

Abstract
Aberrant accumulation of beta-amyloid (Aβ) is thought to be an early event in a biological cascade that eventually leads to Alzheimer’s disease (AD). Along these lines, many clinically normal (CN) older individuals have evidence of beta-amyloid (Aβ) accumulation, which may be indicative of preclinical AD. However, relationships between Aβ and “downstream” AD markers are often inconsistent across studies. These inconsistencies may be due to the presence of other age-related processes that also influence AD markers, as well as additional risk factors that interact with Aβ to influence downstream changes. For instance, it is possible that the effect of Aβ is modified by neurodegeneration, genetics, sex-differences and cognitive reserve. Thus, a multivariate approach to determining risk of AD within CN participants may be more appropriate than reliance on Aβ status alone. An understanding of how additional risk factors interact with Aβ to influence an individual’s trajectory towards AD is essential for characterizing preclinical AD and has implications for prevention trials.
Keywords: Aging, Preclinical Alzheimer’s disease, Beta-amyloid, Amyloid PET imaging, Resilience
Introduction
The Amyloid Hypothesis of Alzheimer’s Disease and in Vivo Visualization of Aβ
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder found in ~10% of individuals over age 65 and ~40% of individuals over age 85 (Alz.org 2014; Hebert et al. 2013). AD typically begins with episodic memory impairment, and slowly affects other cognitive domains such as executive function as the disease progresses (Grober et al. 2008). The amyloid hypothesis of AD posits that aberrant accumulation of the Aβ peptide is an early initiating event that eventually leads to clinical impairment (Hardy and Selkoe 2002), and is supported by research spanning multiple fields (Walsh and Selkoe 2007). For instance, autosomal dominant genetic variants that cause early onset AD involve mutations that directly influence the production of the Aβ peptide (Bentahir et al. 2006; Citron et al. 1992). Down syndrome individuals have an extra copy of chromosome 21 (the locus of the amyloid precursor protein gene, which codes for the larger protein that beta-amyloid is cleaved from), produce high amounts of Aβ and have increased risk of developing AD (Lott et al. 2006). Experimental work in mice has shown a relationship between Aβ and memory that is reversed after passive immunization with an Aβ antibody (Dodart et al. 2002). Soluble Aβ has been shown to reduce synaptic strength and number in rodent models (Shankar et al. 2007; Walsh et al. 2002), which is consistent with the observation that neurons from AD patients have 15-35 % fewer synapses than controls (Davies et al. 1987). Overall, there is strong evidence that beta-amyloid is a central feature of AD development.
In sporadic Alzheimer’s disease, for unknown reasons the Aβ peptide undergoes aggregation after it’s cleavage from the amyloid precursor protein and deposits into extracellular Aβ plaques. Although Aβ plaques have traditionally been studied during postmortem analysis of brain tissue using stains such as thioflavin-T, congo red, and Bielschowsky, the ability to measure Aβ plaques in vivo with positron emission tomography (PET) has emerged in the last decade. 11C-PIB (‘Pittsburgh Compound-B’) was one of the first radiotracers to enable this visualization (Klunk et al. 2004), and has become the most widely studied tracer to date. Further advancements have been made to create amyloid imaging agents radiolabeled with 18F rather than 11C, given the longer half-life and thus greater feasibility to use 18F radiotracers in both research and clinical settings. Excitingly, two 18F compounds have been approved by the FDA to determine whether patients have evidence of brain Aβ deposition—florbetapir (Amyvid) (Wong et al. 2010) and flutemetamol (Vizamyl) (Vandenberghe et al. 2010). Although in vivo assessment of aberrant Aβ accumulation can also be accomplished using cerebrospinal fluid (CSF) markers of Aβ (Fagan et al. 2006), the majority of the literature reviewed herein focuses on data utilizing PET amyloid imaging.
Prevalence of Aβ in Clinically Normal Older Individuals
Although Aβ plaques are a salient feature of AD, they are also commonly observed in the brains of clinically normal (CN) older individuals (individuals that function independently and do not show signs of objective cognitive impairment as detected with neuropsychological assessment). This observation has consistently been observed in postmortem studies and has been replicated in amyloid imaging studies. The prevalence of CNs with evidence of elevated Aβ (Aβ+) increases with older age (~10 % of CNs in their 60’s are Aβ+ whereas ~40 % of CNs in their 80’s are Aβ+, Fig. 1). It is noteworthy to emphasize that these proportions describe individuals that have evidence of elevated Aβ that are clinically normal, and do not include individuals of that age that are diagnosed with mild cognitive impairment or dementia. Although the age at which Aβ plaque deposition begins is unknown, examination across the studies depicted in Fig. 1 suggests Aβ positivity is minimal before age 60. Given that quantification of Aβ across post-mortem and amyloid imaging studies specifically measures Aβ plaque levels, it is possible that abnormal production and aggregation of Aβ may still occur before age 60.
Fig. 1.

Prevalence of Aβ+ CNs across the lifespan. Data across multiple studies are plotted by sample age and percent classified as Aβ+ (a total of 3512 subjects from post-mortem studies and 2034 from amyloid-imaging studies are represented). The area of each bubble is scaled by group size, ranging from 12 to 639 CNs (for studies reporting Aβ+ prevalence across multiple age groups, multiple bubbles are used). Although Aβ+ classification is study specific, a consistent pattern emerges. These studies reveal a low proportion of Aβ+ CNs younger than 60, followed by a linear increase in the proportion of Aβ+ CNs after age 60 (~30 % of CNs are Aβ+ at age 75). Post-mortem studies and Aβ+ classification are as follows: A/black = Braak & Braak (CERAD B & C, N=2661) (Braak and Braak 1997); B/red = Kok et al. (CERAD moderate and frequent, N=534) (Kok et al. 2009); C/light blue = Savva et al. (CERAD moderate and severe, N=183) (Savva et al. 2009); D/pink = Bennett et al., Religious Orders Study (CERAD probable and definite, N=98) (Bennett et al. 2006); E/dark blue = Bennett et al., Memory and Aging Project (CERAD probable and definite, N=36) (Bennett et al. 2006). Amyloid PET studies and Aβ+ classification are as follows: 1/purple = Knopman et al. (>1.5 PIB SUVR with gray matter cerebellum, N=806) (Knopman et al. 2014); 2/orange = Morris et al. (>0.18 PIB Binding Potential with gray matter cerebellum, N=241) (Morris et al. 2010); 3/yellow = Johnson et al. 2014 (florbetapir qualitative read; N=201) (Johnson et al. 2014), 4/dark green = Rowe et al. (>1.5 PIB SUVR with gray matter cerebellum, N=183) (Rowe et al. 2013), 5/hot pink = Mormino et al. 2014, Alzheimer’s Disease Neuroimaging Initiative Phase 2 study (>1.126 florbetapir SUVR gray and white matter cerebellum, N = 198) (Mormino et al. 2014), 6/green = Mormino et al. 2014, Harvard Aging Brain Study (>1.196 PIB SUVR gray and white matter cerebellum, N=161) (Mormino et al. 2014); 7/dark red = Mathis et al. 2013 (>1.57 PIB SUVR gray matter cerebellum, N=152) (Mathis et al. 2013); 8/brown = Wirth et al. 2014 (>1.12 PIB DVR gray matter cerebellum, N=92) (Wirth et al. 2014)
The presence of Aβ accumulation within CNs is consistent with models of AD suggesting that Aβ is an early initiating event that eventually leads to “downstream” brain changes and clinical impairment (Hardy and Selkoe 2002; Jack et al. 2013a). Downstream brain changes are thought to encapsulate alterations in synaptic and neuronal function and structure, and are a more direct substrate of cognitive impairment than initiating Aβ accumulation. In humans, imaging proxies for these downstream brain changes span multiple modalities, such as functional magnetic resonance imaging (MRI), structural MRI, and [18 F] fluorodeoxyglucose positron emission tomography (FDG-PET), and will be discussed in detail throughout this review. Consistent with this framework, work from Villemagne and colleagues suggest that it may take upward to 20 years to transition between Aβ levels typically found in Aβ+ CNs compared with Aβ levels found in AD, highlighting the prolonged period over which Aβ accumulation may occur within CNs (Villemagne et al. 2013). To understand whether Aβ accumulation within CNs is indicative of a preclinical AD state, many investigators have examined the association between Aβ and downstream changes within CNs. These studies are summarized in the following sections.
Associations Between Aβ and Cross-Sectional AD Markers Within CNs
Functional Brain Markers
Aβ has been shown to promote synaptic loss (Shankar et al. 2007) and impair long-term potentiation (Walsh et al. 2002). Given these effects on synaptic function, it is possible that the earliest Aβ-related changes within CNs may be detected using measures of brain function, before the high levels of neuronal loss typical of AD patients have occurred. Two widely used imaging modalities that measure brain function have also been used to investigate the association between Aβ and brain function: (1) functional magnetic resonance imaging (fMRI, which measures brain activation during task and rest) and (2) [18 F] fluorodeoxyglucose positron emission tomography (FDG-PET, which measures glucose metabolism during rest).
The majority of work investigating the association between task-related fMRI and Aβ within CNs focuses on memory tasks, although non-memory domains have also been investigated (Hedden et al. 2011). The default mode network (DMN) in particular has received ample attention, given the high degree of overlap between this network and patterns of cortical amyloid deposition (Buckner et al. 2009). Comprised of medial prefrontal, posteromedial (precuneus, posterior cingulate and retrosplenial) and lateral parieto-temporal cortices, as well as the medial temporal lobe (Buckner et al. 2008), the DMN typically deactivates during memory encoding and activates during memory retrieval (Daselaar et al. 2004; Huijbers et al. 2013). Interestingly, Aβ+ CNs show impaired DMN deactivation during memory encoding (Kennedy et al. 2012; Sperling et al. 2009; Vannini et al. 2012, 2013) and impaired activation during memory retrieval (Vannini et al. 2013). However, impaired deactivation within the DMN has also been observed during an attentional control task (Hedden et al. 2011), suggesting that the effect of Aβ on the DMN may not be specific to memory processing. Aβ has also been associated with increased activation in “task positive” regions known to activate during memory encoding (Mormino et al. 2011a; Sperling et al. 2009) (however see (Kennedy et al. 2012)), which echoes previous fMRI experiments showing increased activation in old versus young CNs (Cabeza et al. 2002; de Chastelaine et al. 2011; Park and Reuter-Lorenz 2009; Rosen et al. 2002). Although increased task-related activity has been interpreted as a compensatory response in aging individuals (Cabeza et al. 2002), it is also possible they reflect reductions in neural efficiency (Li et al. 2006; Logan et al. 2002). We showed that increased task positive activation during memory encoding was related to memory performance within Aβ+ CNs (Mormino et al. 2011a), suggesting that these heightened activations are beneficial to Aβ+ CNs.
In addition to task-related patterns of activation, integrity of brain networks during rest has also been related to Aβ status using function connectivity analyses (FC). Functional connectivity (FC) analyses of resting state fMRI data captures brain regions showing correlated, low frequency (<0.1Hz) spontaneous activity, and have been used to define multiple networks with known anatomical connectivity and co-activation during task-related fMRI (Fox and Raichle 2007). Although the biological mechanism underlying these low frequency fluctuations remains unclear, it is possible that resting state networks reflect spontaneous cognitive processes and/or an intrinsic property of the brain’s “baseline” state (Buckner and Vincent 2007; Raichle and Snyder 2007). Similar to task related fMRI experiments, analyses investigating the effect of Aβ on resting state connectivity within CNs have also focused on the DMN. Interestingly, some studies have reported decreased DMN FC (Hedden et al. 2009; Mormino et al. 2011b; Sheline et al. 2010b) while others have additionally reported increased DMN FC in Aβ+ CNs (Mormino et al. 2011b; Sheline et al. 2010b). It is possible that DMN regions are differently affected by regional Aβ deposition, with the medial temporal lobe subsystem (Andrews-Hanna et al. 2010) showing early decreases and regions beyond the medial temporal subsystem showing early increases (Mormino et al. 2011b). A longitudinal functional connectivity study also found regional discrepancies in DMN FC within AD patients. In this study, Damoiseaux and colleagues reported reduced FC in the posterior component of the DMN (comprised predominantly of the posterior cingulate/precuneus and lateral parietal cortex) whereas increased FC was observed in the anterior DMN component (comprised predominantly of the medial prefrontal cortex). Interestingly, anterior DMN areas showing increased FC at baseline subsequently decreased at the follow up visit (Damoiseaux et al. 2012a). This pattern suggests that distinct DMN components may follow different patterns of FC changes throughout the progression of the disease.
A similar mixed pattern has emerged across studies investigating the relationship between Aβ status and glucose metabolism as measured with FDG-PET within CNs. Although one recent large study of 628 CNs age 70 and older showed that elevated Aβ was associated with hypometabolism in ADvulnerable regions (Knopman et al. 2014), smaller studies have found hypermetabolism within Aβ+ CNs (Johnson et al. 2014; Oh et al. 2014).
Overall, Aβ+ CNs tend to show altered patterns on measures of brain function. However, the direction and regional distribution of these changes are often inconsistent across studies. It is possible that some measures of brain function show initial increases in response to Aβ followed by subsequent decreases. It is also possible that increased brain activation and/or metabolism may predate Aβ deposition (Jagust and Mormino 2011), given that Aβ release has been shown to be activity dependent (Cirrito et al. 2005). Specifically, neuronal inefficiencies that occur independently of Aβ may actually promote the deposition of Aβ late in life. Thus, Aβ+ CNs with elevated measures of brain function may be CNs that have recently become Aβ+, whereas Aβ+ CNs that have been Aβ+ for a longer period may begin to show AD-like decreases in functional brain measures. Interesting, recent work by Jack and colleagues revealed increased DMN FC in Aβ− CNs that subsequently became Aβ+ over a short follow up period (1.3 years), offering support for the idea that increased connectivity may predate Aβ deposition (Jack et al. 2013b). Further longitudinal studies will be essential to disentangle the temporal relationship between Aβ and activation patterns within CNs.
Structural Brain Markers
A link between Aβ and cross-sectional structural brain markers within CNs has been difficult to establish, with some studies reporting reduced gray matter in Aβ+ CNs (Becker et al. 2011; Dickerson et al. 2009; Dore et al. 2013; Mormino et al. 2009; Rowe et al. 2010; Storandt et al. 2009; Tosun et al. 2011) while others have not (Chetelat et al. 2010; Schott et al. 2010; Whitwell et al. 2013; Wirth et al. 2013a). These inconsistencies suggest that cross-sectional effects between Aβ and gray matter structure are likely subtle and may vary depending on cohort and the examined gray matter measurement. Whitwell and colleagues demonstrated the influence of measurement by examining gray matter volumes defined with the AAL atlas and gray matter thickness defined using Freesurfer. This analysis failed to find a consistent pattern across techniques when contrasting Aβ+ and Aβ− CNs. However, a consistent pattern across methods emerged when contrasting Aβ+ clinically impaired subjects (mild cognitive impairment and AD patients) to Aβ− CNs (Whitwell et al. 2013). Thus, although associations within CNs may be subtle and influenced by methodological approaches, Aβ-related gray matter reductions may become more evident once clinical impairment has been reached.
Cognition
Given that change in cognition is thought to be the most downstream event in the cascade leading to AD, it is not surprising that strong and consistent associations between Aβ status and cross-sectional cognition are not identified within CN samples. Furthermore, participants must perform within the normal range on neuropsychological screening tests to be included in a CN sample. Thus, the restricted variance in cognitive measures within studies of CNs will limit the ability to detect associations between Aβ and cross-sectional cognitive measures. Nevertheless, many studies have investigated whether associations between Aβ and cross-sectional cognitive measures exist within CNs. Given that episodic memory impairment is an early feature of AD (Grober et al. 2008; Small et al. 2000), many researchers have focused on the relationship between Aβ status and memory processes within CNs. Although some studies report lower memory scores in CNs with higher levels of Aβ compared to lower levels of Aβ (Pike et al. 2011; Rentz et al. 2011), others have not identified an association between Aβ status and memory (Aizenstein et al. 2008; Rodrigue et al. 2012; Storandt et al. 2009). Furthermore, subtle associations across multiple cognitive domains were reported in a large study of 408 CNs (higher levels of Aβ were associated with lower scores in memory, executive function, language and visual spatial function, with subtle albeit significant Spearman rho values ranging from −0.12 to −0.14) (Kantarci et al. 2012). This analysis highlights that the association between Aβ and cross-sectional cognition is subtle and may span multiple cognitive domains.
Additional Factors Contribute to Variance in Cross-Sectional AD Markers Within CNs
Although some biomarker models suggest that functional and structural brain changes relevant to AD occurs downstream to initiating Aβ accumulation (Hardy and Selkoe 2002; Jack et al. 2013a), other studies suggest that pathways promoting Aβ and these brain changes initially occur independently among CNs (Jack et al. 2013b; Wirth et al. 2013a). Although these early brain changes likely encapsulate alterations in brain function and structure (as discussed in previous sections of this review), recent emphasis in the field has been placed on markers of “neurodegeneration” (ND) (which are typically assessed with structural MRI and FDG-PET, as well as CSF tau) (Jack et al. 2012). Models suggesting that ND occurs downstream to Aβ versus models suggesting these processes initially occur independently are not mutually exclusive—Aβ and ND may initially begin via separate pathways, but at some point this association may strengthen as ND related to Aβ becomes more pronounced. The potential independence of pathways promoting Aβ and ND has been highlighted by attempts to classify older CNs into preclinical AD stages as recently proposed by the National Institute on Aging and the Alzheimer’s Association workgroup (Sperling et al. 2011). This staging approach classifies CNs into groups based on joint Aβ and ND status: Stage 0 is defined as Aβ−/ND−, Stage 1 as Aβ+/ND−, and Stage 2 as Aβ+/ND+. An additional category of Aβ−/ND+ CNs was initially not included within this staging criteria and subsequently labeled as “Suspected Non-Alzheimer’s disease Pathology” (SNAP) by Jack and colleagues (Jack et al. 2012). To classify CNs as ND+, proxy measures of neurodegeneration that are greatly compromised in AD have been used (such as hippocampus volume, glucose metabolism in cortical regions known to be vulnerable to Alzheimer’s disease, and CSF tau). The proportions classified across preclinical stages are remarkably similar across cohorts (Jack et al. 2012; Mormino et al. 2014; Vos et al. 2013; Wirth et al. 2013c), with approximately 40-50 % classified as Stage 0 (Aβ−/ND−), 10-15 % as Stage 1 (Aβ+/ND−), and 15 % as Stage 2 (Aβ+/ND+). Interestingly, approximately 25 % of CNs are classified as SNAP (Aβ−/ND+), emphasizing that many Aβ− CNs have evidence of ND.
It is likely that ND in the absence of elevated Aβ is influenced by non-Aβ factors in aging (Fjell et al. 2013; Jagust 2013). Given that the majority of CNs above age 60 will have neurofibrillary tangle (NFT) pathology in the medial temporal lobe (Braak and Braak 1997; Nelson et al. 2012), it is likely a proportion of the variance captured by markers of ND reflects tangle pathology (Jagust et al. 2009; Whitwell et al. 2008). The prevalence of NFTs and Aβ across the lifespan is consistent with two independent pathways, such that NTFs begin early in adulthood and are common in mid-life (although restricted to the medial temporal lobe) whereas Aβ accumulation is very uncommon until after age 60 (Fig. 1). Thus, although both NFT and Aβ pathologies are central to AD development, these processes likely occur independently during the early stages of AD and may account for the weak association between Aβ and ND within CNs. Non-AD pathologies are also likely contributors to ND within CNs, such as infarcts (Bennett et al. 2006), Lewy bodies (Bennett et al. 2006), TDP-43 (Wilson et al. 2013b), and/or hippocampal sclerosis (Barker et al. 2002). Furthermore, a sizeable subset of amnestic mild cognitive impairment (MCI) subjects demonstrate non-AD pathologies affecting the medial temporal lobe (Jicha et al. 2006), and associations between neuroimaging ND markers and non-AD pathologies have been established. For instance, markers of cerebrovascular disease have been correlated with reductions in gray matter volume and glucose metabolism (DeCarli et al. 1995), whereas cortical gray matter reductions are prominent in patients with autopsy confirmed frontotemporal lobar degeneration with TDP-43 inclusions (Whitwell et al. 2010). In addition to associations between hippocampus volume and non-AD pathologies (Jack et al. 2002), hippocampus volume has also been associated with markers of inflammation (Marsland et al. 2008), stress (Lupien et al. 1998), and the use of estrogen replacement therapy (Eberling et al. 2004), highlighting that the hippocampus in particular may be vulnerable to many distinct processes. Overall, these pervasive associations across multiple factors highlight that imaging markers of ND are not specific for AD processes.
It is also possible that “normal aging” processes in the absence of underlying pathologies, such as synaptic alterations (Morrison and Hof 1997) contribute to the signal captured in neuroimaging markers of ND. Along these lines, associations between chronological age and markers of gray matter (Raz et al. 2004; Sowell et al. 2003) as well as glucose metabolism (Knopman et al. 2014) have been established. Importantly, these relationships are oftentimes gradual and consistent throughout the lifespan, occurring before the age at which abnormal levels of Aβ accumulation are apparent. Recent work directly investigating characteristics of older Aβ− CNs with evidence of ND have found associations with white matter hyperintensities (Wirth et al. 2013c) as well as with sex (such that Aβ− men are more likely to show ND than Aβ− women) (Mormino et al. 2014), offering additional insights into non-Aβ factors that may promote ND. Overall, it is likely that imaging markers used to assess AD-like ND within CNs are not specific to Aβ, but rather are influenced by a multitude of non-Aβ processes as well as normal aging (Jack et al. 2014). Thus, the contributions of multiple processes to ND in aging would likely obscure the ability to isolate effects between Aβ and cross-sectional AD markers within CNs (Fig. 2).
Fig. 2.

Multiple factors contribute to “downstream” brain changes in the trajectory towards Alzheimer’s disease. Although many models of AD suggest that Aβ is an initiating event in a cascade that triggers downstream brain changes and cognitive decline (as depicted by the red boxes and arrows), emerging evidence suggests that multiple non-Aβ factors influence these downstream brain changes. These additional factors include non-Aβ pathologies (arrows 1 & 2), environmental factors such as stress and estrogen replacement therapy (arrow 3) as well as “normal” aging processes (arrow 4). Regardless of which factors contribute to downstream brain changes, the convergence of these changes with Aβ accumulation has been shown to accelerate further downstream changes (arrow 5)
Associations Between Aβ and Longitudinal Changes in AD Markers
Associations between Aβ and longitudinal change in imaging and cognitive markers tend to be more consistent than associations between Aβ and cross-sectional measures. Discrepancies between cross-sectional and longitudinal analyses may be due to the limited range of values selected at baseline inherent to CN samples. As discussed in the above section, it is also possible that effects related to Aβ are superimposed upon changes influenced by many other age-related processes, many of which may occur gradually over the lifespan. For these reasons, isolation of Aβ-related effects in aging may be more readily detectable with longitudinal designs.
Longitudinal Structural Brain Markers
Many studies have reported that Aβ+ CNs undergo faster rates of atrophy as measured with different gray matter measures, such as voxel based morphometry (Chetelat et al. 2012), gray matter thickness (Dore et al. 2013), gray matter volumes (Storandt et al. 2009), as well as in the boundary shift integral (a method that assesses volume loss by determining the amount a structure’s boundary has shifted across longitudinally collected structural MRI measurements) (Schott et al. 2010). However, the spatial pattern of these changes is not consistent across studies, especially with respect to whether Aβ accumulation within CNs is specifically associated with atrophy of medial temporal lobe structures. For instance, some studies have reported longitudinal atrophy in the medial temporal lobe (Chetelat et al. 2012; Schott et al. 2010), whereas other work suggest that medial temporal lobe atrophy increases at a similar rate in CNs with and without evidence of Aβ positivity (Fjell et al. 2009, 2014). Given the prolonged period of Aβ accumulation within CNs (Villemagne et al. 2013), it is possible that different brain regions show varying degrees of vulnerability throughout the period at which Aβ accumulation occurs. For instance, frontoparietal atrophy has been indentified in Aβ− CNs showing early signs of Aβ accumulation whereas a more distributed pattern of atrophy was present in CNs that had already surpassed the threshold of Aβ positivity. Interestingly, atrophy in the hippocampus was not observed within Aβ+ CNs, but was present in Aβ+ AD patients (Mattsson et al. 2014). Studies that simultaneously investigate the time course of both Aβ and ND will be able to establish whether a sequential involvement of brain regions tracks with Aβ accumulation in CNs.
Longitudinal Cognition
Studies assessing the relationship between Aβ and either retrospective or subsequent cognitive and functional decline within CNs have converged to reveal greater risk of decline within Aβ+ CNs. Some studies have found cognitive decline that is specific to memory (Lim et al. 2013a), whereas decline in non-memory domains have been indentified in other studies (Snitz et al. 2013; Wirth et al. 2013b). Specifically, Lim and colleagues reported an association between Aβ status and a composite measure of verbal memory (Logical Memory delayed recall, California Verbal Learning Test delayed recall and California Verbal Learning Test d’) as well as a composite measure of visual memory (Rey Complex Figure Test 30 min delayed recall, CogState One Card Learning task, and CogState One Back task), whereas no association with Aβ status was found with composite measures of executive function (Stroop, Letter Fluency, and Category Fluency Switching), language (Category Fluency and Boston Naming Test), attention (Digit Symbol, CogState Detection task, and CogState Identification task) or visuospatial function (Rey Complex Figure Test Copy, and Clock Drawing) (Lim et al. 2013a). Conversely, Snitz and colleagues reported an association between Aβ status with cognitive decline on individual measures of visual memory (immediate and delayed recall of the Rey Complex Figure Test 30), as well as on a measure of executive function (Trials B), attention (Trials A) and language (Category Fluency) (Snitz et al. 2013). Wirth and colleagues also found associations between Aβ status and cognitive decline that was not specific to memory when examining a composite measure of non-memory tests (Stroop, Controlled Oral Word Association Test, Trails A, and Digit Symbol) (Wirth et al. 2013b). Inconsistencies across these studies may be due to differences in sample sizes (Lim et al.: N=390, Snitz et al.: N=194, Wirth et al.: N=38), as well as differences in the selection criteria used within each cohort [for instance, the cohort used by Lim and colleagues enriches for the APOE4+ genotype and the presence of subjective cognitive complaints (Rowe et al. 2010), whereas the cohort used by Snitz and colleagues specifically enrolls CNs older than other studies investigating the relevance of Aβ within CNs (72–96 years of age)].
In addition to associations across memory and non-memory domains, Aβ+ CNs also show decline in measures of global cognitive function, such as in the MMSE (Resnick et al. 2010) and ADAS-cog (Doraiswamy et al. 2012; Landau et al. 2012b). Furthermore, Aβ+ CNs have greater risk of progression on functional measures, such as on the clinical dementia rating scale (Roe et al. 2013) and progression to mild cognitive impairment and dementia (Villemagne et al. 2011). Overall, Aβ positivity within CNs increases risk of subsequent decline on both cognitive and functional measures. However, future studies are necessary to elucidate whether specific cognitive domains are selectively affected by Aβ status within CNs, and whether these domain specific relationships may be modified by APOE4 status, subjective cognitive complaints, and chronological age.
Factors that may Promote Resilience to Damaging Effects of Beta-Amyloid
An additional concept that may obscure relationships between Aβ and markers of AD within CNs is the notion of resilience to damaging effects of Aβ. Specifically, it is possible that additional factors interact with Aβ to determine whether and how fast an individual will progress along the trajectory towards AD. Thus, it is possible that heterogeneity exists within the Aβ+ group. In a simplified scenario, Aβ+ CNs may be categorized into 2 types—preclinical-Aβ+ and resilient-Aβ+ (in actuality, it is more likely that a continuum of resilience exists with most cases falling between these 2 categories). In this framework, preclinical-Aβ+ CNs are on the trajectory towards AD, and are concurrently cognitively normal because they are at the beginning of the AD trajectory. Longitudinal follow-up of these CNs would reveal cognitive decline and progression to AD. Conversely, longitudinal follow-up of resilient-Aβ+ CNs would reveal that these Aβ+ CNs have remained clinically normal, suggesting the presence of protective factors within this subgroup. The remainder of this review will discuss emerging evidence that heterogeneity within Aβ+ CNs exists.
Greater Atrophy and Decline in CNs that are Both Positive for Aβ and Neurodegeneration
Recent work has suggested that longitudinal atrophy is greatest in CNs that have evidence of both Aβ accumulation and neurodegneration (Fig. 2). For instance, Desikan and colleagues found greater entorhinal cortex atrophy in Aβ+ CNs that also had elevated quantities of CSF phospho-tau (Desikan et al. 2011). Consistent with this finding, data from the Mayo Clinic Study of Aging suggests that CNs that are both Aβ+ and ND+ at baseline show higher rates of medial temporal lobe gray matter loss and hypometabolism (Knopman et al. 2013) as well as atrophy as measured using a multivariate score derived from multiple brain regions known to be vulnerable to AD (the medial temporal lobe as well as cortical regions including lateral temporal cortex, precuneus, angular gyrus and occipital gyrus) (Jack et al. 2014). Likewise, studies investigating decline in function and cognition have also found greatest risk in CNs positive for both Aβ and ND. Specifically, Aβ+/ND+ CNs are more likely to progress to MCI (Knopman et al. 2012; Rowe et al. 2013) and from Clinical Dementia Rating (CDR) 0 to 0.5 (Desikan et al. 2012; Vos et al. 2013) than CNs negative on both markers as well as CNs positive for either Aβ or ND. A similar pattern has emerged when examining longitudinal cognition, such that Aβ+/ND+ CNs show the greatest rate of cognitive decline over time (Mormino et al. 2014; Wirth et al. 2013b).
As discussed earlier in this review, the association between Aβ and cross-sectional ND is often inconsistent and likely obscured by additional non-Aβ factors in aging (Fjell et al. 2014; Jack et al. 2013b; Jagust 2013). Thus, the observation that Aβ+/ND+ CNs are most at risk for decline is consistent with the existence of heterogeneity within Aβ+ CNs by suggesting that two separate processes must converge to direct an individual towards AD. A number of possible mechanisms exist that may underlie a synergistic interaction between Aβ and ND. For instance, it is possible that ND renders neurons more susceptible to toxic effects of Aβ. Either process alone may be insufficient to alter cognition, but the “double hit” of both processes may promote decline by overwhelming compensatory processes. Given that neurofibrillary tangles (NFTs) have been shown to mediate toxic effects of Aβ (Ittner and Gotz 2011), increased risk in Aβ+/ND+ CNs may reflect the co-occurrence of Aβ and NFT pathologies (assuming that ND captures NFT pathology to some extent (Jagust et al. 2009; Whitwell et al. 2008)). Although Aβ and NFT pathologies occur on distinct time scales, the spread of NFTs from medial temporal lobe regions to neocortex may occur in conjunction with late life Aβ accumulation. Thus, late life Aβ may induce NFT spread, which in turn promotes toxicity and cognitive decline. Regardless of the mechanism underlying increased risk in Aβ+/ND+ CNs, factors that reduce the likelihood of ND may offer resilience to damaging affects of Aβ and enable these Aβ+ CNs to remain clinically normal.
Genetic Factors Accelerate the Effect of Aβ
Genetic factors, such as polymorphisms for genes encoding the Apolipoprotein E (APOE) and brain-derived neurotrophic factor (BDNF) proteins, have also been shown to interact with Aβ to accelerate longitudinal changes. We recently showed an interaction between the APOE4+ genotype and Aβ status in predicting cognitive decline in a large group of 490 CNs, such that APOE4+/Aβ+ CNs showed greater decline over a median follow up period of 1.5 years than all other CN groups (APOE4−/Aβ−, APOE4+/Aβ−, and APOE4+/Aβ+) (Mormino et al. 2014). Although the APOE4 genotype is known to influence AD risk through increased Aβ accumulation (Kok et al. 2009; Reiman et al. 2009), this genotype also effects neuronal integrity through Aβ-independent mechanisms (via synaptogensis, synaptic plasticity, tau phosphorylation, mito-chondrial activity, neuroinflammation, and neurodevelopment (Wolf et al. 2013)). These Aβ-independent mechanisms are consistent with human imaging studies that have revealed reduced glucose metabolism (Knopman et al. 2014; Reiman et al. 2004) and gray matter (Alexopoulos et al. 2011; Shaw et al. 2007) in APOE4+ CNs before the age at which Aβ accumulation occurs. Hypometabolism and reduced resting state connectivity have also been shown in older APOE4+ CNs lacking evidence of fibrillar Aβ accumulation (the mean age in the study by Jagust and Landau was 77 years whereas the mean age in the study by Sheline and colleagues was 62 years) (Jagust and Landau 2012; Sheline et al. 2010a). It is also possible that Aβ and APOE4 in conjunction impart greater levels of neuronal toxicity, given that the apoE4 protein is less effective than apoE3/2 in responding to neuronal injury (Mahley and Huang 2012). Potential mechanisms by which APOE4 status may influence the AD trajectory is depicted in Fig. 3.
Fig. 3.
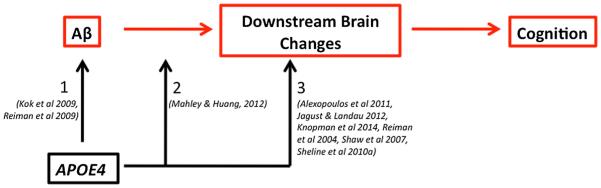
Influence of APOE4 status on the trajectory towards Alzheimer’s disease. The presence of the APOE4 allele may affect an individual’s risk of AD through multiple mechanisms. Specifically, the APOE4+ genotype is consistently associated with greater levels of Aβ accumulation (arrow 1), suggesting that this genotype increases the chance of an individual becoming Aβ+ and entering the AD trajectory. However, there is also evidence that APOE4 interacts with Aβ status to impart greater synaptic damage when these events co-occur (arrow 2). Finally, the APOE4 allele may exert detrimental effects by directly affecting downstream brain changes in the absence of Aβ (arrow 3), making the individual more susceptible to AD in late life when additionally confronted with elevated Aβ
Similar to the increased risk identified in APOE4+/Aβ+ CNs, in a study of 165 CNs followed over 3 years, Yen et al. demonstrated that Aβ+ CNs that also have the val66met BDNF polymorphism show greater rates of hippocampal atrophy and cognitive decline (Lim et al. 2013b). Although this polymorphism is not associated with greater levels of Aβ accumulation, it results in decreased production of the BDNF protein and impairment of neuronal and synaptic growth (Egan et al. 2003). Thus, BDNF genotype is another factor that may dictate an individual’s ability to tolerate underlying levels of Aβ.
Sex Differences and Risk of AD
Given that AD is more common in women than men (Zhang et al. 1990), sex differences may also interact with Aβ to effect rates of atrophy and decline within CNs. Although the mechanism underling women’s heightened AD risk is unknown, it may be influenced by factors such as estrogen related changes during menopause and/or increased inflammatory processes (Carter et al. 2012), either of which may make women more susceptible to toxic effects of Aβ. Although the potential interaction between sex and Aβ in influencing downstream AD changes is currently understudied among amyloid imaging studies, it has been shown that women’s elevated AD risk is modified by both the presence of the APOE4 allele and age (Duara et al. 1996; Farrer et al. 1997). Likewise, decreased resting state functional connectivity within APOE4+ CNs is exacerbated in women CNs (Damoiseaux et al. 2012b). Given that the APOE4 allele and age are both associated with higher levels of Aβ, it is possible that interactions between sex and either APOE4 or age is mediated by elevated Aβ levels. Along these lines, Pike and colleagues demonstrated that only women Aβ+ CNs showed lower scores on a composite measure of episodic memory (comprised of the delayed recall portions of the California Verbal Learning Test, the Rey Complex Figure Test, and the Logical Memory Test from the Wechsler Memory Scale), whereas an effect of Aβ on this composite memory score was not present within men (Pike et al. 2011). Another study found greater entorhinal cortex atrophy in women that was independent of both Aβ and APOE4 status within CNs (Holland et al. 2013). Thus, brain changes related to sex differences in aging may make women more susceptible to AD in the presence of concurrent Aβ accumulation.
Reserve and Risk of AD
Within the reserve framework, an individual with higher reserve is able to remain clinically normal for longer than an individual with lower reserve despite equivalent levels of underlying pathology (Stern 2012). Individuals with high reserve are thought to have either greater quantities of neurons and synapses (“brain reserve”), and/or a better ability to employ alternative strategies or compensatory mechanisms (“cognitive reserve”) than individuals with low reserve.
Although estimating reserve differs across studies, proxies such as education, occupation and engagement in cognitively stimulating activities have all been shown to decrease risk of AD (Stern et al. 1994; Verghese et al. 2003). Amyloid imaging studies investigating reserve within CNs have revealed stronger relationships in low reserve participants between Aβ status and memory (as measured with delayed recall from the Memory Capacity Test) (Rentz et al. 2010) as well as associations between reserve and a global measure of cognition above and beyond what is explained by Aβ status and markers of ND (Vemuri et al. 2012). The latter study by Vemuri and colleagues is consistent with a large postmortem study of non-demented individuals that found associations between past and current cognitive activity with retrospective global cognitive decline that was independent of multiple age-related pathologies (Aβ, but also neurofibrillary tangles, infarcts and Lewy bodies; associations with cognitive activity were also seen with decline in episodic memory, semantic memory, working memory, and visuospatial memory when these cognitive domains were examined separately) (Wilson et al. 2013a). These studies suggest that high levels of reserve may exert protective effects by masking downstream cognitive decline related to Aβ. However, high levels of reserve have been shown to be associated with reduced levels of Aβ within CNs (Landau et al. 2012a), which is consistent with animal work showing increased clearance of Aβ in mice exposed to enriching environments (Costa et al. 2007). Thus, cognitive reserve may exert protective effects both through influencing quantities of pathology, and also by preventing decline related to pathology. Interestingly, individuals with high reserve show steeper rates of decline after AD diagnosis than AD individuals with low reserve (Teri et al. 1995), suggesting that once the protection inferred by reserve is surpassed, the extensive underlying pathology exerts a large effect.
Conclusion
Evidence across multiple laboratories suggests that accumulation of Aβ within CNs is consequential. Specifically, Aβ+ CNs show alterations in functional brain measures, increased brain atrophy, as well as subtle reductions in cognition and increased risk of subsequent decline. Although inconsistencies exist across studies investigating the relevance of Aβ in aging, it is likely that Aβ-related effects are superimposed upon effects related to multiple other age-related processes, making Aβ-related effects difficult to isolate with CN cohorts. Furthermore, detrimental effects of Aβ may be dampened or exacerbated by additional factors, enabling some individuals to be resilient while other CNs show heightened vulnerability to this pathology. Thus, the relevance of Aβ within CNs is complex and involves multiple factors. To fully elucidate these complex relationships, it is essential to investigate large cohorts that are sufficiently powered to test interactions across multiple variables at once. An important remaining question pertains to the extent to which heterogeneity within Aβ+ CNs exists. Specifically, it will be critical to establish the average time for an Aβ+ CN to progress to AD, and how much variance exists around this estimate. Do some Aβ+ CNs progress within 5 years while others progress in 25 years? If so, what factors influence this variability? As we increase our understanding of these complexities, we can build multivariate models that combine Aβ status with additional relevant factors to derive individualized risk estimates (similar to approaches that have been used to determine individualized breast cancer risk (Amir et al. 2010)). Overall, the complex interactions between Aβ and additional risk factors will help elucidate the relevance of Aβ to the aging brain and inform strategies geared towards AD prevention.
Acknowledgments
E. Mormino has received funding from NIH grant F32-AG044054 and P01-AG036694.
References
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Archives of Neurology. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulos P, Richter-Schmidinger T, Horn M, Maus S, Reichel M, Sidiropoulos C, et al. Hippocampal volume differences between healthy young apolipoprotein E epsilon2 and epsilon4 carriers. Journal of Alzheimer's Disease. 2011;26:207–210. doi: 10.3233/JAD-2011-110356. [DOI] [PubMed] [Google Scholar]
- Alz.org Alzheimer’s Disease Facts and Figures. 2014.
- Amir E, Freedman OC, Seruga B, Evans DG. Assessing women at high risk of breast cancer: a review of risk assessment models. Journal of the National Cancer Institute. 2010;102:680–691. doi: 10.1093/jnci/djq088. [DOI] [PubMed] [Google Scholar]
- Andrews-Hanna JR, Reidler JS, Sepulcre J, Poulin R, Buckner RL. Functional-anatomic fractionation of the brain's default network. Neuron. 2010;65:550–562. doi: 10.1016/j.neuron.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Disease and Associated Disorders. 2002;16:203–212. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- Becker JA, Hedden T, Carmasin J, Maye J, Rentz DM, Putcha D, et al. Amyloid-beta associated cortical thinning in clinically normal elderly. Annals of Neurology. 2011;69:1032–1042. doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, et al. Presenilin clinical mutations can affect gammasecretase activity by different mechanisms. Journal of Neurochemistry. 2006;96:732–742. doi: 10.1111/j.1471-4159.2005.03578.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiology of Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Annals of the New York Academy of Sciences. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. Journal of Neuroscience. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Vincent JL. Unrest at rest: default activity and spontaneous network correlations. NeuroImage. 2007;37:1091–1096. doi: 10.1016/j.neuroimage.2007.01.010. discussion 1097-9. [DOI] [PubMed] [Google Scholar]
- Cabeza R, Anderson ND, Locantore JK, McIntosh AR. Aging gracefully: compensatory brain activity in high-performing older adults. NeuroImage. 2002;17:1394–1402. doi: 10.1006/nimg.2002.1280. [DOI] [PubMed] [Google Scholar]
- Carter CL, Resnick EM, Mallampalli M, Kalbarczyk A. Sex and gender differences in Alzheimer's disease: recommendations for future research. Journal of Women's Health (2002) 2012;21:1018–1023. doi: 10.1089/jwh.2012.3789. [DOI] [PubMed] [Google Scholar]
- Chetelat G, Villemagne VL, Pike KE, Baron JC, Bourgeat P, Jones G, et al. Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain. 2010;133:3349–3358. doi: 10.1093/brain/awq187. [DOI] [PubMed] [Google Scholar]
- Chetelat G, Villemagne VL, Villain N, Jones G, Ellis KA, Ames D, et al. Accelerated cortical atrophy in cognitively normal elderly with high beta-amyloid deposition. Neurology. 2012;78:477–484. doi: 10.1212/WNL.0b013e318246d67a. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Costa DA, Cracchiolo JR, Bachstetter AD, Hughes TF, Bales KR, Paul SM, et al. Enrichment improves cognition in AD mice by amyloid-related and unrelated mechanisms. Neurobiology of Aging. 2007;28:831–844. doi: 10.1016/j.neurobiolaging.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Damoiseaux JS, Prater KE, Miller BL, Greicius MD. Functional connectivity tracks clinical deterioration in Alzheimer's disease. Neurobiology of Aging. 2012a;33:828. doi: 10.1016/j.neurobiolaging.2011.06.024. e19-828 e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoiseaux JS, Seeley WW, Zhou J, Shirer WR, Coppola G, Karydas A, et al. Gender modulates the APOE epsilon4 effect in healthy older adults: convergent evidence from functional brain connectivity and spinal fluid tau levels. Journal of Neuroscience. 2012b;32:8254–8262. doi: 10.1523/JNEUROSCI.0305-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daselaar SM, Prince SE, Cabeza R. When less means more: deactivations during encoding that predict subsequent memory. NeuroImage. 2004;23:921–927. doi: 10.1016/j.neuroimage.2004.07.031. [DOI] [PubMed] [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. Journal of Neurological Sciences. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- de Chastelaine M, Wang TH, Minton B, Muftuler LT, Rugg MD. The effects of Age, memory performance, and callosal integrity on the neural correlates of successful associative encoding. Cerebral Cortex. 2011;21:2166–2176. doi: 10.1093/cercor/bhq294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarli C, Murphy DG, Tranh M, Grady CL, Haxby JV, Gillette JA, et al. The effect of white matter hyperintensity volume on brain structure, cognitive performance, and cerebral metabolism of glucose in 51 healthy adults. Neurology. 1995;45:2077–2084. doi: 10.1212/wnl.45.11.2077. [DOI] [PubMed] [Google Scholar]
- Desikan RS, McEvoy LK, Thompson WK, Holland D, Brewer JB, Aisen PS, et al. Amyloid-beta-associated clinical decline occurs only in the presence of elevated P-tau. Archives of Neurology. 2012;69:709–713. doi: 10.1001/archneurol.2011.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, McEvoy LK, Thompson WK, Holland D, Roddey JC, Blennow K, et al. Amyloid-beta associated volume loss occurs only in the presence of phospho-tau. Annals of Neurology. 2011;70:657–661. doi: 10.1002/ana.22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cerebral Cortex. 2009;19:497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nature Neuroscience. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Doraiswamy PM, Sperling RA, Coleman RE, Johnson KA, Reiman EM, Davis MD, et al. Amyloid-beta assessed by florbetapir F 18 PET and 18-month cognitive decline: a multicenter study. Neurology. 2012;79:1636–1644. doi: 10.1212/WNL.0b013e3182661f74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore V, Villemagne VL, Bourgeat P, Fripp J, Acosta O, Chetelat G, et al. Cross-sectional and longitudinal analysis of the relationship between Abeta deposition, cortical thickness, and memory in cognitively unimpaired individuals and in Alzheimer disease. JAMA Neurology. 2013;70:903–911. doi: 10.1001/jamaneurol.2013.1062. [DOI] [PubMed] [Google Scholar]
- Duara R, Barker WW, Lopez-Alberola R, Loewenstein DA, Grau LB, Gilchrist D, et al. Alzheimer's disease: interaction of apolipoprotein E genotype, family history of dementia, gender, education, ethnicity, and age of onset. Neurology. 1996;46:1575–1579. doi: 10.1212/wnl.46.6.1575. [DOI] [PubMed] [Google Scholar]
- Eberling JL, Wu C, Tong-Turnbeaugh R, Jagust WJ. Estrogen- and tamoxifen-associated effects on brain structure and function. NeuroImage. 2004;21:364–371. doi: 10.1016/j.neuroimage.2003.08.037. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Annals of Neurology. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB. Brain changes in older adults at very low risk for Alzheimer's disease. Journal of Neuroscience. 2013;33:8237–8242. doi: 10.1523/JNEUROSCI.5506-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB. hat is normal in normal aging? Effects of aging, amyloid and Alzheimer's disease on the cerebral cortex and the hippocampus. Prog Neurobiol. 2014 doi: 10.1016/j.pneurobio.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, et al. One-year brain atrophy evident in healthy aging. Journal of Neuroscience. 2009;29:15223–15231. doi: 10.1523/JNEUROSCI.3252-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nature Review Neuroscience. 2007;8:700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer's disease. Journal of International Neuropsychological Society. 2008;14:266–278. doi: 10.1017/S1355617708080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80:1778–1783. doi: 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden T, Van Dijk KR, Becker JA, Mehta A, Sperling RA, Johnson KA, et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. Journal of Neuroscience. 2009;29:12686–12694. doi: 10.1523/JNEUROSCI.3189-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden T, Van Dijk KR, Shire EH, Sperling RA, Johnson KA, Buckner RL. Failure to Modulate Attentional Control in Advanced Aging Linked to White Matter Pathology. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland D, Desikan RS, Dale AM, McEvoy LK. Higher rates of decline for women and apolipoprotein E epsilon4 carriers. AJNR - American Journal of Neuroradiology. 2013;34:2287–2293. doi: 10.3174/ajnr.A3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers W, Schultz AP, Vannini P, McLaren DG, Wigman SE, Ward AM, et al. The encoding/retrieval flip: interactions between memory performance and memory stage and relationship to intrinsic cortical networks. Journal of Cognitive Neuroscience. 2013;25:1163–1179. doi: 10.1162/jocn_a_00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Gotz J. Amyloid-beta and tau-a toxic pas de deux in Alzheimer's disease. Nature Review Neuroscience. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr., Dickson DW, Parisi JE, Xu YC, Cha RH, O'Brien PC, et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002;58:750–757. doi: 10.1212/wnl.58.5.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurology. 2013a;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr., Knopman DS, Weigand SD, Wiste HJ, Vemuri P, Lowe V, et al. An operational approach to National Institute on Aging-Alzheimer's Association criteria for preclinical Alzheimer disease. Annals of Neurology. 2012;71:765–775. doi: 10.1002/ana.22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr., Wiste HJ, Knopman DS, Vemuri P, Mielke MM, Weigand SD, et al. Rates of beta-amyloid accumulation are independent of hippocampal neurodegeneration. Neurology. 2014 doi: 10.1212/WNL.0000000000000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr., Wiste HJ, Weigand SD, Knopman DS, Lowe V, Vemuri P, et al. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology. 2013b;81:1732–1740. doi: 10.1212/01.wnl.0000435556.21319.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. Vulnerable neural systems and the borderland of brain aging and neurodegeneration. Neuron. 2013;77:219–234. doi: 10.1016/j.neuron.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Landau SM. Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. Journal of Neuroscience. 2012;32:18227–18233. doi: 10.1523/JNEUROSCI.3266-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193–1199. doi: 10.1212/WNL.0b013e3181bc010c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Mormino EC. Lifespan Brain Activity, β-Amyloid, and Alzheimer's Disease. Trends in Cognitive Science. 2011;15:520–526. doi: 10.1016/j.tics.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha GA, Parisi JE, Dickson DW, Johnson K, Cha R, Ivnik RJ, et al. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Archives of Neurology. 2006;63:674–681. doi: 10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Christian BT, Okonkwo OC, Oh JM, Harding S, Xu G, et al. Amyloid burden and neural function in people at risk for Alzheimer's Disease. Neurobiology of Aging. 2014;35:576–584. doi: 10.1016/j.neurobiolaging.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Lowe V, Przybelski SA, Weigand SD, Senjem ML, Ivnik RJ, et al. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology. 2012;78:232–240. doi: 10.1212/WNL.0b013e31824365ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy KM, Rodrigue KM, Devous MD, Sr., Hebrank AC, Bischof GN, Park DC. Effects of beta-amyloid accumulation on neural function during encoding across the adult lifespan. NeuroImage. 2012;62:1–8. doi: 10.1016/j.neuroimage.2012.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Annals of Neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Jr., Wiste HJ, Lundt ES, Weigand SD, Vemuri P, et al. F-fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Jr., Wiste HJ, Weigand SD, Vemuri P, Lowe V, et al. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology. 2012;78:1576–1582. doi: 10.1212/WNL.0b013e3182563bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Jr., Wiste HJ, Weigand SD, Vemuri P, Lowe VJ, et al. Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly persons with beta-amyloidosis. JAMA Neurology. 2013;70:1030–1038. doi: 10.1001/jamaneurol.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Annals of Neurology. 2009;65:650–657. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- Landau SM, Marks SM, Mormino EC, Rabinovici GD, Oh H, O'Neil JP, et al. Association of lifetime cognitive engagement and low beta-amyloid deposition. Archives of Neurology. 2012a;69:623–629. doi: 10.1001/archneurol.2011.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Annals of Neurology. 2012b;72:578–586. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SC, Brehmer Y, Shing YL, Werkle-Bergner M, Lindenberger U. Neuromodulation of associative and organizational plasticity across the life span: empiricale vidence and neurocomputational modeling. Neuroscience and Biobehavioral Reviews. 2006;30:775–790. doi: 10.1016/j.neubiorev.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Lim YY, Maruff P, Pietrzak RH, Ames D, Ellis KA, Harrington K, et al. Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer's disease. Brain. 2013a;137:221–231. doi: 10.1093/brain/awt286. [DOI] [PubMed] [Google Scholar]
- Lim YY, Villemagne VL, Laws SM, Ames D, Pietrzak RH, Ellis KA, et al. BDNF Val66Met, Abeta amyloid, and cognitive decline in preclinical Alzheimer's disease. Neurobiology of Aging. 2013b;34:2457–2464. doi: 10.1016/j.neurobiolaging.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Logan JM, Sanders AL, Snyder AZ, Morris JC, Buckner RL. Under-recruitment and nonselective recruitment: dissociable neural mechanisms associated with aging. Neuron. 2002;33:827–840. doi: 10.1016/s0896-6273(02)00612-8. [DOI] [PubMed] [Google Scholar]
- Lott IT, Head E, Doran E, Busciglio J. Beta-amyloid, oxidative stress and down syndrome. Current Alzheimer Research. 2006;3:521–528. doi: 10.2174/156720506779025305. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, et al. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nature Neuroscience. 1998;1:69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron. 2012;76:871–885. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsland AL, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR. Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biological Psychiatry. 2008;64:484–490. doi: 10.1016/j.biopsych.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis CA, Kuller LH, Klunk WE, Snitz BE, Price JC, Weissfeld LA, et al. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Annals of Neurology. 2013;73:751–761. doi: 10.1002/ana.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Insel PS, Nosheny R, Tosun D, Trojanowski JQ, Shaw LM, et al. Emerging beta-Amyloid Pathology and Accelerated Cortical Atrophy. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2014.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Betensky RA, Hedden T, Schultz AP, Amariglio RE, Rentz DM, et al. Synergistic effect of beta-amyloid and neurodegeneration on cognitive decline in clinically normal participants. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2014.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Betensky RA, Hedden T, Schultz AP, Ward A, Huijbers W, et al. Amyloid and APOE4 interact to influence short-term decline in preclinical Alzheimer’s disease. Neurology. 2014 doi: 10.1212/WNL.0000000000000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Brandel MG, Madison CM, Marks S, Baker SL, Jagust WJ. Aβ deposition in aging is associated with increases in brain activation during successful memory encoding. Cerebral Cortex. 2011a doi: 10.1093/cercor/bhr255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Smiljic A, Hayenga AO, Onami SH, Greicius MD, Rabinovici GD, et al. Relationships between betaamyloid and functional connectivity in different components of the default mode network in aging. Cerebral Cortex. 2011b;21:2399–2407. doi: 10.1093/cercor/bhr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of Neurology. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. Journal of Neuropathology and Experimental Neurology. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Habeck C, Madison C, Jagust W. Covarying alterations in Abeta deposition, glucose metabolism, and gray matter volume in cognitively normal elderly. Human Brain Mapping. 2014;35:297–308. doi: 10.1002/hbm.22173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DC, Reuter-Lorenz P. The adaptive brain: aging and neurocognitive scaffolding. Annual Review of Psychology. 2009;60:173–196. doi: 10.1146/annurev.psych.59.103006.093656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike KE, Ellis KA, Villemagne VL, Good N, Chetelat G, Ames D, et al. Cognition and beta-amyloid in preclinical Alzheimer's disease: data from the AIBL study. Neuropsychologia. 2011;49:2384–2390. doi: 10.1016/j.neuropsychologia.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Snyder AZ. A default mode of brain function: a brief history of an evolving idea. NeuroImage. 2007;37:1083–1090. doi: 10.1016/j.neuroimage.2007.02.041. discussion 1097-9. [DOI] [PubMed] [Google Scholar]
- Raz N, Gunning-Dixon F, Head D, Rodrigue KM, Williamson A, Acker JD. Aging, sexual dimorphism, and hemispheric asymmetry of the cerebral cortex: replicability of regional differences in volume. Neurobiology of Aging. 2004;25:377–396. doi: 10.1016/S0197-4580(03)00118-0. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentz DM, Amariglio RE, Becker JA, Frey M, Olson LE, Frishe K, et al. Face-name associative memory performance is related to amyloid burden in normal elderly. Neuropsychologia. 2011;49:2776–2783. doi: 10.1016/j.neuropsychologia.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentz DM, Locascio JJ, Becker JA, Moran EK, Eng E, Buckner RL, et al. Cognition, reserve, and amyloid deposition in normal aging. Annals of Neurology. 2010;67:353–364. doi: 10.1002/ana.21904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74:807–815. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigue KM, Kennedy KM, Devous MD, Sr., Rieck JR, Hebrank AC, Diaz-Arrastia R, et al. beta-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology. 2012;78:387–395. doi: 10.1212/WNL.0b013e318245d295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe CM, Fagan AM, Grant EA, Hassenstab J, Moulder KL, Maue Dreyfus D, et al. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology. 2013;80:1784–1791. doi: 10.1212/WNL.0b013e3182918ca6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen AC, Prull MW, O'Hara R, Race EA, Desmond JE, Glover GH, et al. Variable effects of aging on frontal lobe contributions to memory. Neuroreport. 2002;13:2425–2428. doi: 10.1097/00001756-200212200-00010. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Bourgeat P, Ellis KA, Brown B, Lim YY, Mulligan R, et al. Predicting Alzheimer disease with beta-amyloid imaging: results from the Australian imaging, biomarkers, and life-style study of ageing. Annals of Neurology. 2013;74:905–913. doi: 10.1002/ana.24040. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of Aging. 2010;31:1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. New England Journal of Medicine. 2009;360:2302–2309. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- Schott JM, Bartlett JW, Fox NC, Barnes J. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Abeta1-42. Annals of Neurology. 2010;68:825–834. doi: 10.1002/ana.22315. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. Journal of Neuroscience. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, et al. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. Lancet Neurology. 2007;6:494–500. doi: 10.1016/S1474-4422(07)70106-0. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D'Angelo G, et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Abeta42. Journal of Neuroscience. 2010a;30:17035–17040. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biological Psychiatry. 2010b;67:584–587. doi: 10.1016/j.biopsych.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small BJ, Fratiglioni L, Viitanen M, Winblad B, Backman L. The course of cognitive impairment in preclinical Alzheimer disease: three- and 6-year follow-up of a population-based sample. Archives of Neurology. 2000;57:839–844. doi: 10.1001/archneur.57.6.839. [DOI] [PubMed] [Google Scholar]
- Snitz BE, Weissfeld LA, Lopez OL, Kuller LH, Saxton J, Singhabahu DM, et al. Cognitive trajectories associated with beta-amyloid deposition in the oldest-old without dementia. Neurology. 2013;80:1378–1384. doi: 10.1212/WNL.0b013e31828c2fc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW. Mapping cortical change across the human life span. Nature Neuroscience. 2003;6:309–315. doi: 10.1038/nn1008. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the national institute on aging and the Alzheimer's association workgroup. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O'Keefe K, O'Brien J, Rentz DM, Pihlajamaki M, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurology. 2012;11:1006–1012. doi: 10.1016/S1474-4422(12)70191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y, Gurland B, Tatemichi TK, Tang MX, Wilder D, Mayeux R. Influence of education and occupation on the incidence of Alzheimer's disease. JAMA. 1994;271:1004–1010. [PubMed] [Google Scholar]
- Storandt M, Mintun MA, Head D, Morris JC. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Archives of Neurology. 2009;66:1476–1481. doi: 10.1001/archneurol.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teri L, McCurry SM, Edland SD, Kukull WA, Larson EB. Cognitive decline in Alzheimer's disease: a longitudinal investigation of risk factors for accelerated decline. Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 1995;50A:M49–M55. doi: 10.1093/gerona/50a.1.m49. [DOI] [PubMed] [Google Scholar]
- Tosun D, Schuff N, Shaw LM, Trojanowski JQ, Weiner MW. Relationship between CSF biomarkers of Alzheimer's disease and rates of regional cortical thinning in ADNI data. Journal of Alzheimer's Disease. 2011;26(Suppl 3):77–90. doi: 10.3233/JAD-2011-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R, Van Laere K, Ivanoiu A, Salmon E, Bastin C, Triau E, et al. 18 F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Annals of Neurology. 2010;68:319–329. doi: 10.1002/ana.22068. [DOI] [PubMed] [Google Scholar]
- Vannini P, Hedden T, Becker JA, Sullivan C, Putcha D, Rentz D, et al. Age and amyloid-related alterations in default network habituation to stimulus repetition. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini P, Hedden T, Huijbers W, Ward A, Johnson KA, Sperling RA. The ups and downs of the posteromedial cortex: age- and amyloid-related functional alterations of the encoding/retrieval flip in cognitively normal older adults. Cerebral Cortex. 2013;23:1317–1328. doi: 10.1093/cercor/bhs108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Roberts RO, Lowe VJ, et al. Effect of lifestyle activities on Alzheimer disease biomarkers and cognition. Annals of Neurology. 2012;72:730–738. doi: 10.1002/ana.23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese J, Lipton RB, Katz MJ, Hall CB, Derby CA, Kuslansky G, et al. Leisure activities and the risk of dementia in the elderly. New England Journal of Medicine. 2003;348:2508–2516. doi: 10.1056/NEJMoa022252. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurology. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Annals of Neurology. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SJ, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurology. 2013;12:957–965. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. Journal of Neurochemistry. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Jack CR, Jr., Parisi JE, Senjem ML, Knopman DS, Boeve BF, et al. Does TDP-43 type confer a distinct pattern of atrophy in frontotemporal lobar degeneration? Neurology. 2010;75:2212–2220. doi: 10.1212/WNL.0b013e31820203c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71:743–749. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Tosakulwong N, Weigand SD, Senjem ML, Lowe VJ, Gunter JL, et al. Does amyloid deposition produce a specific atrophic signature in cognitively normal subjects? Neuroimage Clinical. 2013;2:249–257. doi: 10.1016/j.nicl.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Boyle PA, Yu L, Barnes LL, Schneider JA, Bennett DA. Life-span cognitive activity, neuropathologic burden, and cognitive aging. Neurology. 2013a;81:314–321. doi: 10.1212/WNL.0b013e31829c5e8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Yu L, Trojanowski JQ, Chen EY, Boyle PA, Bennett DA, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurology. 2013b;70:1418–1424. doi: 10.1001/jamaneurol.2013.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Haase CM, Villeneuve S, Vogel J, Jagust WJ. Neuroprotective pathways: lifestyle activity, brain pathology, and cognition in cognitively normal older adults. Neurobiol Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Madison CM, Rabinovici GD, Oh H, Landau SM, Jagust WJ. Alzheimer's disease neurodegenerative biomarkers are associated with decreased cognitive function but not beta-amyloid in cognitively normal older individuals. Journal of Neuroscience. 2013a;33:5553–5563. doi: 10.1523/JNEUROSCI.4409-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Oh H, Mormino EC, Markley C, Landau SM, Jagust WJ. The effect of amyloid beta on cognitive decline is modulated by neural integrity in cognitively normal elderly. Alzheimers Dement. 2013b;9:687–698. doi: 10.1016/j.jalz.2012.10.012. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Villeneuve S, Haase CM, Madison CM, Oh H, Landau SM, et al. Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people. JAMA Neurology. 2013c;70:1512–1519. doi: 10.1001/jamaneurol.2013.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AB, Valla J, Bu G, Kim J, Ladu MJ, Reiman EM, et al. Apolipoprotein E as a beta-amyloid-independent factor in alzheimer's disease. Alzheimer's Research & Therapy. 2013;5:38. doi: 10.1186/alzrt204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DF, Rosenberg PB, Zhou Y, Kumar A, Raymont V, Ravert HT, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18 F-AV-45 (florbetapir [corrected] F 18) Journal of Nuclear Medicine. 2010;51:913–920. doi: 10.2967/jnumed.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MY, Katzman R, Salmon D, Jin H, Cai GJ, Wang ZY, et al. The prevalence of dementia and Alzheimer's disease in Shanghai, China: impact of age, gender, and education. Annals of Neurology. 1990;27:428–437. doi: 10.1002/ana.410270412. [DOI] [PubMed] [Google Scholar]