregated toxic high molecular weight Aβ oligomers in vivo and in vitro. Oligomer preparations were positive for conformation-sensitive anti-oligomer antibody A11 by dot blot [30]. As expected, this preparation was toxic in vitro to neuroblastoma cells as evident by excessive lactate dehydrogenase (LDH) leakage into media, but the addition of HDL resulted in dose-dependent protection from pre-formed Aβ42 oligomer (Fig. 2A). Next, we investigated the impact of co-stirring freshly dissolved (not pre-aggregated) Aβ with HDL for 48 hours, and found that with this protocol, HDL exacerbated toxicity (Fig. 2B), suggesting that such preparations may be useful for continuous pump infusion. We then incubated the preparation for 2 weeks at 37°C and observed it retained its toxicity over this interval (Fig. 2C). Immunoblotting these preparations on a Tris-Tricine gel with anti-Aβ antibody 6E10 revealed that the soluble fractions of the preparations containing HDL exhibited a greater concentration of soluble high molecular weight oligomers, even after 2 weeks (Fig. 2D). Without HDL, soluble oligomers were greatly reduced by 2 weeks incubation compared to the initial preparation (not shown). Morphological examination of cells (Figures 2E-2H) revealed that, compared to vehicle (HDL, Fig. 2E), Aβ alone slightly increased the number of pyknotic cells (Fig. 2F), while the co-aggregated Aβ-HDL preparation caused more dramatic shrinkage and pyknosis (Fig. 2G). Pyknosis was less pronounced after treatment with the pre-aggregated Aβ-HDL preparation (Fig. 2H).
Fig. 2. Differential effect of HDL on Aβ toxicity in AβPP SwN2A neuroblastoma cells, depending on whether Aβ42 is pre-aggregated.

N2A neuroblastoma cells were incubated for 48h with different mass ratios of HDL:Aβ and toxicity determined by maximum LDH release and morphological assessment. A. HDL caused dose-dependent protection from toxicity caused by pre-aggregated Aβ. B. But if unaggregated Aβ was co-stirred (co-aggregated) with different concentrations of HDL for 48 hours and then applied to N2A cells, HDL exacerbated Aβ toxicity. C. Toxicity of freshly made preparations of unaggregated Aβ (co-stirred with HDL) was compared to that of a preparation incubated for 2 weeks at 37°C. D. Aβ alone (not shown) and co-stirred Aβ-HDL preparations (fresh and incubated for 2 weeks) were spun down, and the soluble fraction was immunblotted with 6E10. E–H. Neuroblastoma cells were stained non-specifically with diaminobenzidine (not quenched with H202) to increase contrast and visualization of cytoplasm to reveal morphological or pyknotic changes. Magnification bar = 100 μm. Values are shown as the mean ± SD. * p < 0.05, ** p < 0.01 and *** p < 0.001 represents a significant difference between vehicle (HDL) and treatment with Aβ + HDL (co-aggregated) or with preaggregated Aβ +HDL.
After 5 weeks of infusion animals, infused with s pre-aggregated Aβ-HDL preparation demonstrated significant elevations in the synaptic proteins PSD-95 and NR2B, a type of subunit of the ionotropic N-methyl D aspartate (NMDA) receptor (Fig. 3A). We previously described to be selectively and dramatically reduced after Aβ infusion[19]. After 15 weeks of infusion these proteins were significantly diminished (Fig. 3B) relative to vehicle-treated animals.
Fig. 3. Delayed post-synaptic protein loss in the hippocampus after pre-aggregating Aβ is co-infused with HDL.

Rats were infused with pre-aggregated Aβ which was shown to be A11 positive on dot blot and neurotoxic at 100 nM in N2A cells. HDL was added in the pump to further attenuate aggregation and rats as previously described. Rats were sacrificed at 5 (A) and 15 weeks post infusion (B). Hippocampal lysates were immunobloted for post- synaptic proteins, normalized to β-actin and quantified. Values are shown as the mean ± SD. * p < 0.05 represent a significant difference between vehicle (HDL) and treatment (HDL +pre-aggregated Aβ).
Epidemiological, in vivo, and in vitro data all suggest a role for copper in AD pathogenesis. Therefore, we also investigated the impact of copper concentrations upon the toxicity of our infusion preparations. Incorporating a high dose of copper (5.6 mM) precipitated the Aβ so the addition of the remaining soluble fraction to cells was not toxic (data not shown). In contrast, lower copper doses, ranging from 2 to 30 μM, enhanced Aβ-HDL toxicity (Fig. 4A). At these concentrations, copper did not appear to disrupt oligomer (A11-immunoreactive) bands in the soluble fraction (Fig. 4B). Similar results were obtained with 6E10 (data not shown).
Fig. 4. Low dose copper enhances toxicity in N2A neuroblastoma cell without destabilizing soluble oligomers.

To determine the copper effect on Aβ, N2A neuroblastoma cells were incubated for 48h with different doses of copper with and without HDL. A. At lower concentrations (0.5–30 μM) copper exacerbated toxicity caused by pre-aggregated Aβ or Aβ co-stirred with HDL. B. These preparations were electrophoresed on Western blot and determined oligomeric band by conformationally specific anti-oligomer antibody A11. Values are shown as the mean ± SD. * p < 0.05, ** p < 0.01 and *** p < 0.001 represents a significant difference between vehicle (HDL+ copper) and treatment (co-aggregated Aβ with HDL or co-aggregated Aβ with HDL and copper).
We infused the Aβ/HDL/copper preparation into 15 month old rats that were fed either a control diet or a high fructose diet for 4 months to induce insulin resistance [52]. Behavioral testing on the Morris water maze was performed 5–7 weeks post initiation of infusion. Average swim speeds in the two groups were similar [Aβ = 26.7 cm/s, vehicle = 28.4 cm/s; F (1, 14) = 1.19, p = 0.29]. However, across all 5 days of testing, the Aβ group exhibited significantly longer latency to the hidden platform than the vehicle group [Fig. 5A; F (1, 12) = 5.93; p = 0.031], but there was no effect of diet [data not shown; F (1, 12) = 0.18; p = 0.68]. The poorer performance of the Aβ group appeared to be attributable to significantly greater proportion of thigmotaxic (i.e. wall-hugging) swimming patterns during the acquisition trials [Fig. 5B; F (1, 14) = 7.70, p = 0.015].
Fig. 5. Behavioral effect of co-aggregated copper- Aβ-HDL by Morris Water Maze.
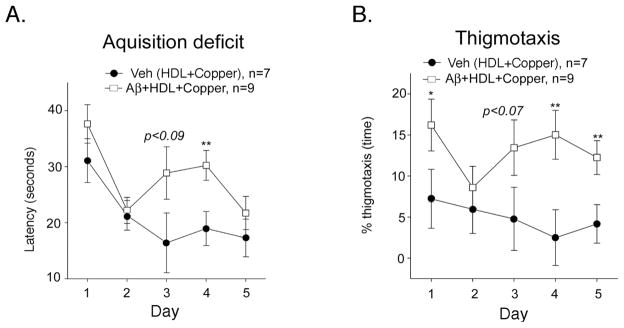
Co-aggregated Aβ-HDL-Copper or vehicle (copper-HDL) was icv infused into Sprague Dawley rats and behavior conducted between 5–7 weeks post infusion. There were no differences in performance to find a visible platform or in swim speeds (not shown), but Aβ-HDL-Copper-infused animals showed acquisition deficits to find a hidden platform (A) and an increase in thigmotaxis (B). Values are shown as the mean ± SEM. * p < 0.05 and ** p < 0.01 represent a significant differences between vehicle (HDL+copper) and treatment (Aβ co-aggregated with HDL and copper).
These rats were subsequently euthanized and their hippocampi were analyzed for post-synaptic NR2B levels (Fig. 6A). Analysis showed a diet x treatment interaction (p < 0.01). There was no Aβ-dependent NR2B depletion among animals maintained on the regular diet. However, NR2B levels were significantly reduced in insulin-resistant animals maintained on the high-fructose diet (Fig. 6B).
Fig. 6. Impact of insulin resistance on Aβ infusion-induced loss of post-synaptic protein NR2B.

Male Sprague Dawley rats were fed a high fructose diet to induce insulin resistance or a regular diet for 4 months, and then at 15 months of age infused with vehicle or Aβ co-aggregated with HDL and copper. Animals were sacrificed 6 weeks later at completion of behavioral analysis and representative lanes from a Western of hippocampal lysate is shown for the post-synaptic protein NR2B and β-actin (A). Western quantification of diet dependent effect on NR2B normalized to β-actin (B). 2×2 ANOVA (Treatment x Diet) revealed significant treatment-diet interaction (p < 0.01). RD; Regular Diet, HFD; High Fructose Diet. Values are shown as the mean ± SD. * p < 0.05 represent a significant difference between vehicle (HDL +copper) and treatment (Aβ co-aggregated with HDL and copper).
Similar hippocampal amyloid plaque densities were seen in Aβ infused animals maintained on either the regular and high-fructose diets (Fig. 7A). However, the difference in Aβ staining was more robust in the regular diet group (p < 0.05), possibly because the high-fructose diet may have increased endogenous Aβ deposition in the vehicle-treated group. Aβ infusion also significantly increased microgliosis in the hippocampus (Fig. 7B). This response was more robust in the animals in the high-fructose diet group (p < 0.01).
Fig. 7. Impact of insulin resistance on Aβ infusion-induced Aβ deposition and microgliosis (Iba-1).

Rats were fed a high fructose diet to induce insulin resistance or a regular diet, and then icv- infused with Aβ co-aggregated with HDL and copper or vehicle (HDL+copper). Sections were immunostained with monoclonal antibody against Aβ (10G4). Representative micrographs of hippocampus are shown, and Aβ burden quantified using image analysis (A). Sections were also stained for antibody to microglia (Iba-1). Representative sections are shown and quantified by image analysis (B). Magnification bar = 100 μm. Values are shown as the mean ± SD. * p < 0.05, ** p < 0.01 and *** p < 0.001 represents a significant difference between vehicle (HDL +copper) and treatment (Aβ co-aggregated with HDL and copper) within diet.
DISCUSSION
The primary advantages of intracerebroventricular (icv) Aβ infusion models of AD over AβPP and/or PS1 transgenic models include the ability to control amyloid dosing, produce neurotoxicity, and avoid the high costs of colony maintenance and patent rights associated with transgenic animals. Many different chronic icv infusion models of AD that incorporate mini-osmotic pumps have been used in the past decade for pre-clinical studies, but inconsistent reproducibility and characterization of the Aβ preparations, particularly in regards to potential aggregation within the pump, have limited widespread use and broad acceptance of any particular model. In the work presented here, we have attempted to optimize the production of toxic Aβ preparations that efficiently produce measurable cognitive and synaptic deficits and remain stable within a chronically implanted infusion pump. Our results indicate that assessing the neurotoxicity of these preparations in vitro can help identify those that are most likely to exhibit greater efficacy in vivo.
In vivo performance of infused Aβ preparations appears to be critically dependent on their formulation. In Aβ42 preparations that are not pre-aggregated, the addition of increasing doses of HDL diminished Aβ deposition, but enhanced toxicity. The inverse relationship between Aβ deposition and neurotoxicity is consistent with published data demonstrating that soluble Aβ42 oligomers are toxic, block long-term potentiation, and induce cognitive deficits. These findings also raise important questions regarding the utility of using amyloid plaque burden in AβPP transgenic models as the primary outcome measure used to predict the potential translational value of candidate drugs.
Our data indicate that HDL restricts Aβ aggregation and are consistent with prior work [51]. Inhibition of Aβ aggregation by HDL represents a potential underlying mechanism for its protective effects in vitro that are reported here and elsewhere [15]. However, another study demonstrated that when HDL is incubated for 5 days at 37°C with a shorter peptide, Aβ40, it can promote increased Aβ fibrillization [59]. These radically different results led us to explore the impact of Aβ pre-aggregation on neurotoxicity.
When we compared preparations of HDL with pre-aggregated Aβ42 or freshly dissolved Aβ42, we found that the freshly dissolved Aβ42 preparation exhibited much greater Aβ toxicity, even after 2 weeks at 37°C. This more neurotoxic preparation contained a significant proportion of oligomeric species that remained stable over an extended period. These results suggest that HDL plays a critical role modulating Aβ toxicity through its regulation of Aβ aggregation.
We then explored the impact of pre-aggregated Aβ in our Aβ-HDL infusion model. After 5 weeks of infusion with this preparation, markers of synaptic function were surprisingly elevated (Fig. 3A). However, after 15 weeks of infusion, post-synaptic protein levels were significantly reduced in the Aβ group relative to the vehicle group (Fig. 3B). Thus, the addition of HDL to pre-aggregated Aβ appeared to delay the onset of synaptic dysfunction in vivo, which is consistent with the neuroprotective effects of this preparation in vitro. Similarly, delayed deficits in cognition have also been described, particularly when Aβ42 preparations are used [47].
Inconsistencies in Aβ preparations make it difficult to create a reproducible infusion model of AD and highlight the importance of designing preparations in which stability and toxicity of Aβ can be pre-tested in vitro to ensure consistent in vivo results. The mechanisms underlying the protective effects of pre-formed Aβ aggregates remain elusive, and in particular, the divergent impact of HDL on Aβ toxicity depending on whether the Aβ preparation is pre-aggregated or freshly dissolved may seem paradoxical. However, the effects of HDL on Aβ42 resemble those seen with ApoJ, which increases Aβ42 aggregation and toxicity under some conditions [49], but exerts neuroprotective effects by inhibiting Aβ42 aggregation under other conditions [2, 41].
We also explored the effect of copper on Aβ-related toxicity in our infusion model. Copper is known to regulate Aβ aggregation and toxicity [1], and increased copper intake appears to increase dementia risk, particularly in conjunction with diets high in trans-fats [46]. Furthermore, even relatively low levels of copper in drinking water can increase AD pathology in other animal models, particularly if in combination with high cholesterol [58,71]. Based on this, we hypothesized that variations in copper levels in the water supplies of different laboratories might be related to the differences in the efficacy of Aβ infusion preparations between groups. The addition of copper to our preparations stabilized soluble Aβ oligomers and exacerbated amyloid toxicity in vitro across a range of levels that are acceptable in drinking water.
The impact of insulin resistance on accelerating onset of post-synaptic loss and exacerbating microgliosis may suggest that insulin resistant diabetes, a major risk factor for AD [7,12], increases vulnerability to Aβ toxicity and may dysregulate microglial responses. In addition, these results may suggest that use of insulin resistant diets such as the one used here, a high fructose diet, may facilitate modeling neurodegeneration in the disease, particularly since it has already been shown to increase amyloidogenesis in AβPP transgenic models [26].
In conclusion, we have shown that the outcomes in Aβ infusion experiments are critically dependent on details of the protocol. We have improved our Aβ42 in vivo infusion model, leading to more rapid onset of deficits by optimizing HDL and copper concentrations. Because copper in the water has been implicated as a risk factor for AD that interacts with dietary lipid intake, there may be an interaction with lipoprotein status in humans. We have successfully used this HDL + copper co-aggregation paradigm to evaluate an environmental risk factor for AD, insulin resistance. With further optimization, this model system, which can reveal significant Aβ-dependent neurodegeneration and cognitive deterioration in a short time, is likely to prove useful in understanding the mechanisms and time course for Aβ-dependent neurodegeneration and the role of environmental risk factors and efficacy of neuroprotective drugs.
Acknowledgments
This work was supported by NIH grants RO1AG685 and AG21795.
Footnotes
The authors state that there are no financial conflicts of interests.
References
- 1.Atwood CS, Perry G, Zeng H, Kato Y, Jones WD, Ling KQ, Huang X, Moir RD, Wang D, Sayre LM, Smith MA, Chen SG, Bush AI. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry. 2004;43:560–568. doi: 10.1021/bi0358824. [DOI] [PubMed] [Google Scholar]
- 2.Calero M, Tokuda T, Rostagno A, Kumar A, Zlokovic B, Frangione B, Ghiso J. Functional and structural properties of lipid-associated apolipoprotein J (clusterin) Biochem J. 1999;344(Pt 2):375–383. [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puolivali J, Lesne S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- 4.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 5.Cleary J, Hittner JM, Semotuk M, Mantyh P, O’Hare E. b-amyloid (1-40) effects on behavior and memory. Brain Res. 1995;682:69–74. doi: 10.1016/0006-8993(95)00323-i. [DOI] [PubMed] [Google Scholar]
- 6.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 7.Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer’s Disease. Exp Gerontol. 2007;42:10–21. doi: 10.1016/j.exger.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 8.Craft JM, Van Eldik LJ, Zasadzki M, Hu W, Watterson DM. Aminopyridazines attenuate hippocampus-dependent behavioral deficits induced by human beta-amyloid in a murine model of neuroinflammation. J Mol Neurosci. 2004;24:115–122. doi: 10.1385/JMN:24:1:115. [DOI] [PubMed] [Google Scholar]
- 9.Craft JM, Watterson DM, Frautschy SA, Van Eldik LJ. Aminopyridazines inhibit beta-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol Aging. 2004;25:1283–1292. doi: 10.1016/j.neurobiolaging.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Craft JM, Watterson DM, Hirsch E, Van Eldik LJ. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J Neuroinflammation. 2005;2:15. doi: 10.1186/1742-2094-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craft JM, Watterson DM, Marks A, Van Eldik LJ. Enhanced susceptibility of S-100B transgenic mice to neuroinflammation and neuronal dysfunction induced by intracerebroventricular infusion of human beta-amyloid. Glia. 2005;51:209–216. doi: 10.1002/glia.20194. [DOI] [PubMed] [Google Scholar]
- 12.Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–152. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- 13.Dall’Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA, Lara DR. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25–35)-induced cognitive deficits in mice. Exp Neurol. 2007;203:241–245. doi: 10.1016/j.expneurol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Fang F, Liu GT. Protective effects of compound FLZ on beta-amyloid peptide-(25-35)-induced mouse hippocampal injury and learning and memory impairment. Acta Pharmacol Sin. 2006;27:651–658. doi: 10.1111/j.1745-7254.2006.00347.x. [DOI] [PubMed] [Google Scholar]
- 15.Farhangrazi ZS, Ying H, Bu G, Dugan LL, Fagan AM, Choi DW, Holtzman DM. High density lipoprotein decreases beta-amyloid toxicity in cortical cell culture. Neuroreport. 1997;8:1127–1130. doi: 10.1097/00001756-199703240-00013. [DOI] [PubMed] [Google Scholar]
- 16.Frautschy S, Cole GM, Baird A. Phagocytosis and deposition of vascular b-amyloid in rat brains injected with Alzheimer b-amyloid. Am J Pathol. 1992;140:1389–1399. [PMC free article] [PubMed] [Google Scholar]
- 17.Frautschy SA, Baird A, Cole GM. Effects of injected Alzheimer –amyloid cores in rat brain. Proc Natl Acad Sci USA. 1991;88:8362–8366. doi: 10.1073/pnas.88.19.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frautschy SA, Yang F, Calderón L, Cole GM. Rodent models of Alzheimer’s disease: rat Aβ infusion approaches to amyloid deposits. Neurobiol Aging. 1996;17:311–321. doi: 10.1016/0197-4580(95)02073-x. [DOI] [PubMed] [Google Scholar]
- 19.Frautschy SA, Hu W, Kim P, Miller SA, Chu T, Harris-White ME, Cole GM. Phenolic anti-inflammatory antioxidant reversal of Abeta-induced cognitive deficits and neuropathology. Neurobiol Aging. 2001;22:993–1005. doi: 10.1016/s0197-4580(01)00300-1. [DOI] [PubMed] [Google Scholar]
- 20.Freir DB, Holscher C, Herron CE. Blockade of long-term potentiation by beta-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J Neurophysiol. 2001;85:708–713. doi: 10.1152/jn.2001.85.2.708. [DOI] [PubMed] [Google Scholar]
- 21.Fu AL, Dong ZH, Sun MJ. Protective effect of N-acetyl-L-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res. 2006;1109:201–206. doi: 10.1016/j.brainres.2006.06.042. [DOI] [PubMed] [Google Scholar]
- 22.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagoplan S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F b-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 23.Gearing M, Mori H, Mirra SS. Abeta-peptide length and apolipoprotein E genotype in Alzheimer’s disease. Ann Neurol. 1996;39:395–399. doi: 10.1002/ana.410390320. [DOI] [PubMed] [Google Scholar]
- 24.Gengler S, Gault VA, Harriott P, Holscher C. Impairments of hippocampal synaptic plasticity induced by aggregated beta-amyloid (25–35) are dependent on stimulation-protocol and genetic background. Exp Brain Res. 2007;179:621–630. doi: 10.1007/s00221-006-0819-6. [DOI] [PubMed] [Google Scholar]
- 25.Head E, Cotman CW, Milgram NW. Canine cognition, aging and neuropathology. Introduction. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24:671–673. doi: 10.1016/s0278-5846(00)00100-7. [DOI] [PubMed] [Google Scholar]
- 26.Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 27.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Ab elevation and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 28.Ishii K, Tamaoka A, Mizusawa H, Shoji S, Ohtake T, Fraser PE, Takahashi H, Tsuji S, Gearing M, Mizutani T, Yamada S, Kato M, St George-Hyslop PH, Mirra SS, Mori H. Abeta1-40 but not Abeta1-42 levels in cortex correlate with apolipoprotein E epsilon4 allele dosage in sporadic Alzheimer’s disease. Brain Res. 1997;748:250–252. doi: 10.1016/s0006-8993(96)01363-7. [DOI] [PubMed] [Google Scholar]
- 29.Jhoo JH, Kim HC, Nabeshima T, Yamada K, Shin EJ, Jhoo WK, Kim W, Kang KS, Jo SA, Woo JI. Beta-amyloid (1-42)-induced learning and memory deficits in mice: involvement of oxidative burdens in the hippocampus and cerebral cortex. Behav Brain Res. 2004;155:185–196. doi: 10.1016/j.bbr.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 30.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 31.Klein WL, Krafft GA, Finch CE. Targeting small Aβ oligomers: the solution to an Alzheimer’s disease conundrum? TINS. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 32.Kong LN, Zuo PP, Mu L, Liu YY, Yang N. Gene expression profile of amyloid beta protein-injected mouse model for Alzheimer disease. Acta Pharmacol Sin. 2005;26:666–672. doi: 10.1111/j.1745-7254.2005.00129.x. [DOI] [PubMed] [Google Scholar]
- 33.Koudinov AR, Berezov TT, Kumar A, Koudinova NV. Alzheimer’s amyloid beta interaction with normal human plasma high density lipoprotein: association with apolipoprotein and lipids. Clin Chim Acta. 1998;270:75–84. doi: 10.1016/s0009-8981(97)00207-6. [DOI] [PubMed] [Google Scholar]
- 34.Kowall NW, Beal MF, Busciglio J, Duffy LK, Yankner BA. An in vivo model for the neurodegenerative effects of b amyloid and protection by substance P. Proc Natl Acad Sci USA. 1991;88:7247–7251. doi: 10.1073/pnas.88.16.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le TV, Crook R, Hardy J, Dickson DW. Cotton wool plaques in non-familial late-onset Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:1051–1061. doi: 10.1093/jnen/60.11.1051. [DOI] [PubMed] [Google Scholar]
- 36.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CC, Yang A, Gallagher M, Ashe KH. A specific amyloid-β assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 37.Levites Y, Jansen K, Smithson LA, Dakin R, Holloway VM, Das P, Golde TE. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J Neurosci. 2006;26:11923–11928. doi: 10.1523/JNEUROSCI.2795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malm T, Ort M, Tahtivaara L, Jukarainen N, Goldsteins G, Puolivali J, Nurmi A, Pussinen R, Ahtoniemi T, Miettinen TK, Kanninen K, Leskinen S, Vartiainen N, Yrjanheikki J, Laatikainen R, Harris-White ME, Koistinaho M, Frautschy SA, Bures J, Koistinaho J. beta-Amyloid infusion results in delayed and age-dependent learning deficits without role of inflammation or beta-amyloid deposits. Proc Natl Acad Sci USA. 2006;103:8852–8857. doi: 10.1073/pnas.0602896103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mann DMA, Iwatsubo T, Pickering-Brown SM, Owen F, Saido TC, Perry RH. Preferential deposition of amyloid b protein (Ab) in the form of Ab40 in Alzheimer’s disease is associated with a gene dosage effect of the apolipoprotein E E4 allele. Neurosci Lett. 1997;221:81–84. doi: 10.1016/s0304-3940(96)13294-8. [DOI] [PubMed] [Google Scholar]
- 41.Matsubara E, Soto C, Governale S, Frangione B, Ghiso J. Apoliprotein J and Alzheimer’s amyloid solubility. Biochem J. 1996;316:671–679. doi: 10.1042/bj3160671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maurice T, Lockhart BP, Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- 43.McDonald MP, Dahl EE, Overmier JB. Effects of exogenous b-amyloid peptide on retention for spatial learning. Behavioral and Neural Biology. 1994;62:60–67. doi: 10.1016/s0163-1047(05)80059-7. [DOI] [PubMed] [Google Scholar]
- 44.McNamara MJ, Gomez-Isla T, Hyman BT. Apolipoprotein E genotype and deposits of Abeta40 and Abeta42 in Alzheimer disease. Arch Neurol. 1998;55:1001–1004. doi: 10.1001/archneur.55.7.1001. [DOI] [PubMed] [Google Scholar]
- 45.Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, Frautschy SA, Cole GM. Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer’s models. Neuropsychopharmacology. 2005;30:1111–1120. doi: 10.1038/sj.npp.1300668. [DOI] [PubMed] [Google Scholar]
- 46.Morris MC, Evans DA, Tangney CC, Bienias JL, Schneider JA, Wilson RS, Scherr PA. Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch Neurol. 2006;63:1085–1088. doi: 10.1001/archneur.63.8.1085. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura S, Murayama N, Noshita T, Annoura H, Ohno T. Progressive brain dysfunction following intracerebroventricular infusion of beta(1-42)-amyloid peptide. Brain Res. 2001;912:128–136. doi: 10.1016/s0006-8993(01)02704-4. [DOI] [PubMed] [Google Scholar]
- 48.Nitta A, Fukuta T, Hasegawa T, Nabeshima T. Continuous infusion of beta-amyloid protein into the rat cerebral ventricle induces learning impairment and neuronal and morphological degeneration. Jpn J Pharmacol. 1997;73:51–57. doi: 10.1254/jjp.73.51. [DOI] [PubMed] [Google Scholar]
- 49.Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti G, Morgan TE, Rozovsky I, Blaine Stine W, Snyder SW, Holzman TF, Krafft GA, Finch CE. Clusterin (apoJ) alters the aggregation of amyloid-peptide (Aβ1-42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp Neurol. 1995;136:22–31. doi: 10.1006/exnr.1995.1080. [DOI] [PubMed] [Google Scholar]
- 50.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 51.Olesen OF, Dago L. High density lipoprotein inhibits assembly of amyloid beta-peptides into fibrils. Biochem Biophys Res Commun. 2000;270:62–66. doi: 10.1006/bbrc.2000.2372. [DOI] [PubMed] [Google Scholar]
- 52.Oron-Herman M, Kamari Y, Grossman E, Yeger G, Peleg E, Shabtay Z, Shamiss A, Sharabi Y. Metabolic Syndrome: Comparison of the Two Commonly Used Animal Models. Am J Hypertens. 2008 doi: 10.1038/ajh.2008.218. [DOI] [PubMed] [Google Scholar]
- 53.Poling A, Paisley-Morgan K, Panos JJ, Kim EM, O’Hare E, Cleary JP, Lesne S, Ashe KH, Porritt M, Baker L. Oligomers of the amyloid-beta protein disrupt working memory: Confirmation with two behavioral procedures. Behav Brain Res. 2008 doi: 10.1016/j.bbr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roch JM, Masliah E, Roch-Levecq AC, Sundsmo MP, Otero DAC, Veinbergs I, Saitoh T. Increase of synaptic density and memory retention by a peptide representing the trophic domain of the amyloid b/A4 protein precursor. Proc Natl Acad Sci USA. 1994;91:7450–7454. doi: 10.1073/pnas.91.16.7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwab C, Hosokawa M, McGeer PL. Transgenic mice overexpressing amyloid beta protein are an incomplete model of Alzheimer disease. Exp Neurol. 2004;188:52–64. doi: 10.1016/j.expneurol.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 57.Sommer B, Sturchler-Pierrat C, Abramowski D, Wiederhold K, Calhoun M, Jucker M, Kelly P, Staufenbiel M. Transgenic approaches to model Alzheimer’s disease. Reviews in the Neurosciences. 2000;11:47–51. doi: 10.1515/revneuro.2000.11.1.47. [DOI] [PubMed] [Google Scholar]
- 58.Sparks DL, Schreurs BG. Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2003;100:11065–11069. doi: 10.1073/pnas.1832769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stanyer L, Betteridge DJ, Smith CC. An investigation into the mechanisms mediating plasma lipoprotein-potentiated beta-amyloid fibrillogenesis. FEBS Lett. 2002;518:72–78. doi: 10.1016/s0014-5793(02)02646-7. [DOI] [PubMed] [Google Scholar]
- 60.Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun MK, Alkon DL. Impairment of hippocampal CA1 heterosynaptic transformation and spatial memory by beta-amyloid(25–35) J Neurophysiol. 2002;87:2441–2449. doi: 10.1152/jn.00230.2001. [DOI] [PubMed] [Google Scholar]
- 62.Szczepanik AM, Ringheim GE. IL-10 and glucocorticoids inhibit Abeta(1–42)- and lipopolysaccharide-induced pro-inflammatory cytokine and chemokine induction in the central nervous system. J Alzheimers Dis. 2003;5:105–117. doi: 10.3233/jad-2003-5205. [DOI] [PubMed] [Google Scholar]
- 63.Tapp PD, Siwak CT, Gao FQ, Chiou JY, Black SE, Head E, Muggenburg BA, Cotman CW, Milgram NW, Su MY. Frontal lobe volume, function, and beta-amyloid pathology in a canine model of aging. J Neurosci. 2004;24:8205–8213. doi: 10.1523/JNEUROSCI.1339-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Townsend M, Cleary JP, Mehta T, Hofmeister J, Lesne S, O’Hare E, Walsh DM, Selkoe DJ. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol. 2006;60:668–676. doi: 10.1002/ana.21051. [DOI] [PubMed] [Google Scholar]
- 65.von Linstow Roloff E, Platt B, Riedel G. No spatial working memory deficit in beta-amyloid-exposed rats. A longitudinal study. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:955–970. doi: 10.1016/s0278-5846(02)00211-7. [DOI] [PubMed] [Google Scholar]
- 66.Waite J, Cole GM, Frautschy SA, Connor DJ, Thal LJ. Solvent Effects on Beta Protein Toxicity In Vivo. Neurobiol Aging. 1992;13:595–599. doi: 10.1016/0197-4580(92)90062-3. [DOI] [PubMed] [Google Scholar]
- 67.Walker LC, Pahnke J, Madauss M, Vogelgesang S, Pahnke A, Herbst EW, Stausske D, Walther R, Kessler C, Warzok RW. Apolipoprotein E4 promotes the early deposition of Abeta42 and then Abeta40 in the elderly. Acta Neuropathol. 2000;100:36–42. doi: 10.1007/s004010051190. [DOI] [PubMed] [Google Scholar]
- 68.Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, Selkoe DJ. The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- 69.Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O’Hare E, Esler WP, Maggio JE, Mantyh PW. Fibrillar -amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998;18:2161–2173. doi: 10.1523/JNEUROSCI.18-06-02161.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winkler J, Connor DJ, Frautschy SA, Behl C, Waite JJ, Cole GM, Thal LJ. Lack of long-term effects after b amyloid protein injections in rat brain. Neurobiol Aging. 1994;16:601–607. doi: 10.1016/0197-4580(94)00054-9. [DOI] [PubMed] [Google Scholar]
- 71.Woodruff-Pak DS, Agelan A, Del Valle L. A rabbit model of Alzheimer’s disease: valid at neuropathological, cognitive, and therapeutic levels. J Alzheimers Dis. 2007;11:371–383. doi: 10.3233/jad-2007-11313. [DOI] [PubMed] [Google Scholar]
- 72.Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, Mc-Conlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 73.Yamada K, Tanaka T, Senzaki K, Kameyama T, Nabeshima T. Propentofylline improves learning and memory deficits in rats induced by beta-amyloid protein-(1-40) Eur J Pharmacol. 1998;349:15–22. doi: 10.1016/s0014-2999(98)00166-6. [DOI] [PubMed] [Google Scholar]
- 74.Yamada K, Tanaka T, Han D, Senzaki K, Kameyama T, Nabeshima T. Protective effects of idebenone and alpha-tocopherol on beta-amyloid-(1-42)-induced learning and memory deficits in rats: implication of oxidative stress in beta-amyloid-induced neurotoxicity in vivo. Eur J Neurosci. 1999;11:83–90. doi: 10.1046/j.1460-9568.1999.00408.x. [DOI] [PubMed] [Google Scholar]
- 75.Yamada K, Tanaka T, Mamiya T, Shiotani T, Kameyama T, Nabeshima T. Improvement by nefiracetam of beta-amyloid-(1-42)-induced learning and memory impairments in rats. Br J Pharmacol. 1999;126:235–244. doi: 10.1038/sj.bjp.0702309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang F, Mak K, Vinters HV, Frautschy SA, Cole GM. Monoclonal antibody to the C-terminus of -amyloid. Neuroreport. 1994;15:2117–2120. doi: 10.1097/00001756-199410270-00032. [DOI] [PubMed] [Google Scholar]
- 77.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 78.Younkin SG. Evidence that Aβ42 is the real culprit in Alzheimer’s disease. Ann Neurol. 1995;37:287–288. doi: 10.1002/ana.410370303. [DOI] [PubMed] [Google Scholar]
- 79.Zavaroni I, Sander S, Scott S, Reaven GM. Effect of fructose feeding on insulin secretion and insulin action in the rat. Metabolism. 1980;29:970–973. doi: 10.1016/0026-0495(80)90041-4. [DOI] [PubMed] [Google Scholar]