

Significance
Gaucher disease (GD) is an inherited metabolic storage disorder characterized by mutations in the gene GBA1 encoding for glucocerebrosidase (GCase). These mutations result in protein misfolding and subsequent premature degradation. Recognition by the heat shock protein (hsp) 90 complex is crucial for targeting of mutant GCase to the proteasome, but the mechanisms governing this association are unclear. This study describes a novel recruitment of Hsp27 to the Hsp90 complex that is specific to misfolded mutant GCase. Both gene knockdown and pharmacologic inhibition of Hsp27 increased GCase levels in patient-derived fibroblasts. Reduction of Hsp27 may circumvent premature protein degradation and represents a viable potential therapeutic strategy in the treatment of protein misfolding disorders.
Keywords: Gaucher disease, heat shock protein, hsp27, hsp90, molecular chaperone
Abstract
Gaucher disease is caused by mutations of the GBA1 gene, which encodes the lysosomal anchored gluococerebrosidase (GCase). GBA1 mutations commonly result in protein misfolding, abnormal chaperone recognition, and premature degradation, but are less likely to affect catalytic activity. In the present study, we demonstrate that the Hsp90/HOP/Cdc37 complex recruits Hsp27 after recognition of GCase mutants with subsequent targeting of GCase mutant peptides to degradation mechanisms such as VCP and the 26S proteasome. Inhibition of Hsp27 not only increased the quantity of enzyme but also enhanced GCase activity in fibroblasts derived from patients with Gaucher disease. These findings provide insight into a possible therapeutic strategy for protein misfolding diseases by correcting chaperone binding and altering subsequent downstream patterns of protein degradation.
Gaucher disease (GD) is an autosomal recessive disorder caused by mutations in the GBA1 gene encoding the lysosomal enzyme, glucocerebrosidase (GCase). Genetic alterations in GBA1 lead to quantitative losses of GCase and accumulation of toxic amounts of glucocerebroside. As a consequence, patients suffer widespread organ and metabolic dysfunction. Among the 360 mutations that have been identified in GD, the majority of nucleotide changes are missense mutations, in which a single amino acid is substituted by another (1, 2). However, instead of catastrophic loss of intrinsic enzymatic catalytic function, most GBA1 mutations lead to changes in the natural conformation of the peptide during protein folding, followed by retention within the endoplasmic reticulum (ER) (3–6). Subsequently, the unnatural peptide conformation affects chaperone recognition, which then stimulates premature protein degradation via mechanisms, involving c-cbl associated proteasomal complexes and the E3 ligases Parkin and Itch (4, 7, 8). Identifying key chaperone proteins that determine GCase proteostasis is a plausible approach for developing targeted therapies in GD especially in cases of disease manifestations in the central nervous system where standard enzyme replacement treatment is ineffective.
Heat shock protein 27 (Hsp27, also known as heat shock protein beta-1, HSPB1) is a chaperone protein of the small heat shock protein group that also includes ubiquitin, α-crystallin, and Hsp20 among others. In addition to its chaperone activities, Hsp27 is involved in thermotolerance, inhibition of apoptosis, and regulation of cell development and differentiation (9). Hsp27 acts as an ATP-independent chaperone by inhibiting protein aggregation and stabilizing partially denatured proteins ensuring proper refolding by the Hsp70 complex. In addition, Hsp27 may be a key player in proteasomal activation. For example, Hsp27 is integral to degradation of phosphorylated I-κBα in the proteasome (10). In addition, Hsp27 has been shown to interact with p27Kip1 and to guide its degradation (11). These findings imply that Hsp27 plays a significant role in the proteasomal degradation of misfolded proteins in mammalian cells.
We explored potential binding partners to mutant GCases using an unbiased mass spectrometric approach (20535800). We found that Hsp27 was a previously unidentified binding partner that specifically recognized two common mutations for type I (N370S/N370S) and type II/III (L444P/L444P) GD, but not its wild-type counterpart. When presented with the mutant GCase, the Hsp90 complex recruited Hsp27 before induction of the VCP/proteasomal degradation pathway. Targeting Hsp27 not only rescued quantitative enzymatic levels but also increased the catalytic activity of GCase in patient derived fibroblasts. These findings suggest that Hsp27 may be a useful target for the treatment of GD.
Results
Identification of Hsp27 That Specifically Recognizes Mutant GCase.
To identify key proteins that recognize mutant GCase, we used immunoprecipitation combined with an unbiased mass spectrometry technique (12). HeLa cells were transfected with wild-type GCase or with mutant N370S GCase. Protein was extracted from the cells and coprecipitated with Dynabead protein G and antibody against DDK epitope-tag (4C5). The precipitated protein was resolved on Bis-Tris gel. Proteins that specifically recognized N370S GCase were harvested and analyzed by mass spectrometry (Fig. 1A). In three independent experiments, we identified a peptide sequence that covered 20% of the Hsp27 sequence, suggesting that Hsp27 was interacting with the N370S GCase mutant. The Hsp27 band was less abundant in the wild-type GCase group, suggesting Hsp27 selectivity for the N370S GCase mutant over the wild type.
Fig. 1.
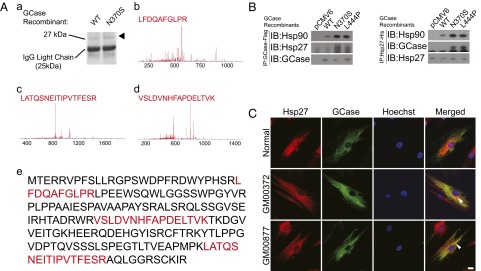
Hsp27 specifically recognizes GCase mutants. (A) Immunoprecipitation assay and unbiased mass spectrometry identified Hsp27 as a specific binding partner of N370S GCase mutants. (a) Silver stain of GCase binding protein. The tested band is indicated (arrow). (b–d) Mass spectrometry readout for 27-kDa band, shown in a. (e) Sequence validation of mass spectrometry read out (red) with Hsp27 amino acid sequence. (B) Immunoprecipitation assay showed that GCase mutants N370S and L444P were specifically recognized by Hsp27 and Hsp90 (Left). Hsp27 exhibited stronger affinity to Hsp90 and GCase mutants (Right). (C) Immunofluorescent staining showed Hsp27 was more likely to colocalize with GCase in fibroblasts derived from GD patients (arrow).
To confirm the interaction between Hsp27 and GCase mutants, we investigated the binding partner of GCase through further immunoprecipitation (Fig. 1B). We found that Hsp27 preferentially recognized two common mutant forms of GCase, type I (N370S/N370S) and type II/III (L444P/L444P). In the same assay we found that Hsp90 exhibited higher affinity for GCase mutants, which was consistent with our previous finding that Hsp90 is an important chaperone protein for the degradation of mutant GCase (13). In addition, through reversed immunoprecipitation assay, we found that Hsp27 was more likely to interact with N370S or L444P GCase mutants compared with the wild-type enzyme (Fig. 1C). We observed increased affinity between Hsp27 and Hsp90 in the presence of GCase mutants, suggesting that these two chaperones may participate in the same functional complex.
We confirmed the specificity of Hsp27 for the mutant GCase by immunofluorescent staining (Fig. 1C). Hsp27 showed punctuated distribution in the cytoplasm of fibroblasts from type I (GM00372) and type II (GM00877) GD patients. Fibroblasts from a normal donor were used as control. Hsp27 was more likely to colocalize with GCase in patient-derived fibroblasts versus cells with normal GBA1, confirming our finding that Hsp27 specifically recognized the mutant form of GCase.
Hsp27 Is Involved in the Hsp90 Chaperone Complex.
The folding and maturation of GCase requires the participation of molecular chaperone/cochaperones. The composition of the chaperone complex may in turn dictate the biologic outcome of successfully folded versus misfolded GCase after posttranslational modification. For example, our previous report showed that TCP1/Hsp70 chaperones were important for the proper folding of GCase (4). In contrast, we have demonstrated that the Hsp90/HOP/Cdc37 complex was integral for proteasomal degradation of unfolded or improperly folded GCase (14). A recent discovery showed that endoplasmic reticulum (ER)-associated Hsp40/ERdj3 was related to GCase degradation and simultaneously enhanced calnexin-associated folding (15). In the present study, the discovery that Hsp27 specifically recognized mutant GCase indicates that Hsp27 is part of the posttranslational complex that determines the destiny of mutant GCase. By using immunoprecipitation assays, we found that Hsp27 physically interacts with the Hsp90/HOP/Cdc37 chaperone complex (Fig. 2A). The affinity of Hsp27 for this chaperone complex depended on Hsp90 chaperone function, because the integration of Hsp27 to the complex was compromised with nonfunctional acetylation mimic mutations (K286Q and K286A) in Hsp90. In contrast, the wild-type or deacetylation mimic Hsp90 mutation (K286R) exhibited similar affinities for Hsp27. The importance of Hsp90 chaperone function in recruiting Hsp27 was further confirmed by pharmacologic inhibition of Hsp90 function with 17-AAG, a derivative of the antibiotic geldanamycin that has been previously studied as an Hsp90 inhibitor in certain tumors (16–18). We found that recognition of GCase mutants by Hsp90/HOP/Cdc37 as well as by Hsp27 were abolished with administration of 17-AAG (Fig. 2B). In addition, we found that Hsp90 recognition of GCase mutants occurs before Hsp27 recruitment, because the genetic silencing of Hsp27 did not alter the affinity of Hsp90 for N370S or L444P GCase mutants (Fig. 2C). Hsp90 recruited wild-type Hsp27, phosphorylation mimic, and dephosphorylation mimic mutants with similar efficiencies, suggesting Hsp27 was recruited by Hsp90 regardless of Hsp27 chaperone function (Fig. 2D).
Fig. 2.

Hsp27 is recruited by Hsp90 chaperone complex. Results of immunoprecipitation assays are shown. (A) Hsp27 was a binding partner to the Hsp90/HOP/Cdc37 chaperone complex. (B) Hsp27 recruitment was abolished by 17-AAG inhibition of Hsp90 chaperone function. (C) Hsp90 consistently recognized GCase mutants after depletion of Hsp27 through siRNA transfection. (D) Hsp90 consistently recognized GCase with different phosphorylation mimic recombinants of Hsp27.
Hsp27 Assists Hsp90 Chaperone Complex for GCase Degradation.
Although Hsp90 governs the posttranslational degradation of mutant GCase, the molecular mechanisms involved in targeting mutant proteins for degradation are unclear. Considering the role of Hsp27 in protein ubiquitination as well as proteasomal degradation, the involvement of Hsp27 in the Hsp90/HOP/Cdc37 complex implied that Hsp27 was functionally related to the degradation of GCase mutants. To confirm this, we first measured Hsp27 recruitment by the Hsp90 complex in the presence of GCase mutants. We found that Hsp27 was more likely to bind with the Hsp90 complex in the presence of GCase mutants N370S or L444P than wild-type GCase (Fig. 3A), suggesting that Hsp27 was actively involved in a Hsp90 complex specific for mutant GCase. We confirmed this with a pull down assay showing that Hsp27 and the Hsp90/HOP/Cdc37 complex were both associated with N370S or L444P GCase mutants (Fig. 3B). The fact that Hsp27 recruitment was significantly diminished in nonfunctional Hsp90 mutants confirmed that proper Hsp90 biologic function was integral to recognition and degradation of mutant GCase.
Fig. 3.

Hsp27 is important for Hsp90-determined GCase degradation. Results of immunoprecipitation assays are shown. (A) There was increased Hsp27 recruitment by Hsp90 in the presence of GCase mutant N370S or L444P. (B) Hsp90 recruitment by Hsp90 was abolished by a nonfunctional Hsp90 mutation. (C) The ubiquitination of GCase mutants was slightly increased in the setting of the Hsp27 dephosphorylation mimic mutant (Hsp27 3A) but was decreased in the presence of the nonfunctional Hsp27 phosphorylation mimic mutant (Hsp27 3D). (D) The ubiquitination of GCase mutants was abolished by a dominant negative Hsp90 recombinant.
GCase mutants are heavily ubiquitinated and degraded (7, 19), a process that depends upon involvement of the Hsp90 complex (13). The mutant-specific recruitment of Hsp27 to the Hsp90 chaperone complex suggested that Hsp27 was related to the ubiquitination and degradation of mutant GCase. Using an immunoprecipitation assay, we showed that N370S or L444P GCase mutants were ubiquitinated suggesting there was rapid turnover of the mutant protein (Fig. 3C). Overexpression of wild-type Hsp27 or of the dephosphorylation mimic mutant (Hsp27 3A) slightly increased GCase ubiquitination. In contrast, decreased protein ubiquitination was seen in the same assay in the presence of the nonfunctional Hsp27 phosphorylation mimic mutant (Hsp27 3D). Hsp27-mediated GCase ubiquitination also depended on Hsp90 chaperone function because the dominant negative variant of Hsp90 showed less efficiency in Hsp27 recruitment and GCase ubiquitination (Fig. 3D). These findings suggested that recognition by Hsp90 was required for Hsp27-associated protein ubiquitination.
Hsp27 Targets GCase Mutants to VCP/Proteasomal Degradation Complex.
We further examined the role of Hsp27 in targeting GCase mutants for degradation by investigating Hsp27 involvement in degradation mechanisms. We found that GCase mutants were more likely to be associated with VCP and the 26S proteasomal complex via the regulatory subunit, 19S (Fig. 4A). The increase in VCP/19S recognition was consistent with our previous finding that VCP and the 26S proteasome were required to degrade GCase mutants (14). In addition, in the present study we found that Hsp27 was more likely to bind with VCP and 19S in the presence of N370S and L444P mutants, suggesting Hsp27 was involved in VCP/26S-dependent GCase recognition (Fig. 4B). We investigated GCase ubiquitination and VCP/19S recognition in the presence of small interference RNA targeting Hsp27. We found that protein ubiquitination was reduced in both N370S and L444P GCase mutants when Hsp27 was depleted through siRNA transfection (Fig. 4C). Similarly, the recruitment of VCP and 19S to GCase mutants was abolished after siRNA knockdown of Hsp90, suggesting that Hsp90 recognition was required for Hsp27/VCP/19S association with GCase mutants (Fig. 4D).
Fig. 4.

Hsp27 targets GCase mutants to proteasomal degradation via VCP/26S degradation pathway. Results of immunoprecipitation assays are shown. (A) There was increased recognition of GCase mutants by VCP and the 19S proteasomal subunit. (B) There was increased VCP and 19S recruitment to Hsp27 in the presence of GCase mutants. (C) The ubiquitination and VCP/19S recognition of GCase mutants were eliminated by depletion of Hsp27 through siRNA. (D) Hsp27, VCP, and 19S binding to GCase was abolished by Hsp90 siRNA transfection.
Targeting Hsp27 Benefits GCase Quantity and Catalytic Activity.
Targeting molecular chaperones has been shown to be effective in rescuing mutant enzymes from degradation, resulting in increased catalytic activity (3, 20). Our data demonstrating the important role of Hsp27 in GCase mutant degradation suggest targeting Hsp27 may be a valuable strategy to rescue GCase activity in GD patients. To test the therapeutic value of targeting Hsp27, we depleted Hsp27 expression in fibroblasts derived from type I (GM00372 and GM00852) and type II/III (GM00877, GM08760 and HS1053) GD patients with siRNA. After two days, the quantity of GCase was measured by Western blot (Fig. 5A and Fig. S1). We found that after Hsp27 siRNA treatment, there was a significant increase (approximately threefold) of GCase levels from initial nondetectable amounts. This rescue of GCase function was confirmed in a fluorometric GCase assay, showing remarkably increased GCase activity upon treatment with Hsp27 siRNA (Fig. 5B).
Fig. 5.

Depletion of Hsp27 benefits GCase quantity and catalytic activity. (A) Western blot showed GCase levels were increased after depleting Hsp27 through siRNA transfection for 2 d. Normal dermal fibroblasts were used as a control. (B) Fluorometric GCase assay showed that depletion of Hsp27 by siRNA (siHsp27) increased the catalytic activity of GCase in patient-derived fibroblasts. (C) Cyclohexamide assay showed the degradation of GCase mutants was changed by Hsp27 siRNA treatment. (D) Densitometry analysis of CHX assay showed that depletion of Hsp27 (siHsp27) stabilized GCase mutants.
We further confirmed that the rescue of GCase quantity and enzymatic activity was through reduction of protein degradation. By using a CHX assay, we found that the efficiency of GCase mutant degradation was significantly reduced after depletion of Hsp27 with siRNA treatment (Fig. 5C). We quantified the degradation dynamic of GCase mutants and showed that protein half-lives increased from 5.76 h to 15.40 h and 0.94 h to 5.35 h for N370S and L444P GCase mutants, respectively (Fig. 5D).
Discussion
A combination of immunoprecipitation assays and unbiased mass spectrometry led to the identification and characterization of Hsp27, a previously unknown chaperone partner in GCase folding and degradation. N370S and L444P GCase mutants were less likely to accomplish correct folding and packaging than normal GCase, and instead, initiated a process characterized by premature degradation. We found that GCase mutants were initially identified by the Hsp90/HOP/Cdc37 chaperone complex. After failure of the mutant proteins to properly fold, this complex recruited Hsp27 and subsequently targeted mutant GCase for ubiquitination and VCP/26S-associated proteasomal degradation. Inhibition of Hsp90 or Hsp27 increased GCase activity in fibroblasts derived from GD patients, suggesting molecular chaperones may be a suitable target for diseases related to protein misfolding.
Protein Misfolding and Human Diseases.
Mutations in human genes are related to a variety of diseases and disorders. Missense mutations, in which one amino acid is substituted for another, characterize the genetic changes behind many diseases. However, most amino acid changes reside distally from the functional domains of the affected protein, and it is unclear how mutations in nonfunctional areas eventually result in the protein dysfunction. Our group among others has studied the significance of protein misfolding as a signal for premature protein degradation. For example, mutations in the SDHB gene are related to the quantitative loss of succinate dehydrogenase in complex II, and therefore, lead to manifestations of human neuroendocrine tumors like paraganglioma or pheochromocytoma (21). Amino acid changes in merlin protein result in quantitative loss of this tumor suppressor, which is related to the onset of neurofibromatosis type II (22). Similarly, the N370S or L444P missense mutation in GCase results in ubiquitination of GCase mutants and quantitative loss of the protein (4). Correction of protein misfolding and degradation may be of significant therapeutic value for these types of genetic abnormalities.
Targeting Chaperones as a Therapy for Protein Misfolding Disorders.
In the present study, we used GD as a disease model to investigate the mechanisms of premature degradation of misfolded proteins. Our previous study showed that cotranslational chaperone recognition is important in determining the biological outcome of GCase (3, 13). The composition of the chaperone complex subsequently dictates correct folding or, conversely, degradation of GCase. For example, Hsp70 and the TRiC/CCT complex are important for the correct folding and maturation of GCase from nascent peptides. When confronting a disease-related mutation such as N370S or L444P in GCase, the nascent peptides are more likely to be recognized by the Hsp90/HOP/Cdc37 chaperone complex. Subsequently, the GCase mutants are restricted to the ER compartment and are removed through ER-associated degradation (23, 24). Taken together, targeting members of the chaperone complex may be a reasonable approach for preventing premature protein degradation and restoring enzymatic activity to therapeutic levels. In the present study, we took advantage of an unbiased mass spectrometric approach and identified Hsp27 as an important participant in the posttranslational modifications of GCase mutants (Fig. 1), which implied a valuable therapeutic target in treating inherited diseases.
Hsp27 Is Recruited and Governed by Hsp90 Complex.
In the present study, we found that recruitment of Hsp27 resulted from recognition of GCase mutants by the Hsp90 complex, a process integral to shifting nascent GCase peptides from folding to degradation. Notably, the Hsp90 complex is a cytoplasmic complex and as such, degradation of mutant GCases by the Hsp90 complex, including Hsp27, occurs in the cytoplasm. The association of Hsp27 with GCase mutants depended on the integrity of Hsp90 chaperone function because inhibition with 17-AAG or nonfunctional mutants of Hsp90 abolished the ability of Hsp27 to recognize GCase (Fig. 2 A and B). On the other hand, although blockage of Hsp27 reduced protein ubiquitination and degradation (Fig. 3C), it did not affect Hsp90 recognition of the mutant protein (Fig. 2 C and D).
Hsp27 Is Important for GCase Degradation.
Our previous study suggested that the association of the Hsp90/HOP/Cdc37 complex with mutant GCase is a hallmark for GCase degradation. However, it remains unclear how an omnipotent chaperone complex such as Hsp90 is able to specifically differentiate GCase mutants from their wild-type counterparts and subsequently target them for degradation. Our discovery of Hsp27 as a GCase mutant-specific binding partner to the Hsp90 complex bridges this gap in understanding of selective protein degradation by an ubiquitous chaperone complex. Blocking Hsp27 function with the phosphorylation mimic mutant or siRNA reduced the activity of degradation machinery like VCP and 26S, and hence diminished protein ubiquitination and degradation (Fig. 4).
Summary
Our present study demonstrated that Hsp27 plays a significant role in the degradation of GCase mutants. Hsp27 was recruited by the Hsp90/HOP/Cdc37 complex when confronted with two common GCase mutants, N370S or L444P. The involvement of Hsp27 was critical for mutant GCase protein elimination via VCP/26S degradation (Fig. 6). Targeting Hsp27 represents a valuable therapeutic approach in GD as this degradation process is unique to GCase mutants. As we have shown, ablation of Hsp27 not only increases protein levels of GCase but also promotes the enzymatic activity in cells derived from GD patients. As such, small molecular compounds that target Hsp27 warrant further study for the treatment of patients with GD.
Fig. 6.

Hsp27 is recruited by Hsp90 and determines the degradation of GCase mutants. GCase mutant peptides are recognized by chaperones/cochaperones after translation. Wild-type GCase normally proceeds to the TRiC/CCT-associated chaperone complex for protein folding, packaging, and lysosomal distribution. The unnatural conformation of the mutant GCase results in protein misfolding and recognition by the Hsp90/Hop/Cdc37 chaperone complex. Hsp27 is recruited by the Hsp90 complex and targets GCase mutants for degradation mechanisms such as VCP and the 26S proteasome. As such, mutant GCase is effectively removed from the cytoplasm through premature degradation.
Materials and Methods
Cell Culture.
Fibroblasts from patients with GD were cultured as described (14). Cells from patients with type I GD (GM00372 and GM00852) and those with type II GD (GM00877 and GM08760) were purchased from Coriell Institute for Medical Research. HS1053 fibroblasts from a type II GD patient were kindly provided by Dr. Seymour Packman (University of California, San Francisco). HeLa cells were obtained from American Type Culture Collection (ATCC). Cells were maintained in Eagle’s minimum essential medium supplemented with 10% FBS (Invitrogen).
DNA Cloning and Site-Directed Mutagenesis.
WT, N370S and L444P GBA1 recombinants were prepared as described (3). Human HSPB1 gene was incorporated into the pCMV6-entry vector and pCMV6-AC-His vector (Origene). Mutagenesis of S15A, S78A, S82A, S15D, S78D, and S82D in Hsp27 was designed as reported (25). Mutagenesis was performed using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent). WT and K286 mutant HSP90AB1 gene was described (13). HSP90AB1 gene was shuttled to pCMV6-AC-HA vector (Origene) using the MluI/AsisI restriction enzyme and T4 ligation kit (New England Biolabs). The nucleotide sequences of constructed plasmids were verified by analyzing the entire coding regions by Sanger’s sequencing. HeLa cells were transfected with plamids, using Lipofectamine 2000 (Invitrogen), 48 h before consequence assays.
Immunoprecipitation.
Cells were collected by trypsin digestion and centrifuged at 200 × g for 10 min. Cells were washed three times in PBS and lysed in Nonidet P-40 lysis buffer supplemented with Halt proteasome inhibitor (Thermo Scientific). For testing protein ubiquitination, cells were treated with 20 μM MG-132 (Sigma) overnight and extracted for protein using RIPA lysis buffer supplemented with 1% SDS and Halt proteasome inhibitor. Total cell lysate was precipitated using the DynaBeads Protein G Immunoprecipitation kit (Invitrogen) and antibodies against Flag-tag, HA-tag or His-tag (Origene). Precipitated protein was eluted and resolved through Western blot analysis.
Western Blot Analysis.
Protein lysate was centrifuged and quantified using a Bio-Rad Protein Assay kit. Protein samples were resolved on a NuPAGE 4–12% Bis-Tris (vol/vol) gel (Invitrogen) and transferred to PVDF membranes (Millipore). The membranes were probed with primary antibodies and detected through an HRP-conjugated species-specific secondary antibody and enhanced chemiluminescence kit (Pierce). The intensity of the image was determined by densitometric analysis using ImageJ software. Primary antibodies used in this study included anti-DDK (Origene), anti-Hsp90 (Cell Signaling Technology), anti-Hsp27 (Cell Signaling Technology), anti-GCase (Abcam), anti-His (Origene), anti-HA (Origene), anti-HOP (Cell Signaling Technology), anti-Cdc37 (Cell Signaling Technology), anti-ubiquitin (Abcam), anti-VCP (Abcam, Cell Signaling Technology), anti-19S (Abcam), and anti-β-actin (Sigma).
Assay of GCase Activity.
The fluorometric GCase enzyme activity assay was performed as described (13).
RNA Interference.
RNA interference was performed as described (26). One hundred picomolar of siRNA oligo was transfected into HeLa cells or patient-derived fibroblasts using Lipofectamine RNAiMAX transfection reagent (Invitrogen). The expressions of targeted genes were analyzed by quantitative PCR (Figs. S2 and S3). A prevalidated siRNA targeting HSP90AB1 was used in the present study (119760, Invitrogen). The siRNA oligos targeting Hsp27 are shown in the supplementary information (Table S1). An AllStar Negative Control siRNA was used as a control (Qiagen).
Cycloheximide Pulse Chase Assay.
HeLa cells were transfected with expression vectors 2 d before protein stability measurement. Cells were exposed to cycloheximide (CHX) (50 μg/mL) for the period as indicated. Total protein was extracted with RIPA lysis buffer supplemented with protease inhibitors, and GCase residues were examined through Western blot and Flag-tag immunoblot.
Supplementary Material
Acknowledgments
The authors thank James A. Shayman, M.D., for his critical suggestions in the preparation of this manuscript. This work was supported by the Intramural Research Program at the National Institutes of Health (NIH).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424288112/-/DCSupplemental.
References
- 1.Horowitz M, Zimran A. Mutations causing Gaucher disease. Hum Mutat. 1994;3(1):1–11. doi: 10.1002/humu.1380030102. [DOI] [PubMed] [Google Scholar]
- 2.Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA) Hum Mutat. 2008;29(5):567–583. doi: 10.1002/humu.20676. [DOI] [PubMed] [Google Scholar]
- 3.Lu J, et al. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc Natl Acad Sci USA. 2011;108(52):21200–21205. doi: 10.1073/pnas.1119181109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J, et al. Decreased glucocerebrosidase activity in Gaucher disease parallels quantitative enzyme loss due to abnormal interaction with TCP1 and c-Cbl. Proc Natl Acad Sci USA. 2010;107(50):21665–21670. doi: 10.1073/pnas.1014376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babajani G, Tropak MB, Mahuran DJ, Kermode AR. Pharmacological chaperones facilitate the post-ER transport of recombinant N370S mutant β-glucocerebrosidase in plant cells: evidence that N370S is a folding mutant. Mol Genet Metab. 2012;106(3):323–329. doi: 10.1016/j.ymgme.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ron I, Horowitz M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet. 2005;14(16):2387–2398. doi: 10.1093/hmg/ddi240. [DOI] [PubMed] [Google Scholar]
- 7.Ron I, Rapaport D, Horowitz M. Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum Mol Genet. 2010;19(19):3771–3781. doi: 10.1093/hmg/ddq292. [DOI] [PubMed] [Google Scholar]
- 8.Maor G, Filocamo M, Horowitz M. ITCH regulates degradation of mutant glucocerebrosidase: implications to Gaucher disease. Hum Mol Genet. 2013;22(7):1316–1327. doi: 10.1093/hmg/dds535. [DOI] [PubMed] [Google Scholar]
- 9.Ciocca DR, Oesterreich S, Chamness GC, McGuire WL, Fuqua SA. Biological and clinical implications of heat shock protein 27,000 (Hsp27): a review. J Natl Cancer Inst. 1993;85(19):1558–1570. doi: 10.1093/jnci/85.19.1558. [DOI] [PubMed] [Google Scholar]
- 10.Parcellier A, et al. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol Cell Biol. 2003;23(16):5790–5802. doi: 10.1128/MCB.23.16.5790-5802.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parcellier A, et al. HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 2006;20(8):1179–1181. doi: 10.1096/fj.05-4184fje. [DOI] [PubMed] [Google Scholar]
- 12.Ho Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415(6868):180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 13.Yang C, et al. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc Natl Acad Sci USA. 2013;110(3):966–971. doi: 10.1073/pnas.1221046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang C, et al. Celastrol increases glucocerebrosidase activity in Gaucher disease by modulating molecular chaperones. Proc Natl Acad Sci USA. 2014;111(1):249–254. doi: 10.1073/pnas.1321341111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan YL, et al. ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher’s disease. Chem Biol. 2014;21(8):967–976. doi: 10.1016/j.chembiol.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimopoulos MA, Mitsiades CS, Anderson KC, Richardson PG. Tanespimycin as antitumor therapy. Clin Lymphoma Myeloma Leukemia. 2011;11(1):17–22. doi: 10.3816/CLML.2011.n.002. [DOI] [PubMed] [Google Scholar]
- 17.Roh JL, Kim EH, Park HB, Park JY. The Hsp90 inhibitor 17-(allylamino)-17-demethoxygeldanamycin increases cisplatin antitumor activity by inducing p53-mediated apoptosis in head and neck cancer. Cell Death Dis. 2013;4:e956. doi: 10.1038/cddis.2013.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Powers MV, et al. Mode of cell death induced by the HSP90 inhibitor 17-AAG (tanespimycin) is dependent on the expression of pro-apoptotic BAX. Oncotarget. 2013;4(11):1963–1975. doi: 10.18632/oncotarget.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bendikov-Bar I, Rapaport D, Larisch S, Horowitz M. Parkin-mediated ubiquitination of mutant glucocerebrosidase leads to competition with its substrates PARIS and ARTS. Orphanet J Rare Dis. 2014;9:86. doi: 10.1186/1750-1172-9-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helquist P, Maxfield FR, Wiech NL, Wiest O. Treatment of Niemann—pick type C disease by histone deacetylase inhibitors. Neurotherapeutics. 2013;10(4):688–697. doi: 10.1007/s13311-013-0217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang C, et al. Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J. 2012;26(11):4506–4516. doi: 10.1096/fj.12-210146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C, et al. Missense mutations in the NF2 gene result in the quantitative loss of merlin protein and minimally affect protein intrinsic function. Proc Natl Acad Sci USA. 2011;108(12):4980–4985. doi: 10.1073/pnas.1102198108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bendikov-Bar I, Horowitz M. Gaucher disease paradigm: from ERAD to comorbidity. Hum Mutat. 2012;33(10):1398–1407. doi: 10.1002/humu.22124. [DOI] [PubMed] [Google Scholar]
- 24.Bendikov-Bar I, Ron I, Filocamo M, Horowitz M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol Dis. 2011;46(1):4–10. doi: 10.1016/j.bcmd.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Knapinska AM, et al. Chaperone Hsp27 modulates AUF1 proteolysis and AU-rich element-mediated mRNA degradation. Mol Cell Biol. 2011;31(7):1419–1431. doi: 10.1128/MCB.00907-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang C, et al. β-Catenin signaling initiates the activation of astrocytes and its dysregulation contributes to the pathogenesis of astrocytomas. Proc Natl Acad Sci USA. 2012;109(18):6963–6968. doi: 10.1073/pnas.1118754109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.