

Significance
A single base substitution in one allele of the PIK3CA gene (encoding the catalytic subunit p110α of PI3K) in a human breast epithelial cell induces a gene expression profile that closely resembles the gene expression profile of basal breast cancer. The mutation also causes extensive remodeling of gene signatures that are not known to be connected to the activity of PI3K. The data show that a cancer-specific mutation that induces a gain of function in PI3K has an unexpectedly deep and broad impact on the phenotypic properties of the cell.
Keywords: RNAseq, SILAC, knock-in, molecular signature, basal breast cancer
Abstract
We have compared the proteome, transcriptome, and metabolome of two cell lines: the human breast epithelial line MCF-10A and its mutant descendant MCF-10A-H1047R. These cell lines are derived from the same parental stock and differ by a single amino acid substitution (H1047R) caused by a single nucleotide change in one allele of the PIK3CA gene, which encodes the catalytic subunit p110α of PI3K (phosphatidylinositol 3-kinase). They are considered isogenic. The H1047R mutation of PIK3CA is one of the most frequently encountered somatic cancer-specific mutations. In MCF-10A, this mutation induces an extensive cellular reorganization that far exceeds the known signaling activities of PI3K. The changes are highly diverse, with examples in structural protein levels, the DNA repair machinery, and sterol synthesis. Gene set enrichment analysis reveals a highly significant concordance of the genes differentially expressed in MCF-10A-H1047R cells and the established protein and RNA signatures of basal breast cancer. No such concordance was found with the specific gene signatures of other histological types of breast cancer. Our data document the power of a single base mutation, inducing an extensive remodeling of the cell toward the phenotype of a specific cancer.
Human cancers carry a multitude of genetic changes (1–4). These mutations can be divided into driver mutations and passenger mutations. Driver mutations confer a selective advantage to the cancer cell, and passenger mutations have no effect on cancer-relevant properties, including proliferation and invasiveness. Most cancers carry mutations in several driver genes. The oncogenic properties of the cancer cell result largely from these driver mutations and their complex interactions. In this cooperative reorganization of the cell, the consequences of individual driver mutations cannot be identified. However, for a fundamental understanding of oncogenesis and for translational goals, knowledge of the specific downstream effects of single cancer mutations is essential.
To understand the global consequences of a single mutation, we have carried out an extensive comparison of two cell lines that differ by a single oncogenic driver mutation. We have used RNA sequencing (RNAseq), stable isotope labeling by amino acids in cell culture (SILAC), and cell biological methods to study the MCF-10A cell line and the knock-in of the H1047R oncogenic mutation of PIK3CA (the gene encoding the catalytic subunit p110α of PI3K) in MCF-10A cells, which was generated by Ben Ho Park (Johns Hopkins University, Baltimore) and provided through his courtesy (5). MCF-10A and MCF-10A-H1047R differ by a single base substitution in one allele of PIK3CA, a difference we have confirmed by RNAseq (Materials and Methods). MCF-10A is an immortalized epithelial cell line isolated from human breast tissue (6). It is capable of continuous growth and shows an abnormal but stable karyotype, modest amplification of c-MYC (v-myc avian myelocytomatosis viral oncogene homolog) (threefold), and homozygous deletion of CDKN2a (p14ARF). It is negative for the expression of the estrogen receptor and the progesterone receptor, and ERBB2 (epidermal growth factor receptor 2) is not amplified (6–8). Both MCF-10A and MCF-10A-H1047R can grow in chemically defined, serum-free medium, facilitating the amino acid substitutions required by SILAC and avoiding the variability introduced by the use of serum in the culture medium (7, 9).
The changes induced in protein and RNA expression by the H1047R mutation document a comprehensive reorganization of the cell, including a shift of the expression patterns toward the signature of basal breast cancer.
Results
Genetic Comparison of the MCF-10A and MCF-10A-H1047R Cell Lines.
MCF-10A and MCF-10A-H1047R are considered isogenic, except for the knock-in mutation of H1047R in one allele of PIK3CA. However, during the creation of the H1047R knock-in or in the course of the subsequent culture, other mutations in cancer-relevant genes could have been introduced or selected for. To investigate this possibility, both cell lines were studied by whole-exome sequencing. The procedures used for exome sequencing are described in SI Materials and Methods. This sequence information was used to determine variant SNPs (single-nucleotide polymorphisms) and insertions and deletions, as well as copy number variations. Variants that are significantly different between the two cell lines are shown in Table S1. Other than PIK3CA-H1047R, there are no mutations in genes that have also been found mutated in a significant fraction of human tumor samples as recorded in the COSMIC database (version 70; cancer.sanger.ac.uk/cancergenome/projects/cosmic/) (10). There are, however, 43 nucleotides mutated between the two cell lines in coding regions spanning 26.8 million nucleotides. These data yield a mutation rate of 1.7 nucleotides per million. These mutations do not affect cancer-relevant genes, but the number of mutations is within the expected range of somatic mutations seen in tumor vs. normal comparisons of breast cancer samples (4). It is surprising that the H1047R mutation should lead to this number of mutations within the few passages that separate the two cell lines and suggests that the H1047R mutation may induce genome instability. Additionally, we determined copy number changes between MCF-10A and MCF-10A-H1047R. An initial analysis for focal deletions and amplifications revealed five small amplifications between the two cell lines (Table S2). None of these regions is known to be significant in human cancer. In an analysis of larger amplifications and deletions using SNP heterozygosity, we found that there are additional alterations in the variant allele frequency in chromosomes 5, 22, and X between MCF-10A-H1047R and the parental MCF-10A cell line (Fig. S1). The entire X chromosome shows a loss of heterozygosity consistent with loss of a complete copy of the chromosome. The 5′ end of chromosome 5 has an alteration that is consistent with amplification of 5p13-15 in MCF-10A-H1047R. Chromosome 22 has two copies near the 5′ end and a single copy throughout the 3′ portion, which is in contrast to amplifications seen in MCF-10A. Therefore, although MCF-10A and MCF-10A-H1047R are not strictly isogenic, none of the genomic differences we have identified can explain the changes in gene expression that we document below.
Changes in Gene Expression.
Knock-in of the H1047R mutation into one allele of the PIK3CA gene encoding the catalytic subunit p110α of PI3K induces changes in gene expression that are reflected in the transcriptome and proteome. These changes were assessed by RNAseq and SILAC. SILAC was performed by culturing the cells with labeled amino acids for three passages. RNAseq data reflect a time series and were collected at four time points over a 24-h period. Sampling times were chosen to assess possible effects of varying growth conditions on gene expression, and indeed revealed an important effect of these conditions on several gene signatures. The cells were initially seeded in DMEM/F-12 growth medium containing 5% (vol/vol) donor horse serum and 20 ng/mL EGF, and were allowed to attach to the plastic substrate for 24 h. The medium was then replaced with serum-free MCDB-170 medium containing 10 ng/mL EGF, and after an additional 24 h of equilibration, the cells received another change of fresh MCDB-170 medium with EGF. At that time, the first samples were taken for RNAseq, followed by additional samples at 6, 12, and 24 h. The changes seen with SILAC and RNAseq amount to an extensive remodeling of the cell; they are complex and reach far beyond the canonical PI3K signaling pathway. Of 12,938 transcripts identified by RNAseq, 1,098 are significantly up-regulated and 986 are down-regulated [, where log2FC represents log2 fold change, and Q (false discovery-corrected P value) < 0.01] (Fig. S2 and Table S3). Likewise, 521 and 853 of 3,982 proteins are significantly up- and down-regulated, respectively (Table S4). In the data as a whole, there is only a very weak positive correlation (r2 = 0.02) between the changes observed in mRNA levels and those changes identified in protein levels (Fig. 1 A and B). This discordance is not unexpected. Although the PI3K pathway leads to the activation of several transcription factors, the changes in protein expression are dominated by the activation of protein translation downstream of TOR (target of rapamycin) through S6, which regulates ribosome biogenesis, and through eIF4E, which directly regulates protein translation. Similar observations were recorded in two studies on the effects of ATP-competitive and allosteric inhibitors of the TOR kinase (11, 12). However, against this background of overall discordance of RNA and protein changes, there are specific gene signatures where the differential effect of H1047R on proteins is broadly concordant with the effect on mRNA expression.
Fig. 1.
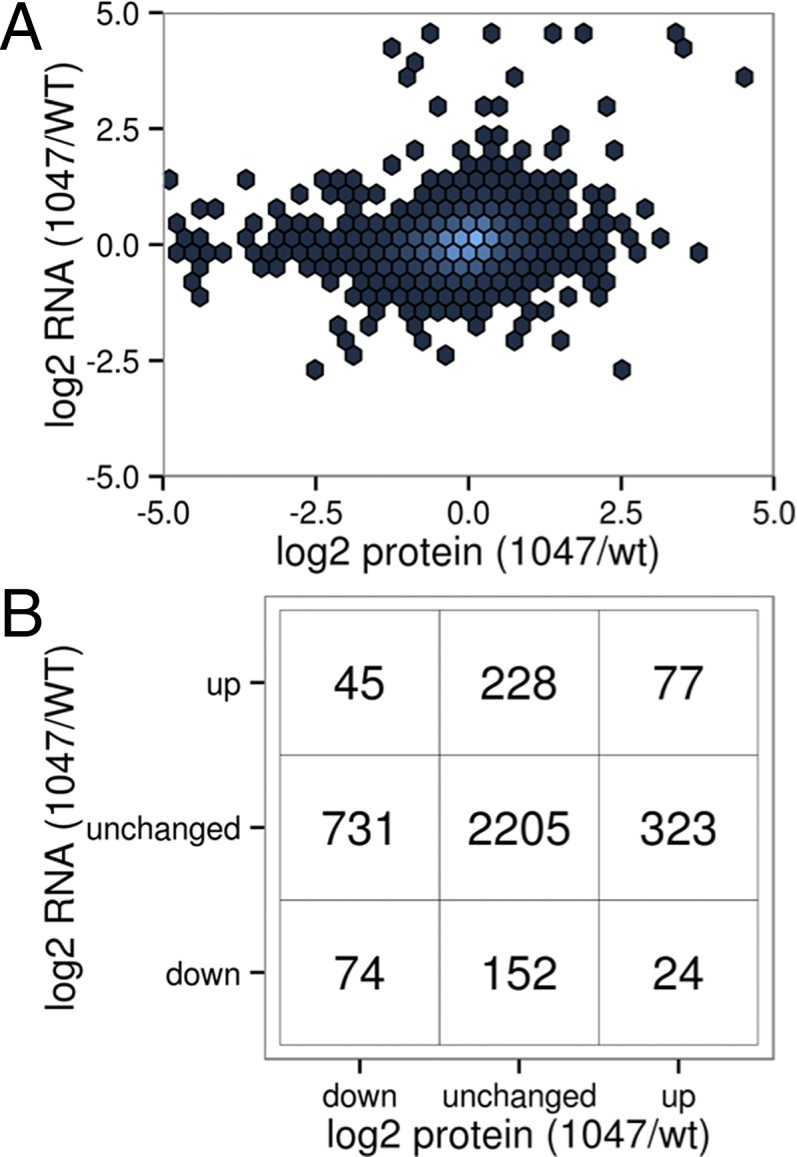
Nonconcordance of changes in RNA and protein. (A) Scatterplot of changes in RNA vs. protein. (B) Contingency table of significant changes (P < 0.001) in RNA and protein levels.
RNAseq and SILAC datasets were subjected to Gene Set Enrichment Analysis (GSEA) (13, 14). This method quantifies the relative enrichment of a gene signature in the MCF-10A-H1047R expression data. Signatures are taken from the publicly available Molecular Signature Database (MSigDB; www.broadinstitute.org/gsea/msigdb). Such signatures typically define specific traits, such as cellular function, specific disease state, chromosomal location, or transcription factor binding sites. A determination is made whether genes of the signature are evenly distributed in the observed gene expression data or are unevenly distributed with a bias toward up-regulated or down-regulated genes in the gene expression data. This bias is quantified as an enrichment score that reflects the relative degree of overrepresentation of the signature in the expression data. Significance is determined by resampling after permutation of the gene labels.
Cancer Gene Signatures.
The expression data for genes differentially expressed in the specific breast cancer subtypes (basal, luminal, ERBB2-positive, and normal-like), were analyzed by GSEA. Selected results are summarized in Table 1 and Fig. 2A, and comprehensive GSEA data are presented in Table S5. MCF-10A-H1047R cells in both RNAseq and SILAC datasets show enrichment for the genes that are overexpressed, as well as the genes that are underexpressed, in basal breast cancer. There is also concordance of RNA and protein expression data with the signature of the normal-like subtype of breast cancer. For the ERBB2-positive subtype of breast cancer, significant concordance is seen only with the protein but not with the RNA expression data. In contrast, the expression profiles on the MCF-10A-H1047R cells show no concordance with the gene signature of the luminal B subtype of breast cancer (Table 1). These data indicate that the H1047R mutation in MCF-10A cells, and hence gain of function in PI3K, is responsible for characteristic phenotypic features of distinct subtypes of breast cancer: primarily basal and, to a lesser extent, normal-like and ERBB2-positive breast cancer. The enrichment for the basal breast cancer signature is dominated by the increased expression of cytokeratins, a family of structural proteins that is not known to be regulated by PI3K (Fig. S3).
Table 1.
GSEA comparing differential RNA and protein expression patterns observed for MCF-10A-H1047R vs. MCF-10A with previously established breast cancer signatures
| Gene set* | Size | NES | P | Dataset |
| SMID BREAST CANCER BASAL UP† | 446 | 1.59 | 0 | RNAseq |
| SMID BREAST CANCER BASAL DN‡ | 434 | −1.2 | 0 | RNAseq |
| SMID BREAST CANCER NORMAL LIKE UP§ | 178 | 1.5 | 0.003 | RNAseq |
| SMID BREAST CANCER ERBB2 UP¶ | 97 | 1.32 | 0.071 | RNAseq |
| SMID BREAST CANCER LUMINAL B UP# | 99 | −1.34 | 0.056 | RNAseq |
| SMID BREAST CANCER LUMINAL B DNǁ | 287 | 1.96 | 0 | RNAseq |
| SMID BREAST CANCER BASAL UP | 149 | 1.51 | 0.006 | SILAC |
| SMID BREAST CANCER BASAL DN | 113 | 1.3 | 0.049 | SILAC |
| SMID BREAST CANCER NORMAL LIKE UP | 32 | 1.72 | 0 | SILAC |
| SMID BREAST CANCER ERBB2 UP | 47 | 1.69 | 0 | SILAC |
| SMID BREAST CANCER LUMINAL B UP | 26 | −1.11 | 0.318 | SILAC |
| SMID BREAST CANCER LUMINAL B DN | 98 | 1.64 | 0 | SILAC |
Size represents number of genes. DN, DOWN; NES, normalized enrichment score.
As listed in the MSigDB (www.broadinstitute.org/gsea/msigdb).
Genes up-regulated in the basal subtype of breast cancer (36).
Genes down-regulated in the basal subtype of breast cancer (36).
Genes up-regulated in the normal-like subtype of breast cancer (36).
Genes up-regulated in ERBB2+ breast cancer (36).
Genes up-regulated in the luminal B subtype of breast cancer (36).
Genes down-regulated in the luminal B subtype of breast cancer (36).
Fig. 2.

GSEA enrichment plots of a gene set composed of genes up-regulated in the basal subtype of breast cancer for RNA expression data (36) (A) and genes involved in cell cycle (37) (B).
Signatures Representing Cellular Functions.
A detailed analysis of the SILAC and RNAseq data revealed extensive effects of the H1047R mutation on diverse cellular functions and activities. Many of these effects are not known to be connected to the activity of PI3K (Table S5). As examples of these changes, we present here the signatures of the cell cycle genes, DNA repair, MYC targets, and the cadherin 1-dependent signature (Table 2). In this context, RNAseq revealed an interesting feature of the H1047R-controlled expression patterns: dependence as well as independence on sampling time. The GSEA results summarized in Figs. 2 and 3 and Table 2 illustrate two strikingly different patterns. Some GSEA results, such as the concordance of the H1047R expression changes with the H1047R expression changes of basal breast cancer or with the CDH1 (cadherin 1) activity, remain constant during the 24-h sampling period, and therefore appear to be independent of growth conditions. In contrast, the enrichment scores for the signature of DNA repair, the cell cycle, MYC targets, and EGF-dependent stimulation change over time from an initial negative score to a positive score, or vice versa (Figs. 2 and 3 and Table 2). Thus, DNA repair and cell cycle genes show negative enrichment scores in the earliest samples, becoming positive at 24 h. For the MYC signatures, we tested two sets of targets: the transcriptionally stimulated and transcriptionally repressed genes. Positive targets that are normally up-regulated when MYC is expressed are down-regulated at the zero time point and up-regulated at the later time points. The reverse is true of the negative MYC targets. These data indicate a transition from low MYC activity early to high activity during the later samplings. Genes transiently responding to EGF start out at a high level, in concordance with the EGF signature immediately upon medium change, and then decline (Fig. 3).
Table 2.
Time-dependent and time-independent gene signatures of MCF-10A-H1047R
| Gene set* | No. of genes | 0 h | 24 h | ||
| NES | P | NES | P | ||
| SMID BREAST CANCER BASAL UP† | 412 | 1.58 | 0 | 1.45 | 0 |
| SMID BREAST CANCER BASAL DN‡ | 391 | −1.18 | 0.043 | −1.04 | 0.333 |
| ONDER CDH1 TARGETS 2 DN§ | 390 | 2.5 | 0 | 1.79 | 0 |
| ONDER CDH1 TARGETS 2 UP¶ | 155 | −1.67 | 0 | −2.29 | 0 |
| REACTOME CELL CYCLE# | 355 | −1.98 | 0 | 1.56 | 0 |
| KAUFFMANN DNA REPAIR GENESǁ | 211 | −1.57 | 0 | 1.72 | 0 |
| DANG REGULATED BY MYC UP** | 64 | −1.75 | 0 | 1.05 | 0.363 |
| DANG REGULATED BY MYC DN†† | 185 | 1.36 | 0.017 | −1.91 | 0 |
| ZWANG CLASS 2 TRANSIENTLY INDUCED BY EGF‡‡ | 33 | 1.78 | 0.01 | −2.17 | 0 |
As listed in the MSigDB (www.broadinstitute.org/gsea/msigdb).
Genes up-regulated in the basal subtype of breast cancer (36).
Genes down-regulated in the basal subtype of breast cancer (36).
Genes down-regulated after E-cadherin knock-down (38).
Genes up-regulated after E-cadherin knock-down (38).
Available at www.reactome.org.
Genes involved in DNA repair (39).
Genes up-regulated by Myc (40).
Genes down-regulated by Myc (40).
Genes transiently induced by EGF (37).
Fig. 3.

Heat map of normalized enrichment scores (NESs) from the GSEA analyses of MCF-10A-H1047R. Although some signatures are time-independent, others show strong time dependence. An explanation of gene sets is provided in Table 2. CDH1, cadherin 1; DN, DOWN.
Metabolic Changes.
Metabolomic analyses of MCF-10A and MCF-10A-H1047R revealed numerous significant differences in metabolic activities between the two cell lines. Selected metabolites are summarized in Table 3, with the complete dataset included in Table S6. Some changes, like the increase in fatty acids, such as linoleic acid (15–17), were anticipated. MCF-10A-H1047R cells show decreases in cholesterol levels despite up-regulation of cholesterol biosynthesis genes (compare Table S5). Additionally, MCF-10A-H1047R cells show large increases in AMP (adenosine monophosphate) but no corresponding elevation in the phosphorylation of AMPK (AMP-activated protein kinase) (Fig. S4), suggesting that MCF-10A-H1047R cells are in a state of energy starvation. Several amino acids are also more abundant in MCF-10A-H1047R cells compared with the parental cells (Table S6). There is also an increase in 5-oxoproline that is indicative of a dysfunction in the gamma-glutamyl cycle, which is important in the production and degradation of glutathione (18).
Table 3.
Select significant changes in metabolites
| Metabolite | FC | P |
| Zymosterol | 0.39 | 7.2E-04 |
| 25-SO4 cholesterol | 0.33 | 2.4E-02 |
| 24-Hydroxycholesterol | 0.28 | 2.2E-02 |
| Linoleic acid | 2.71 | 1.9E-04 |
| Adenosine-5-phosphate | 2.65 | 5.5E-04 |
| Oxoproline | 1.85 | 2.4E-02 |
FC, fold change (H1047R/WT MCF-10A).
Discussion
The “butterfly” effect derives from a concept in chaos theory created by the meteorologist Edward N. Lorenz in a now classical paper that appeared in the Journal of the Atmospheric Sciences in 1963 (19). In essence, this idea states that small changes in a nonlinear system can have very large consequences. Applied to meteorology, Lorenz (19) speculated that the wing flaps of a butterfly could trigger a tornado. The butterfly concept applies perfectly to the single base substitution studied in the MCF-10A cell line: It is the smallest genetic change that can be introduced in a cell, yet it has immense consequences.
The magnitude and extent of the changes seen in MCF-10A-H1047R cells raise the question of whether the process of generating the knock-in cell line has inadvertently also selected for other mutations, amplifications, or deletions that could be responsible for some of the changes observed. The data obtained by exome sequencing and RNAseq show that although there are mutations besides the PIK3CA H1047R that distinguish the two cell lines, none affects a cancer-related gene included in the COSMIC: Cancer Gene Census (cancer.sanger.ac.uk/cancergenome/projects/census/) (10) (Table S1). Additionally, the knock-in cell line has lost one copy of three different chromosomal regions. Although, again, none of these chromosomal regions contain known tumor suppressors, we cannot definitely rule out the possibility that these regions contribute to the overall phenotypes observed. These mutations and deletions could be caused by the knock-in technique, or, alternatively, mutant PI3K could induce some level of genomic instability.
The data from RNAseq and SILAC lead to two unexpected findings: (i) The H1047R mutation in PIK3CA in MCF-10A cells induces extensive remodeling of the cells, and (ii) the protein and RNA signatures of MCF-10A-H1047R cells are concordant with the protein and RNA signatures of basal breast cancer.
The depth and extent of the mutant-induced remodel far exceed the known sphere of signaling that can be tied to PI3K. It is possible that the wide reach of these changes simply reflects our incomplete knowledge of the central importance of PI3K as a regulator of cellular activities and that PI3K is connected much more broadly than is generally appreciated. Mutations in a less critical gene would then have more limited consequences. However, it is also conceivable that some of the H1047R mutant-induced changes are indicators of a novel, general level of broad cellular connectivity. A great diversity of phosphoproteomic changes has also been documented in a recently published comparison of the MCF-10A and MCF-10A-H1047R cell lines (20). Our work is in substantial agreement with that comparison; the two studies are mutually supportive and complementary. Support for a systematic cellular connectivity also comes from investigations on protein kinases (21–23). Molecular mechanisms that could induce the multiple changes identified in MCF-10A cells would include, besides canonical signaling, differential levels of micro-RNAs, long noncoding RNAs, and epigenetic changes. An important additional analysis uses small-molecule inhibitors of PI3K or of TOR to revert the H1047R-associated phenotype in MCF-10A. We have carried out such experiments, which show that inhibitors induce only a partial reversion of the H1047R phenotype. These observations support the suggestion that the MCF-10A-H1047R cells have undergone knock-in–mediated epigenetic changes. This work will be detailed in a separate publication. In this context, it will also be of interest to examine other knock-in mutants (24) in MCF-10A cells and to determine mutant-induced changes in molecular signatures.
Some of the remodeling activities depend on the sampling time for RNAseq. This variability probably reflects conditions of cell culture, including growth factor and nutrient availability and population density. The GSEA data indicate that there are significant differences between WT and mutant cells in the response to such changing conditions. Hence, PIK3CA affects not only the static state of the cell but also the dynamics of response to external stimuli.
Breast cancers, on average, carry about one nonsilent mutation per megabase or around 4,000 per tumor (3). Most of these mutations will be passenger mutations, but a multiplicity of potential driver mutations still remain. It is tempting to assume that in this multiplicity, the effects of a single driver mutation are confined and marginal. However, analysis of the MCF-10A-H1047R cells proves otherwise. A single mutation can drive the cellular expression profile close to the cellular expression profile of a fully developed cancer. Although the single mutation of PIK3CA is insufficient to transform the cell completely, it nevertheless advances the cell substantially toward the gene expression state of basal breast cancer. MCF-10A-H1047R cells become fully transformed in combination with a KRAS mutant knock-in; they are then able to form tumors in nude mice (24). This fact suggests that the consistent mutations in cancer function as the main drivers in oncogenesis, with the more sporadic genetic changes making incremental or no contributions to the process.
The molecular signature of MCF-10A-H1047R cells is concordant with the molecular signature of basal breast cancer and, to a lesser extent, with normal-like and ERBB2-positive breast cancer, but not with the signature of luminal breast cancer. MCF-10A cells are derived from normal human breast epithelium and were originally characterized as luminal ductal cells (25). However, they are negative for expression of the estrogen and progesterone receptors and show marginal expression of ERBB2, thus sharing basic features with basal triple-negative breast cancer. It is therefore conceivable that the gain of function in PI3K triggers a preprogrammed process inherent in MCF-10A cells that inevitably leads to the phenotype of basal breast cancer. The differentiation state of MCF-10A cells may prescribe the type of cancer signature that can be induced by the PI3K mutation. In an isogenic pair of cells representing another state of differentiation, the H1047R mutation could induce a gene signature characteristic of a cancer that typifies this other cell type. In general terms, this observation suggests that the effects of a somatic mutation are determined by the cell type in which the mutation occurs.
Materials and Methods
Cells.
MCF-10A and MCF-10A-H1047R cells were acquired from Ben Ho Park. Cells were grown in DMEM/F-12 supplemented with 5% donor horse serum (Gemini), 0.5 μg/mL hydrocortisone (Sigma), 10 μg/mL insulin (Sigma), 100 ng/mL Cholera Toxin (Sigma), and penicillin/streptomycin/l-glutamine (Sigma). WT cells were supplemented with 20 ng/mL EGF (Repligen). Cells were passaged using 0.25% trypsin EDTA (Life Technologies) at a 1:4 ratio every 3 d. Passage numbers were kept below 10 for all experiments. MCDB-170 was made according to previously published procedure with the following modifications: arginine and lysine were removed from the recipe and added along with the final supplements to facilitate SILAC labeling, and ovine prolactin and bovine pituitary extract were omitted from the final MDS supplement (9).
RNAseq.
Five hundred thousand cells were plated in a 10-cm Petri dish in DMEM/F-12 growth media in triplicate. After 24 h, the media were exchanged to MCDB-170 containing 10 ng of EGF. After a further 24 h, the medium was exchanged again to fresh MCDB-170 with EGF, and cells were collected at 0, 6, 12, and 24 h. RNA was prepared from cells using TRIzol (Life Technologies) extraction. Genomic DNA was removed using Ambion DNA-free. NuGEN Encore reagents were used for library preparation from total RNA samples. One microgram of total RNA input was used for each sample. The libraries were sequenced on an Illumina HiSeq 2000 sequencing system using 100-bp single-ended reads. Raw, as well as processed, data are available online (Gene Expression Omnibus accession no. GSE63452).
SILAC.
Cells were seeded in 10-cm Petri dishes in DMEM/F-12 growth media in triplicate. After 24 h, the media were exchanged to MCDB-170 medium containing either U-12C-14N-lysine and U-12C-14N-arginine for WT cells or U-13C-15N-lysine and U-13C-15N-arginine for H1047R cells. Cells were passaged in this serum-free medium using 0.25% trypsin in PBS followed by neutralization with 0.25% soybean trypsin inhibitor in PBS. Cells were centrifuged for 3 min at 300 × g, and the inactivated trypsin was removed. Cells were resuspended in MCDB-170 and seeded at a 1:4 ratio in new plates. After three passages, cells were collected when about 50% confluent using trypsin and soybean trypsin inhibitor. Cells were counted using a Coulter Z1 counter, and equal numbers of WT and H1047R cells were mixed and centrifuged for 3 min at 300 × g. The cell pellets were lysed with RIPA buffer and used for MS.
MS.
Thirty micrograms of protein extract from the cell lysates was precipitated with a 5× vol of cold acetone. The protein pellets were obtained by centrifuging at 14,000 × g for 10 min at 4 °C, and then solubilized and reduced with 100 mM Tris⋅HCl/8 M urea/5 mM DTT (pH 8.5). Cysteines were alkylated with 10 mM iodoacetamide. The solution was diluted at a 1:4 ratio with 100 mM Tris (pH 8.5) and digested with 1 μg of trypsin at 37 °C overnight. Adding formic acid to 2% terminated the digestion. Nineteen biological replicates were analyzed.
MS and data analysis were performed as previously described (26). Briefly, the protein digest was analyzed using an 11-step MudPIT (multidimensional protein identification technology) (27). In each salt step, peptides were eluted from the C18 microcapillary column over a 2-h chromatographic gradient and electrosprayed directly into an LTQ Velos Orbitrap mass spectrometer (ThermoFisher) with the application of a distal 2.5-kV spray voltage. A cycle of one full-scan mass spectrum (400–1,800 m/z) at a resolution of 60,000, followed by 20 data-dependent MS/MS spectra at 35% of normalized collision energy, was repeated continuously throughout each step of the multidimensional separation.
Gene Expression.
RNAseq data were mapped to HG19 using Bowtie/Tophat aligner (28). Reads were counted using htseq (29) utilizing the GENCODE version 19 gene annotations (30, 31). Analysis of differential expression was performed using edgeR (32) after filtering the data such that a minimum of three samples had more than 0.3 reads per million.
Peptide identification was performed with the Integrated Proteomics Pipeline (IP2; Integrated Proteomics Applications, Inc.; www.integratedproteomics.com/) using ProLuCID (33). The detected SNPs and expressed genes, along with the HG19 genome, were used to create a targeted protein database. The tandem mass spectra were searched against this protein database. A target-decoy database containing the reversed sequences of all of the proteins appended to the target database was used in the database search. Cysteine carboxyamidomethylation was set as a stable modification. Peptide expression ratios were measured using Census. The logarithm of the mean expression ratio was calculated, and the data were shifted such that the mean expression ratio was 0 for all samples. There was a consistent bias in all three SILAC samples for approximately twofold higher expression of proteins in H1047R cells compared with WT cells.
Raw peptides from Census were mapped to genes by creating graphs of peptides to proteins and proteins to gene symbols. The union of these graphs allows the mapping of peptides to gene symbols. Only those peptides with unique mapping to a single gene symbol are used for peptide quantification. The set of uniquely assignable peptide ratios measured by Census was then used to determine the median gene expression ratio using bootstrap statistics (R = 10000). Ninety-five percent confidence intervals for the median were also calculated, along with P values comparing the median expression with the 100 most abundant proteins.
GSEA was performed using GSEA2 (13) with the MsigDB version 4.0 curated gene sets (R = 10,000). GSEA graphs were reproduced in R using ggplot2 by reimplementation of GSEA2 algorithms.
Metabolomics.
Cholesterols and steroids were analyzed by UPLC (ultra performance liquid chromatography)-MS/MS as previously described (34). Other metabolites were analyzed by GC/MS/EI (electron impact ionization) as previously described (35).
Supplementary Material
Acknowledgments
We thank Dr. Ben Ho Park for generously providing the MCF-10A-H1047R cell line used in this study. The metabolomics analysis was carried out at the NIH West Coast Metabolomics Center (University of California, Davis). This work was supported by the NIH under Awards R01 CA078230 and R21 AG039716 (to P.K.V.) and Awards R01 MH067880 and P41 GM103533 (to J.R.Y.). This is Manuscript 28000 of The Scripps Research Institute.
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE63452). The raw reads used for variant calling have been deposited in the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA), www.ncbi.nlm.nih.gov/sra (project SRP050011).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424012112/-/DCSupplemental.
References
- 1.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gustin JP, et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci USA. 2009;106(8):2835–2840. doi: 10.1073/pnas.0813351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soule HD, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6075–6086. [PubMed] [Google Scholar]
- 7.DiRenzo J, et al. Growth factor requirements and basal phenotype of an immortalized mammary epithelial cell line. Cancer Res. 2002;62(1):89–98. [PubMed] [Google Scholar]
- 8.Subik K, et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer (Auckl) 2010;4:35–41. [PMC free article] [PubMed] [Google Scholar]
- 9.Hammond SL, Ham RG, Stampfer MR. Serum-free growth of human mammary epithelial cells: Rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc Natl Acad Sci USA. 1984;81(17):5435–5439. doi: 10.1073/pnas.81.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh AC, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu PP, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subramanian A, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 15.Huffman TA, Mothe-Satney I, Lawrence JC., Jr Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc Natl Acad Sci USA. 2002;99(2):1047–1052. doi: 10.1073/pnas.022634399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19(22):R1046–R1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soliman GA. The integral role of mTOR in lipid metabolism. Cell Cycle. 2011;10(6):861–862. doi: 10.4161/cc.10.6.14930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meister A. The gamma-glutamyl cycle. Diseases associated with specific enzyme deficiencies. Ann Intern Med. 1974;81(2):247–253. doi: 10.7326/0003-4819-81-2-247. [DOI] [PubMed] [Google Scholar]
- 19.Lorenz EN. Deterministic nonperiodic flow. Journal of the Atmospheric Sciences. 1963;20(2):130–141. [Google Scholar]
- 20.Wu X, et al. Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nat Commun. 2014;5:4961. doi: 10.1038/ncomms5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graves LM, Duncan JS, Whittle MC, Johnson GL. The dynamic nature of the kinome. Biochem J. 2013;450(1):1–8. doi: 10.1042/BJ20121456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stuhlmiller TJ, Earp HS, Johnson GL. Adaptive reprogramming of the breast cancer kinome. Clin Pharmacol Ther. 2014;95(4):413–415. doi: 10.1038/clpt.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson GL, Stuhlmiller TJ, Angus SP, Zawistowski JS, Graves LM. Molecular pathways: Adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin Cancer Res. 2014;20(10):2516–2522. doi: 10.1158/1078-0432.CCR-13-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang GM, et al. Single copies of mutant KRAS and mutant PIK3CA cooperate in immortalized human epithelial cells to induce tumor formation. Cancer Res. 2013;73(11):3248–3261. doi: 10.1158/0008-5472.CAN-12-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tait L, Soule HD, Russo J. Ultrastructural and immunocytochemical characterization of an immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6087–6094. [PubMed] [Google Scholar]
- 26.McClatchy DB, Liao L, Lee JH, Park SK, Yates JR., 3rd Dynamics of subcellular proteomes during brain development. J Proteome Res. 2012;11(4):2467–2479. doi: 10.1021/pr201176v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 28.Kim D, et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shern JF, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4(2):216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engström PG, et al. RGASP Consortium Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods. 2013;10(12):1185–1191. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steijger T, et al. RGASP Consortium Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013;10(12):1177–1184. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu T, et al. ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program. Mol Cell Proteomics. 2006;5:S174. [Google Scholar]
- 34.Gaikwad NW. Ultra performance liquid chromatography-tandem mass spectrometry method for profiling of steroid metabolome in human tissue. Anal Chem. 2013;85(10):4951–4960. doi: 10.1021/ac400016e. [DOI] [PubMed] [Google Scholar]
- 35.Fiehn O, et al. Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 2008;53(4):691–704. doi: 10.1111/j.1365-313X.2007.03387.x. [DOI] [PubMed] [Google Scholar]
- 36.Smid M, et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68(9):3108–3114. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 37.Zwang Y, et al. Two phases of mitogenic signaling unveil roles for p53 and EGR1 in elimination of inconsistent growth signals. Mol Cell. 2011;42(4):524–535. doi: 10.1016/j.molcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onder TT, et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68(10):3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 39.Kauffmann A, et al. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene. 2008;27(5):565–573. doi: 10.1038/sj.onc.1210700. [DOI] [PubMed] [Google Scholar]
- 40.Zeller KI, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 2003;4(10):R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.