

Significance
Although loss of fragile X mental retardation protein 1 (FMRP) causes a wide range of abnormalities in both pre- and postsynaptic compartments, the link between various FMRP functions and specific phenotypes in patients has been difficult to establish. Through the study of a novel fragile X mental retardation 1 (FMR1) missense mutation, c.413G > A (R138Q), recently identified in a patient with a partial fragile X syndrome (FXS) phenotype (intellectual disability and seizures), we found that pre- and postsynaptic functions of FMRP are independent. Our findings suggest that loss of a presynaptic, translation-independent function of FMRP is linked with a specific subset of FXS clinical features. Our study thus provides a major step in teasing out the domain-specific functions of FMRP in pre- and postsynaptic compartments, and their contribution to various elements of FXS pathophysiology.
Keywords: fragile X syndrome, missense mutation, FMR1 sequencing, FMRP, BK channels
Abstract
Fragile X syndrome (FXS) results in intellectual disability (ID) most often caused by silencing of the fragile X mental retardation 1 (FMR1) gene. The resulting absence of fragile X mental retardation protein 1 (FMRP) leads to both pre- and postsynaptic defects, yet whether the pre- and postsynaptic functions of FMRP are independent and have distinct roles in FXS neuropathology remain poorly understood. Here, we demonstrate an independent presynaptic function for FMRP through the study of an ID patient with an FMR1 missense mutation. This mutation, c.413G > A (R138Q), preserves FMRP’s canonical functions in RNA binding and translational regulation, which are traditionally associated with postsynaptic compartments. However, neuronally driven expression of the mutant FMRP is unable to rescue structural defects at the neuromuscular junction in fragile x mental retardation 1 (dfmr1)-deficient Drosophila, suggesting a presynaptic-specific impairment. Furthermore, mutant FMRP loses the ability to rescue presynaptic action potential (AP) broadening in Fmr1 KO mice. The R138Q mutation also disrupts FMRP’s interaction with the large-conductance calcium-activated potassium (BK) channels that modulate AP width. These results reveal a presynaptic- and translation-independent function of FMRP that is linked to a specific subset of FXS phenotypes.
Fragile X syndrome (FXS) is the most common single-gene disorder responsible for intellectual disability (ID) in patients (1). Along with cognitive dysfunction, the syndrome typically presents with several other comorbidities, including behavioral and social impairments (anxiety and autism spectrum disorder), neurological defects (seizures and abnormal sleep patterns), and morphological abnormalities (dysmorphic facies and macroorchidism). Most patients inherit the syndrome through a maternal repeat expansion mutation that transcriptionally silences the FMR1 gene and results in loss of the gene product, FMRP.
FMRP has complex multifaceted functions at synapses both in pre- and postsynaptic compartments. As an RNA binding protein, FMRP is best known for its function as a translation regulator in dendrites (2). Loss of FMRP has been linked to various forms of long-term synaptic plasticity defects that depend on local protein synthesis in the postsynaptic neuron (3). In addition to disrupted metabotropic glutamate receptor signaling, which has been shown across multiple brain regions (4–7), FMRP is necessary for activity-dependent protein synthesis downstream of other signaling receptor pathways, including ACh, dopamine, and TrkB (8–10).
Although postsynaptic control of translation is believed to be the dominant function of FMRP, it is unable to explain all of the pathophysiology observed in FXS animal models. For instance, in Drosophila, presynaptic expression of the FMR1 homolog, dfmr1, completely rescues the synaptic overgrowth phenotype at the neuromuscular junction (NMJ) in dfmr1-null mutants (11–13). In rodent brain, FMRP has been found in structures called fragile-X granules, which are present only in axons and presynaptic terminals, and are highly expressed during periods of peak synaptic plasticity (14, 15). In mosaic Fmr1 mice, the presynaptic presence of FMRP is sufficient to rescue synaptic connectivity defects in the hippocampal circuit (16), and in Aplysia, down-regulation of FMRP in presynaptic neurons is sufficient to enhance long-term synaptic depression (17). Furthermore, FMRP regulation of neurotransmitter release at excitatory hippocampal and cortical synapses has been shown to be a cell-autonomous presynaptic and translation-independent process (18, 19).
Both the pre- and postsynaptic functions of FMRP have also been linked to neuronal and circuit hyperexcitability in FXS animal models. Consistent with its role as a protein synthesis regulator, FMRP regulates the expression of a number of dendritic voltage-gated K+ channels in various brain circuits (20–22). In addition to translational regulation, FMRP has been shown to influence neural excitability by directly modulating the activity of a number of presynaptic voltage-gated ion channels. For example, FMRP interacts with the sodium-activated potassium channel, Slack, to control channel gating (23, 24) and directly interacts with the N-type voltage-gated calcium channel, Cav2.2, to control its surface expression (25). FMRP also modulates activity of the large-conductance calcium-activated potassium (BK) channel in hippocampal and cortical excitatory neurons, which is critical for controlling action potential (AP) duration and neurotransmitter release (18). FMRP interacts with the regulatory β4-subunit of the BK channel to increase channel Ca2+ sensitivity and limit AP duration and neurotransmitter release. In brain slices from Fmr1 KO mice, the absence of FMRP leads to excessive AP broadening, which can be acutely rescued with intracellular perfusion of an aminoterminal FMRP fragment containing amino acids 1–298 into the presynaptic neuron. These effects are independent of translation, suggesting a distinct presynaptic function of FMRP unrelated to translational control. However, to what extent the functions of FMRP in pre- and postsynaptic compartments independently contribute to FXS neuropathology remains unknown. Moreover, the multitude and complexity of FMRP functions have made it difficult thus far to establish the link between various FMRP functions and specific phenotypes in patients.
Distinguishing the role of specific FMRP functions in FXS phenotypes could, in principle, be possible if partial loss-of-function mutations were found within the population that contributed to a limited subset of FXS phenotypes. However, to date, only two missense mutations have been reported to cause FXS, and both mutations result in functional null forms of FMRP that phenocopy transcriptionally silenced repeat expansion mutations (26–28).
Here, we have studied the synaptic deficits associated with the FMR1 missense mutation, c.413G > A (R138Q), which was recently identified in a screen of developmentally delayed males who were negative for repeat expansion (29). The identified patient has a history of ID and intractable seizures, but no other features commonly associated with FXS. We show that R138Q is a partial loss-of-function mutation that specifically impairs a presynaptic FMRP function while preserving the translation regulation capabilities of FMRP. Postsynaptically, R138Q-FMRP was able to regulate normal AMPA receptor trafficking, as well as retaining normal polyribosome association and mRNA binding functions. However, presynaptically, neuronally driven expression of the mutant FMRP was unable to rescue synaptic overgrowth at the NMJ in dfmr1-deficient Drosophila. In Fmr1 KO mice, presynaptic R138Q-FMRP was also unable to rescue AP broadening defects in hippocampal and cortical pyramidal neurons. Furthermore, biochemical studies revealed that the R138Q mutation disrupts FMRP’s interaction with BK channels, which mediate the effect of FMRP loss on AP duration. Together, these results demonstrate functions for FMRP in the presynaptic compartment that are distinct from translation regulation, and suggest that a specific FMR1 mutation linked to a limited subset of FXS phenotypes is associated with isolated loss of presynaptic function.
Results
Identification of ID Patient with R138Q Missense Mutation.
In a previous sequencing study of 963 developmentally delayed males, we identified a patient with an R138Q missense mutation in the FMR1 gene without the CGG-repeat expansion (29). This patient has a history of global developmental delay, ID, and intractable seizures but no other behavioral, neurological, or dysmorphic features commonly associated with FXS (Fig. 1A). Molecular FXS testing revealed 45 CGG repeats, which is within the normal range (30); however, full sequencing of the patient’s FMR1 gene revealed a c.413G > A transition that causes the highly conserved Arg (R) 138 residue to be replaced by Gln (Q) (Fig. 1 C and D). Both maternal and paternal sides of the family have a history of learning problems; however, this mutation was passed through the mother, who was found to be a carrier of the R138Q mutation (Fig. 1B). A full patient description and clinical history are provided in SI Case Report.
Fig. 1.
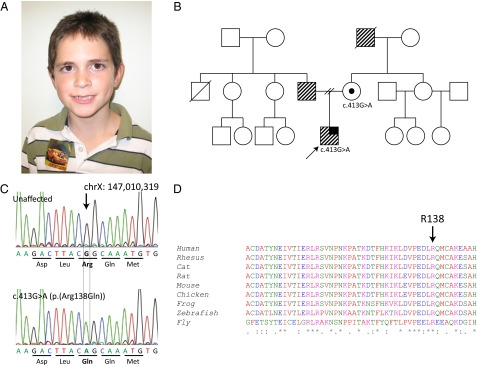
Identification of the ID patient with an R138Q missense mutation. (A) Patient does not have any dysmorphic facial features commonly associated with FXS. (B) Family pedigree of the proband (indicated by arrow). The stripe pattern indicates learning disability, and the solid black line indicates a seizure disorder. The proband’s mother is a carrier of the R138Q mutation. No other family members were available for genetic testing. (C) DNA chromatogram of the unaffected and patient alleles showing the single-nucleotide substitution (NM_002024.5:c.413G > A) that replaces Arg at residue 138 with Gln (R138Q). (D) ClustalW alignment across multiple species of FMRP amino acids 98–147. FMRP at residue 138 is highly conserved from human through Drosophila.
R138Q Mutation Retains Postsynaptic Functions of FMRP in Translation Regulation.
To test the functional significance of the R138Q missense mutation, we previously assayed whether this mutation impairs FMRP’s established role as a protein synthesis regulator. In hippocampal neurons, postsynaptic AMPA receptor trafficking is influenced by FMRP-mediated control of local protein synthesis such that, in the absence of FMRP, there is exaggerated AMPA receptor internalization (31, 32). To determine if the R138Q mutation affects FMRP’s ability to regulate AMPA receptor trafficking, we measured AMPA receptor internalization in Fmr1 KO mouse hippocampal neurons that were infected with either R138Q-FMRP or WT-FMRP lentivirus. We found that AMPA receptor internalization in R138Q-FMRP–infected neurons was not significantly different from WT neurons or WT-FMRP–infected Fmr1 KO neurons, although Fmr1 KO neurons alone were significantly different from all of the other groups as expected (figure S6 of ref. 33). Because regulation of AMPA receptor trafficking is a function of FMRP’s ability to control protein synthesis, this finding suggests that R138Q-FMRP retains its canonical function of regulating mRNA translation.
In line with its role as a protein synthesis regulator, FMRP is known to bind polyribosomes and specific mRNA targets (1, 34). To determine if R138Q-FMRP can bind polyribosomes, we infected Fmr1 KO mouse embryonic fibroblasts (MEFs) with R138Q-FMRP or WT-FMRP lentivirus and probed for FMRP distribution after sucrose gradient fractionation. We found that R138Q-FMRP is robustly present in polyribosome fractions, similar to WT-FMRP (Fig. 2A). To determine if R138Q-FMRP can bind mRNA, we infected Fmr1 KO cortical neurons with R138Q-FMRP or WT-FMRP lentivirus and performed FMRP-RNA coimmunoprecipitation (co-IP) to measure the relative mRNA levels of three validated FMRP targets: Map1b, Dlg4, and CamKIIα (1). We found that R138Q-FMRP associated with these targets to a similar level as WT-FMRP, whereas neurons infected with the functional null G266E-FMRP mutant (26), or GFP alone, did not pull down these mRNAs and were significantly different from WT-FMRP as expected (Fig. 2B; two-way ANOVA with a significant main effect of lentivirus infection; F = 66.57, P < 0.0001, Dunnett’s post hoc analysis: *P < 0.0001 for pairwise comparison with WT-FMRP control group for each mRNA target). Together, these data indicate that the R138Q mutation does not impair FMRP’s postsynaptic function as a translation regulator, as evidenced by rescue of AMPA receptor trafficking, intact polyribosome association, and mRNA pull-down.
Fig. 2.
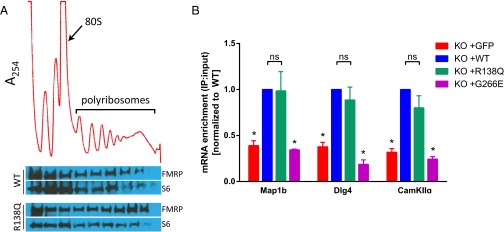
R138Q mutation retains postsynaptic functions of FMRP in translation regulation. (A) Polyribosome assay on Fmr1 KO MEF cells infected with either WT or R138Q-FMRP. (Top) Representative A254 absorbance profile, with the monosome (80S) and polyribosome peaks indicated. (Bottom) FMRP protein distribution showing that R138Q-FMRP associates with polyribosome fractions similar to WT-FMRP. Each fraction corresponds to the same region of the linear sucrose gradient above it. S6 ribosomal protein is shown to verify sample loading in each well. These blots are representative of n = 3 experiments. (B) RNA co-IP on Fmr1 KO cortical neurons infected with GFP, WT-FMRP, R138Q-FMRP, or G266E-FMRP. The relative mRNA enrichment of FMRP targets microtubule-associated protein 1b (Map1b), discs large homolog 4 (Dlg4), and calcium/calmodulin-dependent protein kinase II alpha (CamKIIα) were analyzed by quantitative PCR. Data are represented as mean ± SEM. *P < 0.0001; ns, not significant.
R140Q Mutation Impairs Presynaptic Function in Drosophila.
The preservation of FMRP’s canonical function as a protein synthesis regulator in the above biochemical studies led us to consider that the R138Q mutation might simply be a rare benign variant without any functional consequences. To test this possibility further, the function of R138Q-FMRP was assessed in an animal model. Drosophila provides a uniquely powerful system for studying FMRP function because there is only a single ortholog from the fragile X-related gene family, dfmr1 (35); therefore, subtle phenotypes are more likely to be revealed due to loss of compensatory action from the fragile X-related protein FXR1 or FXR2. The Drosophila equivalent of the R138Q mutation is R140Q according to ClustalW alignment (Fig. 1D).
One particularly robust phenotype is the overelaboration of synaptic branching and number of boutons at the NMJ in dfmr1-null flies (11). To determine if R140Q-FMRP is able to rescue synaptic overgrowth at the Drosophila NMJ, we used Da-Gal4 to drive ubiquitous expression of upstream activation sequence (UAS)-dfmr1-R140Q in dfmr1-deficient Drosophila. To our surprise, we found that R140Q-FMRP was unable to rescue overgrowth of NMJ length or branching, unlike WT-FMRP (Fig. S1). Because these NMJ structural defects are known to be a function of presynaptic FMRP (13), we also used Elav-Gal4 to drive panneuronal expression of UAS-dfmr1-R140Q in dfmr1-deficient Drosophila. We found that presynaptic specific expression of R140Q-FMRP was also unable to rescue overgrowth of NMJ length or branching, unlike WT-FMRP (Fig. 3; one-way ANOVA for NMJ length and branching; F = 272.5 and F = 68.32, respectively; P < 0.0001, Tukey’s post hoc analysis: ****P < 0.0001 for comparison with dfmr1−/−). Quantitative PCR assays for dfmr1 on whole larvae show two- to threefold higher expression of R140Q-dfmr1 compared with WT-dfmr1 (Fig. S2), indicating that the lack of rescue in NMJ overgrowth was not due to inadequate R140Q-FMRP expression.
Fig. 3.

R140Q mutation impairs presynaptic function in Drosophila. (A) Representative micrographs of the NMJ in dfmr1 heterozygous WT flies (dfmr1−/+, indistinguishable from homozygous WT), dfmr1-deficient mutants (dfmr1−/−), and dfmr1-deficient mutants with either UAS-dfmr1-WT– or UAS-dfmr1-R140Q (Drosophila equivalent of R138Q)–driven expression. Quantification of NMJ length (B) and branching (C) in dfmr1-deficient flies with neuronally driven (Elav-Gal4) expression of WT or R140Q-FMRP. Data are represented as mean ± 95% confidence interval. ****P < 0.0001. (Also Figs. S1 and S2.)
R138Q Mutation Impairs Presynaptic Function in Mouse Central Neurons.
To validate the presynaptic specific loss of function we observed with R138Q-FMRP in Drosophila, we sought to corroborate these findings within a mammalian system. We previously discovered that FMRP modulates AP duration via BK channels in hippocampal and cortical excitatory neurons in a cell-autonomous presynaptic manner (18). We showed that excessive AP broadening in Fmr1 KO neurons can be acutely rescued with presynaptic intracellular perfusion of a commercially available aminoterminal FMRP fragment consisting of amino acids 1–298 and, importantly, that these effects were independent of translation. To determine if R138Q-FMRP can rescue AP broadening in Fmr1 KO neurons, we created and purified the same aminoterminal FMRP fragment consisting of amino acids 1–298 with and without the R138Q mutation (R138Q-FMRP298 and WT-FMRP298, respectively). We then performed AP rescue experiments in current-clamped CA3 hippocampal pyramidal cells in acute slices from 15- to 17-d-old Fmr1 KO mice. AP trains were evoked by repetitive injection of a 1-ms current to evoke a 25-AP train at 62.5 Hz [with 16-ms interstimulus intervals (ISIs)]. An FMRP fragment was introduced into the cells via a patch pipette using a microperfusion system that permitted us to control the exact time point at which perfusion was initiated. Only recordings in which AP duration could be recorded before and after FMRP fragment perfusion were used in this analysis. As in our previous studies, perfusion of WT-FMRP298 into CA3 neurons of Fmr1 KO mice rapidly rescued excessive AP broadening both at baseline and during the train (Fig. 4 A and C; n = 6; paired Student’s t test: P = 0.012 for baseline APs, P = 0.008 for the averaged last two APs in the train). In contrast, intracellular perfusion of an R138Q-FMRP298 fragment was unable to reduce AP broadening in these neurons [Fig. 4 B and C; n = 6; paired Student’s t test: P = 0.40 (baseline AP), P = 0.44 (end of train)]. We note that the rescue effect of the WT-FMRP fragment was measurable within ∼5 min after perfusion initiation. Together with the fact that the FMRP298 fragment lacks the K-homology 2 (KH2) domain critical for association with polyribosomes, this rapid rescue provides further support for our earlier findings that the mechanism of these presynaptic FMRP actions is translation-independent. These results indicate that the R138Q mutation disrupts FMRP’s ability to regulate AP duration in a presynaptic neuron.
Fig. 4.

R138Q mutation impairs presynaptic function in mouse central neurons. Effect of intracellular perfusion of WT-FMRP298 (A) or R138Q-FMRP298 (B) on AP duration in Fmr1 KO CA3 pyramidal neurons before (0.2 Hz) and during 25 stimuli at 62.5 Hz. In each condition, AP duration during trains was normalized to four immediately preceding baseline measurements. (C) Actual AP width in different conditions is quantified. AP duration measurements in WT mice (n = 8) are provided as a reference. (D–F) Same as A–C for Fmr1 KO layer 5 entorhinal cortex (EC) pyramidal neurons, except 25-AP trains at 60 Hz were used. WT AP measurements (n = 9) in F are reanalyzed, in part, from our earlier study (18). (G) In vitro binding assay with His-tagged WT-FMRP298 or R138Q-FMRP298. Brain lysates from WT mice were incubated with His-tagged protein fragments or no bait as a control, and the levels of BK channel α- and β4-subunits bound to FMRP fragments were analyzed by Western blot with the indicated antibodies. (Top and Middle) Representative Western blots from n = 4 experiments (n = 5 for β4-subunit measurements) are shown. (Bottom) Ponceau staining is shown as control for an equal amount of His-tagged WT-FMRP298 or R138Q-FMRP298 used in this assay. (H) Quantification of G. (I–J) Effect of intracellular perfusion of WT-FMRP298 on AP duration at baseline and during 25-AP trains at 60 Hz in Fmr1 KO CA3 pyramidal neurons in the presence of transcription inhibitor actinomycin D (40 μM) with at least 1 h of preincubation. Data are represented as mean ± SEM. *P < 0.05; **P < 0.01.
We have previously shown that FMRP regulation of AP duration is a widespread phenomenon observed in both excitatory hippocampal and cortical neurons. We thus tested whether the inability of R138Q-FMRP298 to rescue the AP broadening defect in Fmr1 KO mice is also observed in cortical pyramidal neurons. AP measurements were performed in 15- to 20-d-old layer 5 cortical pyramidal neurons of the entorhinal cortex of Fmr1 KO mice using the same intracellular perfusion approach described above, except a 25-AP train at 60 Hz was used. Similar to hippocampal neurons, intracellular perfusion of WT-FMRP298 rapidly reduced AP broadening in cortical pyramidal neurons, whereas perfusion of R138Q-FMRP298 did not have a significant effect [Fig. 4 D–F; paired Student’s t test; WT: n = 8, P = 0.015 (baseline), P = 0.028 (end of train); R138Q: n = 7, P = 0.69 (baseline), P = 0.72 (end of train)]. These results support the above evidence that the R138Q mutation disrupts the presynaptic function of FMRP to regulate AP duration in both hippocampal and cortical pyramidal neurons.
R138Q Mutation Disrupts FMRP Interaction with BK Channels.
What is the mechanism by which the R138Q mutation renders FMRP incapable of regulating AP duration? We have previously shown that FMRP regulates AP duration and neurotransmitter release in excitatory pyramidal neurons through FMRP’s interaction with the BK channels, particularly their regulatory β4-subunit (18). The above observations therefore suggest that the R138Q mutation may interfere with FMRP/BK channel interactions. To test this possibility, we used an in vitro binding assay in which His-tagged WT-FMRP298 or R138Q-FMRP298 fragments were used to pull down the BK channel from WT brain lysates. The WT-FMRP298 fragment interacted with the BK channel β4-subunit (Fig. 4G) in agreement with our previous observations (18) and revealed that FMRP/β4 interaction is mediated by the FMRP amino terminus. In contrast to WT-FMRP298, we observed markedly reduced β4-subunit association with R138Q-FMRP298 (Fig. 4 G and H; n = 5; paired Student’s t test, P < 0.0001). We also observed binding of the BK channel α-subunit with WT-FMRP298, and this interaction was slightly but significantly reduced with R138Q-FMRP298 (Fig. 4 G and H; n = 4; paired Student’s t test, P = 0.02). These results indicate that the R138Q mutation reduces FMRP interaction with the BK channels, particularly with the BK β4-subunit.
Presynaptic Effects of R138Q Mutation on AP Duration Are Transcription-Independent.
A recent study has revealed a nuclear function of FMRP in chromatin binding and transcriptional regulation, which was abolished by the R138Q mutation (33). We thus tested whether presynaptic effects of the R138Q mutation could be mediated, in part, by transcriptional changes. Slices were preincubated for at least 1 h and continuously perfused during recordings with the transcription inhibitor actinomycin D (40 μM). AP duration was examined before and after intracellular perfusion of WT-FMRP298 in Fmr1 KO CA3 pyramidal neurons, as described above, to test whether the ability of FMRP to regulate the AP waveform requires transcriptional changes. Preincubation with actinomycin D alone caused a significant increase in the baseline AP width (n = 8; paired Student’s t test, P < 0.001), indicating the effectiveness of the drug application. Most importantly, however, actinomycin D had no effect on the ability of WT-FMRP298 to reduce AP width at baseline or during AP trains [Fig. 4 I and J; 25-AP train at 60 Hz; n = 8; paired Student’s t test; P = 0.02 (baseline), P < 0.001 (end of train)], indicating that FMRP regulation of AP duration in excitatory hippocampal neurons is transcription-independent.
Discussion
The pathophysiology of FXS is complex, involving both pre- and postsynaptic defects. However, the interdependence and specific contributions of pre- and postsynaptic FMRP functions to various FXS phenotypes have been difficult to determine. Through the study of a previously unidentified FMR1 missense mutation, R138Q, we have demonstrated that FMRP plays an important presynaptic role that is independent from its canonical postsynaptic function in mRNA translation. Our experiments indicate that the R138Q mutation retains the postsynaptic translation regulation capabilities of FMRP but is incapable of rescuing presynaptic structural defects at the dfmr1-deficient Drosophila NMJ or of rescuing presynaptic AP broadening defects in Fmr1 KO mouse central neurons. Furthermore, we found that the R138Q mutation strongly reduces FMRP’s ability to interact with the BK channels that mediate FMRP’s regulation of AP duration and glutamate release. The finding that R138Q is a partial loss-of-function mutation that abolishes only a subset of FMRP functions is consistent with the patient’s limited FXS phenotype consisting of ID and seizures, but not any other symptoms commonly associated with FXS. Taken together, these results suggest that pre- and postsynaptic functions of FMRP are independent, and link the isolated loss of presynaptic FMRP function with a specific subset of FXS clinical features.
Insights into FXS Pathophysiology from FMR1 Missense Mutations.
To date, there have only been two other reported missense mutations within FMR1: G266E (26) and I304N (27). Both of these mutations reside within the RNA binding domains of FMRP and result in loss of RNA binding and polyribosome association (26, 28). Studies of these mutations provide strong evidence that FMRP’s inability to regulate protein synthesis is a critical component of FXS pathophysiology, given that both of the patients with the G266E and I304N mutations presented with a very characteristic FXS phenotype, including marked ID, developmental delay, macroorchidism, and dysmorphic facies. Conversely, the R138Q mutation does not reside within an RNA binding domain but, instead, is located at the aminoterminal domain of FMRP. In contrast to the patients with G266E and I304N mutations, the patient with the R138Q mutation only displays ID and seizures, which corresponds well with our observation that R138Q is a partial loss-of-function mutation. These results suggest that presynaptic FMRP function may be specifically connected to ID and seizure pathology in FXS, possibly through the aminoterminal domain.
BK Channel Dysfunction in ID and Seizures.
We have previously shown that FMRP binds to and modulates the activity of BK channels to regulate AP duration and neurotransmitter release (18). These functions are abolished by the R138Q mutation, thereby potentially linking FMRP and BK channel modulation to the ID and seizure phenotype exhibited by the patient with the R138Q mutation. BK channels have, in fact, already been linked to ID and seizures separately in several previous reports. For instance, an autistic patient with severe ID was found to have a balanced translocation resulting in disruption of the KCNMA1 gene that encodes the BK channel (36). Dysregulation of BK channel isoform expression has also been linked to a mild form of autosomal recessive nonsyndromal mental retardation (37). Probably the most compelling evidence comes from another report showing that the SNPs with the highest risk for autism spectrum disorder were found in KCNMB4, the gene encoding the β4-subunit of the BK channel (38).
BK channels have also been implicated in epilepsy and seizure disorders in numerous cases through both loss-of-function and gain-of-function mechanisms (39). Because BK channels play a critical role in repolarizing the membrane potential after AP firing, loss of BK channel function typically results in increased seizure susceptibility (40, 41). In line with loss-of-function mechanisms, pharmacological inhibition of BK channels triggers seizures (42), down-regulation of BK channel expression is associated with the development of temporal lobe epilepsy in rats (43, 44), and a particular KCNMB4 SNP is strongly correlated with temporal lobe epilepsy in humans (45). Interestingly, some gain-of-function mutations in BK channels have also been implicated in seizure etiology, primarily absence epilepsy, by rapidly repolarizing the membrane and allowing for faster firing rates (46, 47). Nevertheless, BK channel activity is clearly intimately connected with seizure pathology. In fact, presynaptic BK channels have recently been shown to localize preferentially to and regulate neurotransmitter release at glutamatergic, but not GABAergic, synapses, further supporting their critical role in modulating circuit hyperexcitability (48). Whether FMRP/BK channel interactions are directly responsible for seizure and/or ID phenotypes and whether targeting BK channels could be a useful component of FXS therapy remains to be determined.
Aminoterminal Domain and FMRP Function.
Our studies suggest that this translation-independent presynaptic function of FMRP, abolished by the R138Q mutation, depends on an intact aminoterminal domain. Indeed, an aminoterminal fragment of FMRP (residues 1–298) was sufficient to rescue excessive AP broadening in Fmr1 KO cortical and hippocampal neurons, as well as mediating FMRP binding with BK channel subunits. This 298-aa fragment is composed of the FMRP aminoterminal domain (amino acids 1–215) in addition to the full KH1 and a small portion of the KH2 RNA binding domains. Although we cannot rule out the possibility that FMRP’s presynaptic function may be mediated by the KH1 domain, it is highly unlikely, given that we have previously shown the presynaptic effects of FMRP on AP duration and glutamate release are independent of translation, and therefore unlikely to involve any RNA binding domain function (18). Furthermore, we have shown that the R138Q mutation does not disrupt FMRP’s RNA binding ability yet does impair presynaptic FMRP function at the Drosophila NMJ and in mouse hippocampal and cortical neurons. Thus, we propose that the aminoterminal domain mediates FMRP’s presynaptic function, yet the exact locus that is responsible for FMRP’s modulation of AP duration or interaction with BK channels will require further studies.
The FMRP aminoterminal domain also mediates interaction of FMRP with a number of its binding partners (49–52) and contains two tandem Tudor/Agenet motifs (53, 54) that mediate a nuclear FMRP function in chromatin binding and DNA damage response (33). Interestingly, the R138Q mutation also abolished nucleosome binding, indicating this mutation may play a role in the pathophysiology of FXS through transcriptional changes (33). We note, however, that our observations of the very rapid effects of WT-FMRP298 perfusion on AP duration (within ∼5 min) and that WT-FMRP298 is fully capable of rescuing AP duration in the presence of the transcription inhibitor actinomycin D indicate that FMRP regulation of the AP waveform is transcription-independent. The effects of the R138Q mutation on this presynaptic FMRP function are thus unlikely to be mediated by transcriptional changes. Regardless, our work suggests a previously unidentified function for the aminoterminal domain of FMRP in the pathophysiology of FXS by modulating AP duration in a cell-autonomous presynaptic manner and presumably contributing to circuit hyperexcitability via interactions with the BK channels. In summary, our study of the R138Q missense mutation in FMR1, associated with a limited subset of FXS clinical features, provides a first step in teasing out the domain-specific functions of FMRP in pre- and postsynaptic compartments, and their contribution to various elements of FXS pathophysiology.
Experimental Procedures
Constructs.
For lentivirus production, full-length human aminoterminal Flag-tagged FMR1 was cloned into the FUGW plasmid with and without the R138Q mutation and was sent to the Emory University Viral Vector Core for lentivirus production. For Drosophila transgene injection, the Drosophila equivalent of the human R138Q mutation, c.419_420delinsAG (R140Q), was introduced into the pUAST-dfmr1 plasmid. For protein expression, the Emory Custom Cloning Core Facility cloned truncated WT and R138Q human FMR1 (residues 1–298), with and without an aminoterminal 6xHis tag, into a modified pET28b expression vector.
Animals.
Control and Fmr1 KO mice on a C57BL/6J background were used for cell culture experiments. Control and Fmr1 KO mice on an FVB background were used for electrophysiology experiments. All animal procedures conformed to the guidelines approved by the Washington University Animal Studies Committee and the Emory University Institutional Animal Care and Use Committee guidelines. This study was approved by the Emory University Internal Review Board.
Cell Culture.
Immortalized MEFs were generated from Fmr1 KO mice and maintained in culture with DMEM (Corning) supplemented with 10% (vol/vol) FBS (HyClone). Primary cortical neuronal cultures were generated from embryonic day 16.5 Fmr1 KO mice. Neurons were plated directly onto poly-l-lysine–coated (0.2 mg/mL) dishes and maintained in culture with Neurobasal medium (Gibco) supplemented with B27 (Invitrogen).
Polyribosome Profiling and Western Blotting.
Fmr1 KO MEFs were transduced with lentivirus at ∼40–50% confluency for 16 h and collected for polyribosome assay 24 h after virus removal. Immediately before harvest, cells were treated with cycloheximide for 15 min at 37 °C. Cell lysates were clarified and loaded on top of 15–45% wt/wt linear sucrose gradients, centrifuged at 247,600 × g for 2 h at 4 °C using a SW41Ti rotor (Beckman Coulter), and fractionated into 10 × 1.1-mL fractions with continuous monitoring at OD254. For Western blotting, 500 μL of each fraction was concentrated with Amicon Ultra 30K centrifugal filters (Millipore) and processed for Western blotting using standard techniques. Enhanced chemiluminescence signal was detected using SuperSignal Femto Max Sensitivity Substrate (Thermo Scientific).
RNA Co-IP and Quantitative RT-PCR.
Fmr1 KO primary cortical neurons were transduced with lentivirus at 10 days in vitro (DIV) for 18 h and collected for RNA co-IP 72 h after virus removal. Neuronal lysates were separated into input and IP fractions, and the IP fractions were incubated with EZview Red Anti-Flag M2 Affinity Gel (Sigma) for 2 h at 4 °C. RNA was extracted from both input and IP samples, and processed for quantitative RT-PCR. Before RNA extraction, a portion of the input and IP samples was saved for Western blotting to verify that lentiviral infection produced equal FMRP expression and pull-down across samples.
For quantitative RT-PCR, total RNA was reverse-transcribed with the SuperScript III First Strand Synthesis System for RT-PCR (Invitrogen), PCR was performed with iQ SYBR Green Supermix (BioRad), and mRNA was quantified using the standard curve method for relative quantification. See Table S1 for a list of primers. The ratio between IP and input mRNA quantification was normalized to the WT value for each experiment to allow comparison across different experiments.
Drosophila NMJ Immunostaining and Analysis.
All flies were maintained under standard culture conditions. Transgenic dfmr1 WT and R140Q mutant flies were generated by standard P-element transgene injection (Bestgene, Inc.) into a w1118 strain and then crossed with either a Da-GAL4 or Elav-GAL4 line (nos. 8641 and 458, respectively; Bloomington Stock Center). Third-stage wandering larvae were dissected and immunostained for antidiscs large primary antibody (4F3; Developmental Studies Hybridoma Bank) using standard techniques. NMJ length and number of branches were measured from muscles 6/7 of abdominal segment 2 or 3. Approximately 30 larvae were analyzed per genotype.
Brain Slice Preparation.
Both male and female 15- to 20-d-old mice were used. Brain slices were prepared as we described previously (18). Briefly, after being deeply anesthetized with CO2, mice were decapitated and their brains were dissected out in ice-cold artificial cerebral spinal fluid (ACSF) solution. Horizontal brain slices (350 μm), including the hippocampus, were cut using a vibrating microtome (Leica). Slices were initially incubated at 35 °C for 1 h for recovery and then kept at room temperature until use.
AP Recordings and Analysis.
APs were recorded using an Axopatch 700B amplifier (Molecular Devices) in whole-cell configuration from hippocampal CA3 pyramidal neurons or layer 5 pyramidal neurons in the entorhinal cortex, as we described previously (18). All recordings were performed at near-physiological temperatures (33–34 °C). APs were evoked by repetitive injection of a 1-ms current to evoke a 25-AP train at 60 Hz or 62.5 Hz. Sixty-hertz trains have fractional 16.67-ms ISIs, whereas 62.5-Hz trains have 16-ms ISIs. Because of software rounding of submillisecond timing to a whole millisecond, every third ISI is 1 ms shorter in 60-Hz stimulus trains, causing apparent periodicity in the data (55). This periodicity was no longer present in 58.8-Hz trains (constant 17-ms ISIs) (55) or 62.5-Hz trains (constant 16-ms ISIs; Fig. 4 A–C). This effect is very small (<2%) and is present in all recordings at 60 Hz. Membrane potential was set at −65 mV by automatic slow current injection to ensure stability of the resting potential and to prevent spontaneous AP firing. AP duration during bursts was normalized to an averaged duration of four low-frequency baseline APs (0.2 Hz) that were evoked before each burst. FMRP fragments were introduced into the neurons via patch pipette using a microperfusion system (Bioscience Tools). Only recordings in which no significant AP amplitude rundown was observed after FMRP perfusion were used in this analysis. Recordings were filtered at 2 kHz, digitized at 20 kHz, acquired using custom software written in LabView (National Instruments), and analyzed using programs written in MATLAB (MathWorks).
Protein Expression and Purification.
WT and R138Q FMRP fragments (residues 1–298), with or without an additional 6xHis tag immediately aminoterminal to FMRP, were expressed in Escherichia coli as 6xHis-SUMO fusion proteins (56). Protein expression was induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside for 16 h. The fusion proteins were then isolated on a nickel-charged HiTrap chelating column (GE Healthcare) and eluted with 500 mM imidazole, and the 6xHis-SUMO tag was removed by overnight incubation with yeast ubiquitin-like–specific protease 1 (Ulp1; purified in-house) at 4 °C. The cleaved proteins were further purified by a HiTrap-Q column (GE Healthcare) and Superdex 75 (16/60) sizing column (GE Healthcare).
Pull-Down Assay.
Whole mouse brain from WT C57BL/6J mice was homogenized on ice in lysis buffer, clarified by centrifugation at 20,000 × g, and precleared with HisPur Cobalt Resin (Pierce) for 1 h at 4 °C. His-tagged WT-FMRP298 or R138Q-FMRP298 fragments were then incubated with brain lysate overnight at 4 °C. HisPur Cobalt Resin was added and incubated for 1 h at 4 °C, followed by centrifugation at 350 × g for 1 min. Beads were washed five times with washing buffer before elution of bound proteins with 1× SDS/PAGE loading buffer and analysis by standard Western blotting.
Additional details of experimental procedures are available in SI Experimental Procedures.
Supplementary Material
Acknowledgments
We thank the Emory Viral Vector Core for lentivirus production and the Emory Custom Cloning Core for engineering constructs. This work was supported, in part, by NIH Award NS091859 from the National Institute of Neurological Disorders and Stroke and the Eunice Kennedy Shriver National Institute of Child Health and Human Development in support of the Emory National Fragile X Research Center (to S.T.W.) and by NIH Grant R01 NS089449 and a FRAXA Foundation grant (both to V.A.K.). H.H. and X.C. were supported by NIH Grant R01 GM049245-21.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423094112/-/DCSupplemental.
References
- 1.Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 2.Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60(2):201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99(11):7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Fukushima H, Kida S, Zhuo M. Ca2+/calmodulin-dependent protein kinase IV links group I metabotropic glutamate receptors to fragile X mental retardation protein in cingulate cortex. J Biol Chem. 2009;284(28):18953–18962. doi: 10.1074/jbc.M109.019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Todd PK, Malter JS, Mack KJ. Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res Mol Brain Res. 2003;110(2):267–278. doi: 10.1016/s0169-328x(02)00657-5. [DOI] [PubMed] [Google Scholar]
- 6.Suvrathan A, Hoeffer CA, Wong H, Klann E, Chattarji S. Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci USA. 2010;107(25):11591–11596. doi: 10.1073/pnas.1002262107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huber KM. The fragile X-cerebellum connection. Trends Neurosci. 2006;29(4):183–185. doi: 10.1016/j.tins.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci. 2007;27(43):11624–11634. doi: 10.1523/JNEUROSCI.2266-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, et al. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron. 2008;59(4):634–647. doi: 10.1016/j.neuron.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 10.Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30(46):15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang YQ, et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107(5):591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 12.Pan L, Zhang YQ, Woodruff E, Broadie K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14(20):1863–1870. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- 13.Gatto CL, Broadie K. Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development. 2008;135(15):2637–2648. doi: 10.1242/dev.022244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christie SB, Akins MR, Schwob JE, Fallon JR. The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J Neurosci. 2009;29(5):1514–1524. doi: 10.1523/JNEUROSCI.3937-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akins MR, Leblanc HF, Stackpole EE, Chyung E, Fallon JR. Systematic mapping of fragile X granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions. J Comp Neurol. 2012;520(16):3687–3706. doi: 10.1002/cne.23123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanson JE, Madison DV. Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J Neurosci. 2007;27(15):4014–4018. doi: 10.1523/JNEUROSCI.4717-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Till SM, Li HL, Miniaci MC, Kandel ER, Choi YB. A presynaptic role for FMRP during protein synthesis-dependent long-term plasticity in Aplysia. Learn Mem. 2011;18(1):39–48. doi: 10.1101/lm.1958811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng PY, et al. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron. 2013;77(4):696–711. doi: 10.1016/j.neuron.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel AB, Hays SA, Bureau I, Huber KM, Gibson JR. A target cell-specific role for presynaptic Fmr1 in regulating glutamate release onto neocortical fast-spiking inhibitory neurons. J Neurosci. 2013;33(6):2593–2604. doi: 10.1523/JNEUROSCI.2447-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strumbos JG, Brown MR, Kronengold J, Polley DB, Kaczmarek LK. Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci. 2010;30(31):10263–10271. doi: 10.1523/JNEUROSCI.1125-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci. 2011;31(15):5693–5698. doi: 10.1523/JNEUROSCI.6661-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HY, et al. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron. 2011;72(4):630–642. doi: 10.1016/j.neuron.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown MR, et al. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci. 2010;13(7):819–821. doi: 10.1038/nn.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, et al. Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J Neurosci. 2012;32(44):15318–15327. doi: 10.1523/JNEUROSCI.2162-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferron L, Nieto-Rostro M, Cassidy JS, Dolphin AC. Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat Commun. 2014;5:3628. doi: 10.1038/ncomms4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myrick LK, et al. Fragile X syndrome due to a missense mutation. Eur J Hum Genet. 2014;22(10):1185–1189. doi: 10.1038/ejhg.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Boulle K, et al. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet. 1993;3(1):31–35. doi: 10.1038/ng0193-31. [DOI] [PubMed] [Google Scholar]
- 28.Zang JB, et al. A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet. 2009;5(12):e1000758. doi: 10.1371/journal.pgen.1000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins SC, et al. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A. 2010;152A(10):2512–2520. doi: 10.1002/ajmg.a.33626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu YH, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67(6):1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 31.Nakamoto M, et al. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci USA. 2007;104(39):15537–15542. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27(20):5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alpatov R, et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell. 2014;157(4):869–881. doi: 10.1016/j.cell.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stefani G, Fraser CE, Darnell JC, Darnell RB. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci. 2004;24(33):7272–7276. doi: 10.1523/JNEUROSCI.2306-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wan L, Dockendorff TC, Jongens TA, Dreyfuss G. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol Cell Biol. 2000;20(22):8536–8547. doi: 10.1128/mcb.20.22.8536-8547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laumonnier F, et al. Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am J Psychiatry. 2006;163(9):1622–1629. doi: 10.1176/ajp.2006.163.9.1622. [DOI] [PubMed] [Google Scholar]
- 37.Higgins JJ, Hao J, Kosofsky BE, Rajadhyaksha AM. Dysregulation of large-conductance Ca2+-activated K+ channel expression in nonsyndromal mental retardation due to a cereblon p.R419X mutation. Neurogenetics. 2008;9(3):219–223. doi: 10.1007/s10048-008-0128-2. [DOI] [PubMed] [Google Scholar]
- 38.Skafidas E, et al. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol Psychiatry. 2014;19(4):504–510. doi: 10.1038/mp.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.N’Gouemo P. Targeting BK (big potassium) channels in epilepsy. Expert Opin Ther Targets. 2011;15(11):1283–1295. doi: 10.1517/14728222.2011.620607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu H, et al. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21(24):9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9(3):181–194. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- 42.Young KL, et al. Tremorgenic mycotoxin intoxication with penitrem A and roquefortine in two dogs. J Am Vet Med Assoc. 2003;222(1):52–53. doi: 10.2460/javma.2003.222.52. [DOI] [PubMed] [Google Scholar]
- 43.Pacheco Otalora LF, et al. Down-regulation of BK channel expression in the pilocarpine model of temporal lobe epilepsy. Brain Res. 2008;1200:116–131. doi: 10.1016/j.brainres.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ermolinsky B, Arshadmansab MF, Pacheco Otalora LF, Zarei MM, Garrido-Sanabria ER. Deficit of Kcnma1 mRNA expression in the dentate gyrus of epileptic rats. Neuroreport. 2008;19(13):1291–1294. doi: 10.1097/WNR.0b013e3283094bb6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cavalleri GL, et al. Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: a case-control study. Lancet Neurol. 2007;6(11):970–980. doi: 10.1016/S1474-4422(07)70247-8. [DOI] [PubMed] [Google Scholar]
- 46.Brenner R, et al. BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8(12):1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- 47.Du W, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37(7):733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- 48.Martire M, et al. Pre-synaptic BK channels selectively control glutamate versus GABA release from cortical and hippocampal nerve terminals. J Neurochem. 2010;115(2):411–422. doi: 10.1111/j.1471-4159.2010.06938.x. [DOI] [PubMed] [Google Scholar]
- 49.Bardoni B, et al. 82-FIP, a novel FMRP (fragile X mental retardation protein) interacting protein, shows a cell cycle-dependent intracellular localization. Hum Mol Genet. 2003;12(14):1689–1698. doi: 10.1093/hmg/ddg181. [DOI] [PubMed] [Google Scholar]
- 50.Bardoni B, Schenck A, Mandel JL. A novel RNA-binding nuclear protein that interacts with the fragile X mental retardation (FMR1) protein. Hum Mol Genet. 1999;8(13):2557–2566. doi: 10.1093/hmg/8.13.2557. [DOI] [PubMed] [Google Scholar]
- 51.Schenck A, Bardoni B, Moro A, Bagni C, Mandel JL. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci USA. 2001;98(15):8844–8849. doi: 10.1073/pnas.151231598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siomi MC, Zhang Y, Siomi H, Dreyfuss G. Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them. Mol Cell Biol. 1996;16(7):3825–3832. doi: 10.1128/mcb.16.7.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maurer-Stroh S, et al. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci. 2003;28(2):69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- 54.Ramos A, et al. The structure of the N-terminal domain of the fragile X mental retardation protein: a platform for protein-protein interaction. Structure. 2006;14(1):21–31. doi: 10.1016/j.str.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 55.Deng P-Y, et al. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron. 2013;78(1):205. doi: 10.1016/j.neuron.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lan F, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448(7154):718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.