

Abstract
Anti-inflammatory and anti-apoptotic effects of polydatin (PD) have been demonstrated in our previous studies. Recently, we have found that PD treatment can ameliorate burn-induced acute lung injury (ALI). In the present study, we hypothesized that PD may provide protective effect against LPS-induced ALI through reducing inflammation and apoptosis. Rats were respectively pretreated with PD at doses of 15, 30 and 45 mg/kg weight, followed by intratracheal administration of lipopolysaccharide (LPS). LPS-challenged rats exhibited significant lung injury characterized by the deterioration of histopathology, pulmonary microvascular hyperpermeability, wet-to-dry weight ratio, and oxygenation index, which was attenuated by PD (30 and 45 mg/kg) treatment. Moreover, PD (30 and 45 mg/kg) treatment inhibited LPS-induced inflammatory response, as evidenced by the downregulation of lung myeloperoxidase activity, total cells and PMNs in bronchoalveolar lavage fluid, and the systemic levels of the pro-inflammatory cytokines. Furthermore, PD (30 and 45 mg/kg) treatment remarkably improved LPS-induced increase in TUNEL (deoxynucleotidyl transferase dUTP nick end labeling) staining-positive cells, caspase 3 activity, Bax over-expression and Bcl-2 down-expression. In conclusion, these results demonstrate that PD (30 and 45 mg/kg) treatment attenuates LPS-induced ALI through reducing lung inflammation and apoptosis.
Keywords: Acute lung injury, polydatin, apoptosis, inflammation
Introduction
Acute lung injury (ALI) is a frequent complication following sepsis in critically ill patients and is associated with high rates of morbidity and mortality [1-3]. Endotoxin is thought to be the most important pathogen that leads to the development of ALI [4,5]. Excessive cytokine-mediated inflammation plays a fundamental role in the pathogenesis of ALI [6,7]. Increased levels of local and systemic inflammatory mediators, such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β and IL-6, and activated leukocytes, lead to systemic inflammatory response syndrome (SIRS) [8,9]. Moreover, increasing evidence has suggested that apoptosis may also play an important role in the pathophysiology of ALI [10,11].
Polydatin (PD) is a monocrystalline drug that can be isolated from the herb Polygonum cuspidatum, used in traditional Chinese remedies [12]. It is mainly used in the treatment of hemorrhagic shock, burns, and ischemia-reperfusion injury [13,14]. The Sino Food and Drug Administration has approved a clinical trial on the effects of PD on hypotension after severe shock, and the trial has now entered the second stage.
In the previous research, we have demonstrated PD can attenuate burn-induced lung injury [8]. Moreover, the influence of PD on inflammatory processes and apoptosis has been demonstrated efficiently in several models [15-17]. In the present study, we hypothesized that PD may provide protective effect against LPS-induced ALI through reducing inflammation and apoptosis.
Materials and methods
Animals
The experimental protocols were approved by the Animal Care and Use Committee of the Southern Medical University, Guangzhou, China. The care of animals was in accordance with the National Institutes of Health guidelines and with those of the Chinese National ones. Male Sprague-Dawley rats weighing 180-220 g were purchased from the Experimental Animal Center at South Medical University and allowed to acclimatize for a week before being used. Animals had ad libitum access to chow and water.
Animal model
As previously described, ALI was induced by intratracheal administration of LPS. Briefly, animals were intramuscularly anesthetized with an injection of sodium pentobarbital (30 mg/kg). Rats were placed in a supine position on a warming device and the trachea was surgically exposed by a cervical middle line incision in the skin, and LPS (5 mg/kg body weight; Escherichia coli 0111:B4; Sigma, St. Louis, Mo, USA) was slowly injected into the trachea of each rat. Control rats were intratracheally given 0.5 ml of sterile normal saline (NS).
Experimental design
Animals were randomly divided into five groups.
1. Control (sham) group, in which the rats were anesthetized and intratracheally given 0.5 ml of sterile NS. 2. LPS + NS group, in which 0.5 ml NS was given via caudal vein 30 min before LPS injection. 3. LPS + PDs group, in which a small dose of PD (15 mg/kg) was given via caudal vein 30 min before LPS injection. 4. LPS + PDM group, in which a medium dose of PD (30 mg/kg) was given via caudal vein 30 min before LPS injection. 5. LPS + PDL group, in which a large dose of PD (45 mg/kg) was given via caudal vein 30 min before LPS injection.
Rats were sacrificed at 24 post-LPS. The oxygenation index (PaO2/FIO2), histopathologic changes, number of total cells and polymorphonuclear neutrophils (PMNs) in bronchoalveolar lavage fluid (BALF), lung microvascular permeability, wet-to-dry (W/D) weight ratio, and myeloperoxidase (MPO) activity were measured. To investigate the underlying mechanisms of PD treatment in LPS-induced ALI, the inflammation (TNF-α, IL-1β, IL-6, high-mobility group box 1 [HMGB1]) and apoptosis (terminal deoxynucleotidyl transferase dUTP nick end labeling [TUNEL] staining, Lipid peroxidation [LPO], Bax, Bcl-2 expression, and caspase-3 activity) were detected.
Oxygenation index (PaO2/FiO2) analysis
At 24 h after ALI (or sham), animals were anesthetized and given endotracheal intubation with a 20-gauge catheter. The animals were mechanically ventilated with pure oxygen at 7 mL/kg (120 breaths/min). After 20 min-ventilation, the arterial blood was obtained from carotid artery and measured using a commercial blood gas analyzer (model ABL8000; Radiometer Copenhagen, Westlake, Ohio).
Histologic examination
Lungs were harvested 24 h after LPS injection. The right middle lobes of the lungs were fixed with 10% formalin, embedded in paraffin, and sectioned to 4-μm thickness. After deparaffinization and rehydration, the sections were stained with hematoxylin and eosin. The pathological sections were observed in a blinded fashion.
Lung microvascular permeability assay
The permeability assay was performed as previously described [8]. Briefly, EB (20 mg/kg weight; Sigma, USA) was injected intravenously through the femoral vein. Thirty minutes later, the animals were sacrificed, and a midline thoracotomy was performed. Then, the superior and inferior vena cava were ligated, the aorta was transected, and 20 ml normal saline solution was injected into the right ventricle at a pressure of 20 cm H2O to wash out the pulmonary intravascular content. A sample of lung tissue was weighed, homogenized, and immersed in N,N-dimethylformamide (Sigma, USA). The homogenate was incubated at room temperature for 48 h. Eluted EB was measured at 620 nm using an automatic microplate reader (SpectraMax M5; Molecular Devices, Sunnyvale, CA, USA), and the amount was expressed as micrograms per 100 mg dry tissue.
Measurement of wet lung/dry lung weight (W/D) ratio
The harvested wet lung was weighed and then placed in an oven for 48 h at 80°C and then weighed again when it was dried. The ratio of wet lung to dry lung weight was calculated.
Cell counts and protein concentration in BALF
Rats were killed, a median sternotomy was performed, and the trachea was isolated by blunt dissection; then, a suitable small-caliber tube was inserted into the airway and secured. Phosphate-buffered saline (PBS) (pH 7.2) was infused slowly into the lungs and bronchoalveolar lavage fluid (BALF) was withdrawn via the tube. The fluid recovery rate was more than 90%. Lavage samples were centrifuged at 1,500 g for 10 min at 4°C. T The sedimented cells were resuspended in PBS and subjected to cell counting. The slides were visualized using Wright-Giemsa staining (Sigma, USA), and PMNs were counted in a double-blind fashion. Total protein concentration in the BALF was determined using a standard commercial kit (Sigma, USA).
Enzyme-linked immunosorbent assay
Arterial blood was withdrawn from rats at 24 h after ALI (or sham) and allowed to remain for 30 min; it was then centrifuged at 3,000 rpm for 10 min to obtain serum. The levels of TNF-α, IL-1β, IL-6 and HMGB1 in serum were measured using commercially available enzyme-linked immunosorbent assay (ELISA) kits (all kits are from Sigma, USA) according to the manufacturers’ instructions. All spectrophotometric readings were performed with an automatic microplate reader (SpectraMax M5).
Myeloperoxidase activity assay
Lungs were harvested, rinsed, homogenized and centrifuged. Supernatants were collected and subjected to ELISA for determination of MPO activity using the myeloperoxidase (MPO) activity colorimetric assay kit (Biovision, California, USA).
TUNEL staining
Lung histopathological slides were dewaxed and incubated with proteinase K. Slides were stained using a terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) kit (Biovision, USA), counterstained with Hoechst 33258 (Sigma, USA) and examined under a fluorescence microscope (ECLIPSE FNl, Nikon, Tokyo, Japan). Apoptotic cells appeared fluorescent green and were counted per 10 visual fields at a 200 × magnification.
Measurement of lipid peroxides
The serum was obtained as described above. Lipid peroxides (LPOs) were measured using a commercially available kit (Cayman Chemical Co., Ann Arbor, Michigan, USA) according to the manufacturer’s instructions. The LPO content was read at 500 nm using an automatic microplate reader (SpectraMax M5).
Caspase-3 activity
The lung tissue were homogenized in the lysate provided in the kit and centrifuged (10,000 × g for 60 min at 4°C), and the supernatant (cytosolic fraction) was collected and subjected to a protein assay (BCA method). Then, the caspase-3 activity was measured using the caspase-3/CPP32 fluorometric assay kit (Biovision, USA) in keeping with the manufacturer’s instructions.
Western blot analysis for Bcl-2-Bax
The lung tissues were homogenized and analyzed for Bcl-2-Bax by western blotting. Protein concentrations were determined using the BCA method. An equal amount of protein was loaded onto 10% sodium dodecyl sulphate polyacrylamide gel for electrophoresis. After electrophoresis, proteins were electroblotted onto polyvinylidene fluoride membranes and blotted with primary antibodies against Bcl-2-Bax (Abcam, UK) and beta-actin (Tianjin Sungene Biotech Co., Ltd. Tianjin, China). Membranes were then incubated with the horseradish peroxidase-tagged secondary antibody (Tianjin Sungene Biotech Co., China), and protein expression was detected using an enhanced chemiluminescence reagent.
Statistical analysis
All variables are presented as mean ± SD. Differences between groups were determined using one-way ANOVA with the LSD multiple-comparison test and Student’s t-test when appropriate. Values were considered significant at P < 0.05, and n represents the number of animals.
Results
PD attenuates LPS-induced lung injury
Rats in the LPS + NS group showed accumulation of a large number of neutrophils in the intra- and interalveolar space, a thickened alveolar wall, less alveolar space, interstitial congestion, and these alterations were markedly attenuated by PD treatment (30 and 45 mg/kg, not 15 mg/kg) (Figure 1). Moreover, LPS-challenged rats showed the significant increase in lung microvascular permeability, evidenced by elevated EB content in lung tissue compared with control group (P < 0.05), then decreased in PD treatment group (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 2A). Meanwhile, the W/D ratio in the LPS-challeged animals was significantly increased compared with control group, which was also decreased by PD treatment (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 2B). In addition, the PaO2/FiO2 was significantly decreased in LPS-challenged rats, which was improved by PD treatment (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 2C). These results demonstrate that PD treatment significantly improves the lung histopathology and lung function in LPS-challenged rats.
Figure 1.
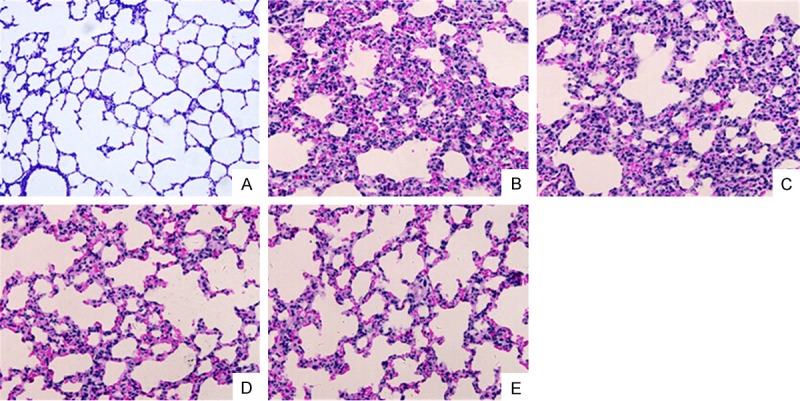
PD attenuates histopathological changes in lung (200 × magnification). Comparison of histopathological changes using HE staining: after LPS stimulation, lung in the LPS + NS group showed a thickened alveolar wall, edema, less alveolar space and obvious inflammatory cell infiltration. Medium and large dose of PD significantly prevented the histopathological changes caused by LPS. A. Control group. B. LPS+NS group. C. LPS+PDs group. D. LPS+PDM group. E. LPS+PDL group.
Figure 2.
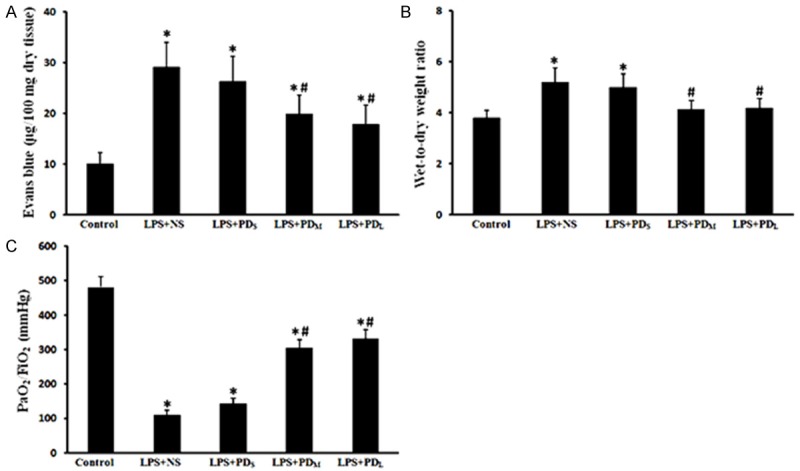
PD prevented pulmonary microvascular permeability, edema and dysfunction. Increased EB and W/D values, decreased PaO2/FIO2 were detected in the LPS + NS group, which were improved by medium and large dose of PD treatment. A. Evans blue (EB) content in lungs. B. Wet-to-dry (W⁄D) lung weight ratio. C. Oxygenation index (PaO2/FIO2). Data are presented as mean ± SD (n = 6 in each group). *P < 0.05 vs. sham group; #P < 0.05 vs. LPS + NS group.
PD reduced the cells and protein in the BALF
Rats in the LPS + NS group showed the significant increase in total cells, PMNs, and total protein in the BALF compared with control group (P < 0.05), which were markedly reduced by PD treatment (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 3). These data further indicate that PD treatment attenuates LPS-induced lung injury in rats.
Figure 3.
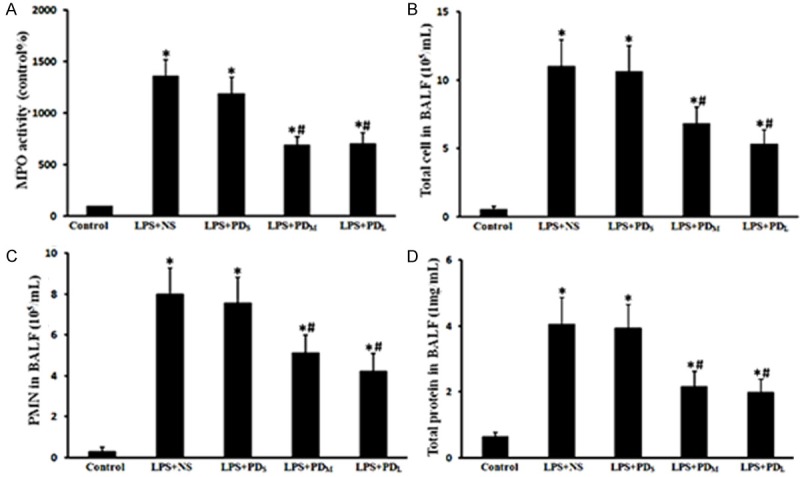
PD reduced the cell counts and protein concentration in the BALF as well as the lung MPO activity. MPO activity in lung, total cells, PMNs and protein concentration in BALF were increased in the LPS + NS group, but were alleviated in the medium and large dose of PD treatment group. A. Lung MPO activity. B. Total cells in BALF. C. Polymorphonuclear neutrophils in BALF. D. Total protein concentration in BALF. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05 vs. sham group; #P < 0.05 vs. LPS + NS group.
PD decreased neutrophils recruitment in lung
Lung MPO activity, an indicator of neutrophil infiltration, was detected. The lung MPO activity of LPS-challenged rats dramatically increased (P < 0.05 vs. control group), which was inhibited by PD treatment (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 4A). These results suggest that PD treatment attenuates the LPS-induced neutrophils recruitment in lung.
Figure 4.
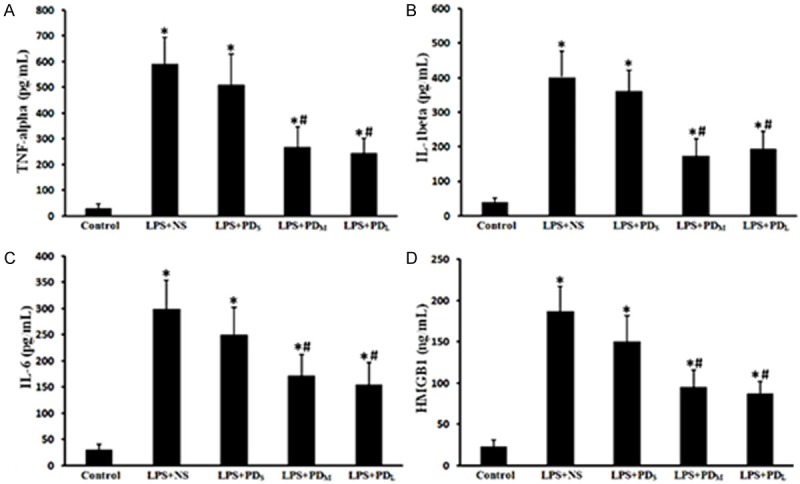
PD downregulated the levels of proinflammatory cytokines and chemokines in the serum. The circulating levels of inflammatory mediators of TNF-α, IL-1β, IL-6 and HMGB1 were elevated in the LPS + NS group, but decreased in the medium and large dose of PD treatment group. A. TNF-α level in serum. B. IL-1β level in serum. C. IL-6 level in serum. D. HMGB1 level in serum. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05 vs. sham group; #P < 0.05 vs. LPS + NS group.
PD downregulated systemic levels of cytokines and chemokines
TNF-α, IL-1β, IL-6 and HMGB1levels in the peripheral blood of LPS-challenged animals were significantly increased (P < 0.05 vs. control group; Figure 4B-E). The increased levels of TNF-α, IL-1β and IL-6 were markedly attenuated by PD treatment (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 4B-E). These results suggest that PD treatment downregulated LPS-induced inflammation in rats.
PD prevented the oxidative stress
To evaluate the oxidative stress following LPS administration, we assayed LPO levels in rat serum. The LPO levels increased remarkably in the LPS-challenged rats (P < 0.05 vs. control group), then reduced by PD treatment (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 5F).
Figure 5.
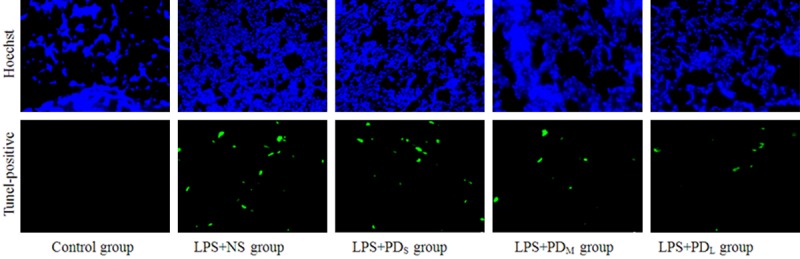
PD prevented the lung cell apoptosis (200 × magnification). The pulmonary cell apoptosis in LPS-challenged rats were measured by Tunel staining. LPS-challenged animals showed a significant increase in Tunel-positive cells, which was reduced by the medium and large dose of PD treatment.
PD inhibited the lung cell apoptosis
We investigated the effects of PD treatment on pulmonary cell apoptosis in LPS-challenged rats by TUNEL staining, a common method for detecting DNA fragmentation. LPS-challenged animals showed a significant increase in apoptotic cells (P < 0.05 vs. control group), which was reduced by PD treatment (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 5A, 5B). Similarly, we found that the caspase 3 activity was significantly increased in the lungs of LPS-challenged animals, which was prevented by PD treatment (P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 5C). Moreover, we investigated the changes of Bcl-2 family of proteins (bax and bcl-2) expression in lung tissue. LPS inhalation resulted in downregulation of the Bcl-2 protein, and PD prevented this decrease (P < 0.05 vs. control group for LPS + NS group; P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 6). Expression of the pro-apoptotic protein Bax was up-regulated by LPS induction and inhibited by PD treatment (P < 0.05 vs. control group for LPS + NS group; P < 0.05 vs. LPS + NS group for PDM and PDL group; P > 0.05 vs. LPS + NS group for PDs group; Figure 7). These results indicate that intratracheal administration LPS induce the lung cell apoptosis, which can be significantly alleviated by PD treatment.
Figure 6.
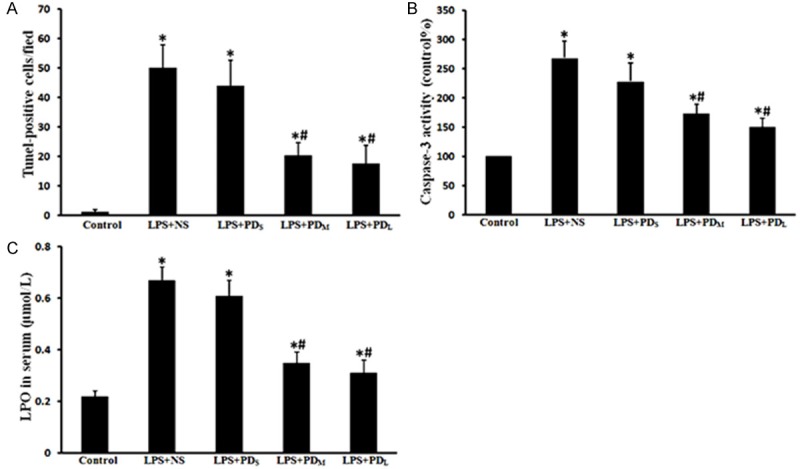
PD ameliorated Tunel-positive cells, caspase-3 activity and oxidative stress. Tunel-positive cells and caspase-3 activity in lung, LPO level in serum were increased in the LPS + NS group, but decreased in the medium and large dose of PD treatment group. A. Graphical representation of TUNEL-positive cells averaged over 10 microscopic fields per animal. B. Caspase-3 activity in lung tissues. C. LPO level in serum. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05 vs. sham group; #P < 0.05 vs. LPS + NS group.
Figure 7.
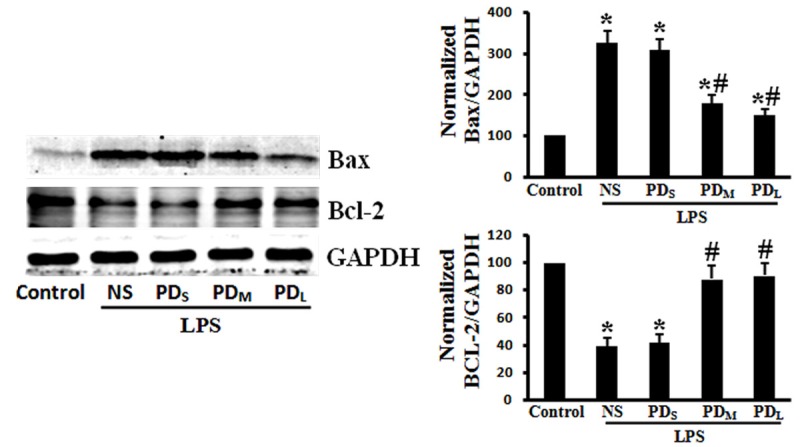
PD improved Bax upregulation and Bcl-2 downregulation. The increased Bax expression and decreased Bcl-x1 expression in the LPS + NS group was ameliorated by the medium and large dose of PD treatment. Data are presented as mean ± SD (n = 6 in each group). *P < 0.05 vs. sham group; #P < 0.05 vs. LPS + NS group.
Discussion
The present study showed that PD attenuated LPS-induced ALI in a rat model, and the protective effects of PD are related to its ability of ameliorating the release of pro-inflammatory molecules and preventing lung cell apoptosis.
LPS is the most important pathogen that leads to the development of ALI and intra-tracheal instillation of LPS has been commonly used to induce an animal model of ALI, so we established an ALI rat model through intra-tracheal injection of LPS (5 mg/kg) in our present study [18,19]. We observed a significant lung injuries and dysfunction after LPS administration, evidenced by deterioration of histopathology, increased vascular permeability and W/D ratio, decreased PaO2/FIO2, which is consistent with other studies [3,20]. In our previous study, we found PD can improve burn injury-induced lung injury in rats. In present study, we investigated the protective effects of PD against LPS-induced ALI by a dose-dependent of PD treatment (Small: 15 mg/kg; Medium: 30 mg/kg; Large: 45 mg.kg). In the current study, we showed that treatment with medium and large dose of PD attenuated the LPS-induced ALI, evidenced by improved histopathological changes, vascular permeability, lung water content, and PaO2/FIO2.
Recruitment of neutrophils is a key event in development of ALI, linking to plasma leakage and deterioration of oxygenation [2,3]. The importance of neutrophils in ALI is supported by studies where lung injury is abolished or reversed by depletion of neutrophils [22,23]. In the present study, rats exposed to LPS exhibited a massive recruitment of inflammatory cells including neutrophils and macrophages in the airways. We found that the total cells, PMNs and total protein in the BALF of LPS-challenged rats were significantly increased, which were attenuated by medium and large dose of PD treatment. Moreover, we investigated lung neutrophil infiltration by measuring the activity of lung MPO, a neutrophil-specific enzyme [24,25]. Medium and large dose of PD treatment prevented the increase in lung MPO activity in LPS-challenged rats. Moreover, proinflammatory cytokines and chemokines play an important role in the initiation and amplification of inflammatory responses [3,26,27]. We found that medium and large dose of PD treatment dramatically prevented the increase in proinflammatory cytokines (TNF-α, IL-1β, IL-6) and cytokines (HMGB1) in the serum of LPS-challenged rats. These results demonstrate that PD treatment ameliorates LPS-induced lung neutrophil infiltration and inflammation.
Recent studies have shown that apoptosis contributes to the pathogenesis of lung injury [28,29,30]. Moreover, apoptosis can amplify inflammatory responses in lung diseases [10]. In this study, apoptotic cells were detected on the basis of positive TUNEL staining. As shown, numerous apoptotic cells were observed in the lungs of LPS-challenged animals. The number of TUNEL-positive lung cells was significantly reduced after medium and large dose of PD treatment. In addition, several apoptosis-related proteins such as Bcl-2, BAX and caspase-3were detected. The Bcl-2 family consists of both antiapoptotic (Bcl-2, Bcl-xL) and proapoptotic (BAK, BAX) factors [31,32]. The antiapoptotic members of this family prevent apoptosis by sequestering proforms of death-driving cysteine proteases or by preventing the mitochondrial apoptogenic factors release. On the contrary, the proapoptotic members trigger the mitochondrial apoptogenic factors release into the cytoplasm through the mitochondrial permeability transition pore, thereby leading to caspases activation [33,34]. Caspase-3 is considered to be the most important of the executioner caspases and is activated by any of the initiator caspases [35,36]. We also found that, in this study, that medium and large dose of PD treatment prevents LPS-induced up-regulation of Bax and caspase-3 activity and down-regulation of Bcl-xl. These results demonstrated that PD treatment prevents lung cell apoptosis in LPS-induced ALI.
The role of oxidative stress in inflammation and apoptosis is well described [37,38]. In the present study, lipid peroxides (LPO) levels were measured to determine the extent of LPS-induced oxidative stress. Increased LPO levels were detected in the serum of LPS-challenged rats, but they reduced with medium and large dose of PD treatment.
In conclusion, we have provided evidence for the first time, of PD-induced significant attenuation of LPS-induced ALI in a rat model. We also found that the potential mechanism of this action is through amelioration of inflammation and apoptosis.
Disclosure of conflict of interest
None.
References
- 1.Singh N. Acute lung injury and acute respiratory distress syndrome. Lancet. 2007;370:383–385. doi: 10.1016/S0140-6736(07)61183-0. [DOI] [PubMed] [Google Scholar]
- 2.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369:1553–1564. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 3.Xie K, Yu Y, Huang Y, Zheng L, Li J, Chen H, Han H, Hou L, Gong G, Wang G. Molecular hydrogen ameliorates lipopolysaccharide-induced acute lung injury in mice through reducing inflammation and apoptosis. Shock. 2012;37:548–555. doi: 10.1097/SHK.0b013e31824ddc81. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Qiang Y, Wang L, Wang G, Yi J, Jing H, Wu H. Repair of lipopolysaccharide-induced acute lung injury in mice by endothelial progenitor cells, alone and in combination with simvastatin. Chest. 2013;144:876–886. doi: 10.1378/chest.12-2429. [DOI] [PubMed] [Google Scholar]
- 5.Zhu T, Wang DX, Zhang W, Liao XQ, Guan X, Bo H, Sun JY, Huang NW, He J, Zhang YK, Tong J, Li CY. Andrographolide protects against LPS-induced acute lung injury by inactivation of NF-kappaB. PLoS One. 2013;8:e56407. doi: 10.1371/journal.pone.0056407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo Z, Li Q, Han Y, Liang Y, Xu Z, Ren T. Prevention of LPS-induced acute lung injury in mice by progranulin. Mediators Inflamm. 2012;2012:540794. doi: 10.1155/2012/540794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu X, Li H, Tang H, Jin Y, Li W, YuSun , PingFeng Hydrogen inhalation ameliorates lipopolysaccharide-induced acute lung injury in mice. Int Immunopharmacol. 2011;11:2130–2137. doi: 10.1016/j.intimp.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Li T, Cai S, Zeng Z, Zhang J, Gao Y, Wang X, Chen Z. Protective Effect of Polydatin Against Burn-Induced Lung Injury in Rats. Respir Care. 2014;59:1412–21. doi: 10.4187/respcare.02831. [DOI] [PubMed] [Google Scholar]
- 9.Fang Y, Xu P, Gu C, Wang Y, Fu XJ, Yu WR, Yao M. Ulinastatin improves pulmonary function in severe burn-induced acute lung injury by attenuating inflammatory response. J Trauma. 2011;71:1297–1304. doi: 10.1097/TA.0b013e3182127d48. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt EP, Tuder RM. Role of Apoptosis in Amplifying Inflammatory Responses in Lung Diseases. J Cell Death. 2010:41–53. doi: 10.4137/JCD.S5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galani V, Tatsaki E, Bai M, Kitsoulis P, Lekka M, Nakos G, Kanavaros P. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): an up-to-date cell-specific review. Pathol Res Pract. 2010;206:145–150. doi: 10.1016/j.prp.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Zhao KS, Jin C, Huang X, Liu J, Yan WS, Huang Q, Kan W. The mechanism of Polydatin in shock treatment. Clin Hemorheol Microcirc. 2003;29:211–217. [PubMed] [Google Scholar]
- 13.Jiang X, Liu W, Deng J, Lan L, Xue X, Zhang C, Cai G, Luo X, Liu J. Polydatin protects cardiac function against burn injury by inhibiting sarcoplasmic reticulum Ca2+ leak by reducing oxidative modification of ryanodine receptors. Free Radic Biol Med. 2013;60:292–299. doi: 10.1016/j.freeradbiomed.2013.02.030. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Song R, Chen Y, Zhao M, Zhao KS. Polydatin--a new mitochondria protector for acute severe hemorrhagic shock treatment. Expert Opin Investig Drugs. 2013;22:169–179. doi: 10.1517/13543784.2013.748033. [DOI] [PubMed] [Google Scholar]
- 15.Ince S, Arslan AD, Neuwirth O, Demirel HH, Denk B, Kucukkurt I, Turkmen R. Protective effect of polydatin, a natural precursor of resveratrol, against cisplatin-induced toxicity in rats. Food Chem Toxicol. 2014;72:147–153. doi: 10.1016/j.fct.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 16.Deng J, Liu W, Wang Y, Dong M, Zheng M, Liu J. Polydatin modulates Ca(2+) handling, excitation-contraction coupling and beta-adrenergic signaling in rat ventricular myocytes. J Mol Cell Cardiol. 2012;53:646–656. doi: 10.1016/j.yjmcc.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Ravagnan G, De Filippis A, Carteni M, De Maria S, Cozza V, Petrazzuolo M, Tufano MA, Donnarumma G. Polydatin, a natural precursor of resveratrol, induces beta-defensin production and reduces inflammatory response. Inflammation. 2013;36:26–34. doi: 10.1007/s10753-012-9516-8. [DOI] [PubMed] [Google Scholar]
- 18.Aulakh GK, Suri SS, Singh B. Angiostatin inhibits acute lung injury in a mouse model. Am J Physiol Lung Cell Mol Physiol. 2014;306:L58–L68. doi: 10.1152/ajplung.00368.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim IK, Rhee CK, Yeo CD, Kang HH, Lee DG, Lee SH, Kim JW. Effect of tyrosine kinase inhibitors, imatinib and nilotinib, in murine lipopolysaccharide-induced acute lung injury during neutropenia recovery. Crit Care. 2013;17:R114. doi: 10.1186/cc12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chien MH, Bien MY, Ku CC, Chang YC, Pao HY, Yang YL, Hsiao M, Chen CL, Ho JH. Systemic human orbital fat-derived stem/stromal cell transplantation ameliorates acute inflammation in lipopolysaccharide-induced acute lung injury. Crit Care Med. 2012;40:1245–1253. doi: 10.1097/CCM.0b013e31823bc89a. [DOI] [PubMed] [Google Scholar]
- 21.Hou S, Ding H, Lv Q, Yin X, Song J, Landén NX, Fan H. Therapeutic effect of intravenous infusion of perfluorocarbon emulsion on LPS-induced acute lung injury in rats. PLoS One. 2014;9:e87826. doi: 10.1371/journal.pone.0087826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah D, Romero F, Stafstrom W, Duong M, Summer R. Extracellular ATP mediates the late phase of neutrophil recruitment to the lung in murine models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306:L152–L161. doi: 10.1152/ajplung.00229.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang H, Hu R, Sun L, Chai D, Cao Z, Li Q. Critical role of toll like receptor4 in hypoxia inducible factor-1alpha activation during trauma /hemorrhagic shock induced acute lung injury after lymph infusion in mice. Shock. 2014;42:271–8. doi: 10.1097/SHK.0000000000000212. [DOI] [PubMed] [Google Scholar]
- 24.Bhargava R, Altmann CJ, Andres-Hernando A, Webb RG, Okamura K, Yang Y, Falk S, Schmidt EP, Faubel S. Acute lung injury and acute kidney injury are established by four hours in experimental sepsis and are improved with pre, but not post, sepsis administration of TNF-alpha antibodies. PLoS One. 2013;8:e79037. doi: 10.1371/journal.pone.0079037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Du Z, Zhou Q, Wang Y, Li J. Remifentanil Attenuates Lipopolysaccharide-Induced Acute Lung Injury by Downregulating the NF-kappaB Signaling Pathway. Inflammation. 2014;37:1654–60. doi: 10.1007/s10753-014-9893-2. [DOI] [PubMed] [Google Scholar]
- 26.Lee I, Dodia C, Chatterjee S, Feinstein SI, Fisher AB. Protection against LPS-induced acute lung injury by a mechanism-based inhibitor of NADPH oxidase (type 2) Am J Physiol Lung Cell Mol Physiol. 2014;306:L635–L644. doi: 10.1152/ajplung.00374.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang Y, Fu XJ, Gu C, Xu P, Wang Y, Yu WR, Sun Q, Sun XJ, Yao M. Hydrogen-rich saline protects against acute lung injury induced by extensive burn in rat model. J Burn Care Res. 2011;32:e82–e91. doi: 10.1097/BCR.0b013e318217f84f. [DOI] [PubMed] [Google Scholar]
- 28.Hirota JA, Hiebert PR, Gold M, Wu D, Graydon C, Smith JA, Ask K, McNagny K, Granville DJ, Knight DA. Granzyme B deficiency exacerbates lung inflammation in mice after acute lung injury. Am J Respir Cell Mol Biol. 2013;49:453–462. doi: 10.1165/rcmb.2012-0512OC. [DOI] [PubMed] [Google Scholar]
- 29.Matsuo S, Yang WL, Aziz M, Jacob A, Wang P. Cyclic arginine-glycine-aspartate attenuates acute lung injury in mice after intestinal ischemia/reperfusion. Crit Care. 2013;17:R19. doi: 10.1186/cc12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Childs EW, Tharakan B, Hunter FA, Tinsley JH, Cao X. Apoptotic signaling induces hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol. 2007;292:H3179–H3189. doi: 10.1152/ajpheart.01337.2006. [DOI] [PubMed] [Google Scholar]
- 31.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 32.Shore GC, Nguyen M. Bcl-2 proteins and apoptosis: choose your partner. Cell. 2008;135:1004–1006. doi: 10.1016/j.cell.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 33.Whaley JG, Tharakan B, Smith B, Hunter FA, Childs EW. (-)-Deprenyl inhibits thermal injury-induced apoptotic signaling and hyperpermeability in microvascular endothelial cells. J Burn Care Res. 2009;30:1018–1027. doi: 10.1097/BCR.0b013e3181bfb825. [DOI] [PubMed] [Google Scholar]
- 34.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 35.Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke. 2003;34:207–213. doi: 10.1161/01.STR.0000047101.87575.3C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grommes J, Vijayan S, Drechsler M, Hartwig H, Morgelin M, Dembinski R, Jacobs M, Koeppel TA, Binnebösel M, Weber C, Soehnlein O. Simvastatin reduces endotoxin-induced acute lung injury by decreasing neutrophil recruitment and radical formation. PLoS One. 2012;7:e38917. doi: 10.1371/journal.pone.0038917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 38.Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–1180. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]