

Abstract
Objectives: This study is conducted to investigate an effect of simvastatin on cigarette smoke-induced COPD. Methods: Rats were exposed to air (control) and cigarette smoke (smoking) in presence and absence of simvastatin. Heart and lung tissues were harvested for histopathologic and morphometric analysis. Body weight of rat, mean liner intercept (MLI), mean alveolar number (MAN), lung function test, mean pulmonary artery pressure (mPAP), right ventricular hypertrophy index (RVHI) and 5-HTT level in serum and BALF were examined in experimental rats, respectively. Results: Application of simvastatin mitigated peribronchiolar inflammation and pulmonary bullae formed in the smoke-exposed lungs with weight gain as compared to the smoking rats (P < 0.05). Simvastatin-treated rats showed slight but significant decreases in MLI and MAN with a partial reversal of lung function decline (all P < 0.05). Treatment with simvastatin resulted in a significant decrease not only in mPAP and RVHI but also in a 5-HTT level in serum and BALF (P < 0.01 or 0.05) with a good correlation between the 5-HTT level and mPAP or RVHI (r = 0.693 and 0.479; 0.675 and 0.508). Conclusion: Simvastatin partly reverses lung function decline and attenuates structural impairments of lung and right ventricle possibly through reducing 5-HTT content in the model of COPD.
Keywords: Simvastatin, cigarette smoke, 5-hydroxytryptamine transporter, peribronchiolar inflammation and pulmonary bullae
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading global cause of morbidity and mortality, and will continue to increase in importance as the world population continues to age [1]. COPD is characterized by airflow limitation that is not fully reversible but usually progressive and associated with an inflammatory response of the lung [2]. It has long been accepted that exposure to cigarette smoke can cause airway inflammation, which subsequently leads to a series of pulmonary structure changes [3]. Moreover, advanced COPD can lead to pulmonary hypertension (PH) due to low oxygen levels in the alveoli and thereby harm heart [4].
5-Hydroxytryptamine (5-HT) is a highly conserved monoamine, which is a major neurotransmitter in the CNS. However, 5-HT level is increased in COPD patients and the development of PH has been shown to be associated with an increased 5-HT level [5]. It has been known that 5-HT transporter (5-HTT) is abundantly expressed in the lung, where it is predominantly located on pulmonary-artery smooth muscle cells [6]. The 5-HTT expression level appears to be considerably higher in human lung than in human brain, suggesting that altered 5-HTT expression may have direct consequences on pulmonary-artery smooth muscle cell function. Furthermore, an increase in 5-HTT gene expression level is associated with COPD [7,8].
Treatment of COPD patients is still a major challenge, with the difficulties at least including permanent destructive enlargement of airspaces and increased smooth muscle hyperplasia in small airway involved in the event. Simvastatin have emerged as a possible disease modifying agent in COPD since it possess pleiotropic effects in addition to their conventional lipid-lowering properties including anti-inflammatory, inhibiting airway smooth muscle proliferation and vascular function-restoring actions for the prevention of cardiovascular disease [9-11].
The current study is aimed at investigating the effect of simvastatin on cigarette smoke-induced COPD with a possible mechanism(s) by which this agent may influence functional and structural impairment of lung and heart in devising therapeutic approaches.
Materials and methods
Animals and model of cigarette smoke
Specific pathogen-free, male SD rats (6 wks) weighing about 100 g were purchased from experimental centre of animals at Hebei Medical University. The rats were housed in an environmentally controlled animal facility of our hospital for the duration of the experiments. All procedures were reviewed and approved by Hospital Research Review Committee.
Rats were divided randomly into three groups of 10 each and the protocol for making animal model of cigarette smoke-induced COPD is modified with different treatments and a smoking period. The animal model of COPD was established with mainstream smoke exposure (20 cigarettes, twice a day with an interval of 4-5 hours) for 4 months in a cigarette smoke chamber. The cigarette smoke-exposed rats were treated with an equivalent volume of phosphate buffered saline (PBS) as smoking group and simvastatin dissolved in PBS by gastric gavage (5 mg/kg, once a day for 4 months) during the period of the challenge, respectively. Rats in the control group were exposed to air and treated with PBS.
Measurement of lung function
Lung function was examined after measuring weight gain of the rats. Briefly, the rats were anaesthetized by an intraperitoneal injection of 10% Chloral hydrate (3 ml/kg) and maintained with an appropriate plane of the anesthesia. The trachea was opened with an inverted T-shaped incision in the position between the 2nd and the 3rd cartilage ring, rapidly intubated, and placed the animal into an apparatus (animal spirometer, (Beijing Rambo Technology Co., Ltd., Beijing China) for measuring the volume of air inspired and expired by the lungs. The one of the exports of the T-typed cannula in the trachea was connected to a pressure transducer applied to a pulmonary mechanics analyzer and another one was used for administration of air to expand the lungs of rats.
A ratio of forced expiratory volume at 0.3 s and forced vital capacity (FEV0.3/FVC), dynamic lung compliance (Cdy), and inspiratory (Ri) and expiratory (Re) resistance were measured with injecting 6.0 ml air into the T-typed cannula in the trachea of the anaesthetized rats and data regarding the parameters of lung function were automatically recorded by the analyzer.
Preparation of peripheral blood, bronchoalveolar lavage fluid and organ specimens
Rat peripheral blood samples (2.0 ml per rat) were collected from each group via cardiac puncture under light anesthesia and transferred to EDTA-K2 collection tubes. The samples were allowed to clot for 45 to 60 min at room temperature and centrifuged at 3000 g for 10 min to separate the serum. The samples were stored at -70°C until use.
Experimental animals were sacrificed by blood loss from opening femoral artery and bronchoalveolar lavage fluid (BALF) was collected with 3.0 ml followed by 2 × 3.0 ml PBS. To remove cells recovered lavage fluid (70-80%) was centrifuged at 300 g for 5 min and the BALF supernatant was obtained with centrifuge and applied for the enzyme-linked immunosorbent assays (ELISA).
Following the collection of BALF, heart and lungs were dissected and blood accumulated in the right atrium was completely removed from the organ. Three lobes in right lung were surgically removed from the rat and immediately snap frozen in liquid nitrogen, then stored at -80°C until processed. Last lobe in the right lung and heart were harvested and placed them in a wide-mouth bottle filled up in formaldehyde (4%) for histopathological examination.
Morphological study
The harvested lobes of lung were fixed in 4% formaldehyde solution for 72 hours. Histopathologic examination of the samples was performed in all groups. Briefly, the lung tissues from groups were embedded in paraffin and cut into a 4-μm section. The lung sections stained with hematoxylin and eosin (H&E) solution were applied to a glass slide and photographed under a light microscope (Olympus, Japan) at a magnification of 400 for morphological analysis.
Mean linear intercept (MLI) and mean alveolar number (MAN) were examined on two glass slides of each group in the same size of the field of view. Using light microscopy, MLI was determined for each region studied on an overlay consisting of horizontal and vertical lines. All intercepts with alveolar septal number (ASN) were counted at the intersection point of the two lines in the central field of the view under microscope. The total length (L) of all the lines together divided by the number of intercepts gives the mean linear intercept for the region studied. A formula is shown as MLI = L/ASN, which is used to estimate an average diameter of a single alveolus in size.
MAN was determined according to alveolar number (AN) in each field of view and a square area (SA) of the field. A formula is shown as MAN = AN/SA (mm2), which is an indicator for density of alveoli.
Measurement of pulmonary artery pressure and ventricular hypertrophy
Mean pulmonary artery pressure (mPAP) was determined by using a technique of right heart catheterization via the right external jugular vein after rats were sacrificed. mPAP was recorded through a pressure transducer connected to the animals and a change in mPAP was obtained in multi-channel physiological recorder (Chengdu Instrument Factory, China).
We dissected the right ventricular free wall from its atrial, septal and valve ring attachments and carefully removed epicardial fat. The right ventricular free wall was then flattened out (endocardial surface down) and blotted on filter paper for mass determination by weight. The left ventricle, including the septum (S), was similarly weighed after removal of atrial and valvular attachments and epicardial fat. A right ventricular hypertrophy index (RVHI) was shown as RVHI = RV/(LV+S).
Determination of 5-HTT in serum and BALF
5-HTT contents in serum and BALF were assessed by ELISA according to the manufacturer’s instructions (BIO-TEK, USA) and all samples were analyzed in one assay per analyte. The detection limit for the primary antibody (murine anti-5-HTT) was 5 ng/ml (Cusabio Biotech Co., Ltd, USA). Briefly, 100 μl of the substrate solution (components?) was added to 100 μl of sample in microtiter plates and incubated for 30 min at room temperature. The reaction was stopped by adding 50 μl of 4 M sulfuric acid, and the OD values were read in a microtiter autoreader at 490 nm.
Statistic analysis
Values were expressed mean ± standard deviation (SD) on the results. Statistical analysis was performed using Statistical Package for the Social Science (SPSS, version 17.0). Comparisons from groups were performed by one-way analysis of variance (ANOVA). Student’s paired t-test was used to compare measurements of individual groups. Significance was accepted at P < 0.05.
Results
Histopathologic analysis of lungs and change in body weight
The lower lobe of right lung was removed from experimental animals exposed to either air or cigarette smoke in presence and absence of simvastatin (5 mg/kg, once a day for 4 months). Sectional tissues of the lung stained with H&E were photographed by light microscope at × 400 magnification and the results are shown in Figure 1A. Morphologically, rats in control group showed a normal structure of lungs with an intact epithelium layer. Those of rats were free of cellular infiltration and mucus observed in the lung sections. With the exposure to cigarette smoke, the histopathologic features showed some of airway epithelium was denuded and/or patchy shedding of epithelial cells on the luminal side of bronchus in the lung with an increase in airway wall thickness and airway narrowing. Peribronchiolar inflammation was clearly observed with a large amount of inflammatory cells infiltration around the airways. Furthermore, an enlargement of alveolar airspaces, destruction of septal walls of alveoli and pulmonary bullae as a morphological change of emphysema were visual due to some alveolar fusion in the sectional tissue. Treat- ment with simvastatin may relieve shedding of epithelial cells and accumulation of inflammatory cells observed around the airway, whereas there was only focal emphysema seen in a limited area of the lungs as compared to the pathological changes in the lungs from the smoking rats.
Figure 1.

Morphological analysis in lungs and change in weight gain. Rats were exposed to air (control) and cigarette smoke (smoking) in presence and absence of simvastatin. Histological structures of lungs, the status of airway epithelial layer and alveolar septa, and inflammatory cell infiltration were observed on frozen sections of lung specimens stained with H&E (A). Histopathologic examination was performed under a light microscopy at a magnification of × 400. Body weight of rats was examined before exposure to cigarette smoke (BW) and after its last exposure (AW), respectively (B). Data were expressed as Mean ± SD (n = 10).
Body weight of rats was examined prior to cigarette smoking and after its last challenge, respectively. Weight gain was obviously different and the results are shown in Figure 1B. In contrast, there were no changes in measurement of body weight of the grouped rats prior to the challenge. Weight gain (g) of the rats after its last challenges was 385.4 in control rats, 348.3 and 364.3 in the smoking rats treated without and with simvastatin, respectively. There was a statistical difference observed in the weight gain between control and smoking rats (P < 0.01). Furthermore, there was a significant difference in weight gain between smoking rats treated with and without simvastatin (P < 0.05). However, there was a significant change in weight gain between control rats and the simvastatin-treated ones (P < 0.01).
Size and number of alveoli
Lungs were surgically removed from the experimental rats and then applied to evaluate a change in size of air spaces in the lungs using the MLI measurement technique. The results are shown in Figure 2A. The average values (mm) of MLI were shown as 41.27 ± 2.74 in control rats, 97.80 ± 6.99 in smoking rats and 90.70 ± 6.84 in the simvastatin-treated rats, respectively. In contrast, enlargement of air spaces in the lungs was obvious in the cigarette smoke-explored rats as compared to either control or the simvastatin-treated rats (P < 0.01 or 0.05). Treatment with simvastatin may slightly but significantly reduce number of air spaces, but there was a significant difference in the MLI measurement between control and the simvastatin-treated rats (P < 0.01).
Figure 2.
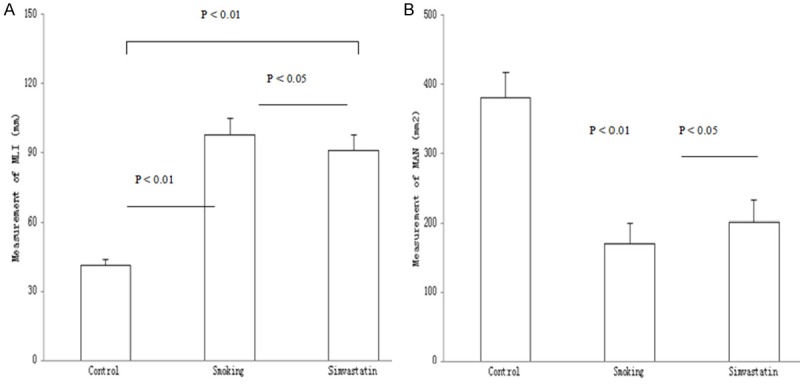
Morphometry of lung. Rats were exposed to air (control) and cigarette smoke (smoking) in presence and absence of simvastatin. Size of air spaces in the lungs was evaluated in the mean linear intercept (MLI) measurement technique (A). The average values of alveolar density (μm2) were examined by calculating alveolar number in microscopic vision of lung tissue (B). Measurements were expressed as Mean ± SD (n = 10).
Alveolar density was examined by calculating MAN in a microscopic vision of the lung tissue and the results are shown in Figure 2B. The average values of alveolar density (alveolar number/mm2) were 381.05 ± 35.76 in control rats, 170.61 ± 29.60 in smoking rats and 201.37 ± 30.43 in the simvastatin-treated rats, respectively. In contrast, there was a statistical difference in the MAN measurement between control and smoking rats (P < 0.01). Treatment with simvastatin may slightly increase MAN in the lungs with a statistical difference as compared to the smoking rats (P < 0.05). However, there was a significant difference in MAN between control and the simvastatin-treated rats (P < 0.01).
Observation of lung function
FEV0.3/FVC and Cdy were determined at the end of the challenge procedure and the results are shown in Figure 3A, 3B. The average values (%) for FVC/FVC0.3 and Cdy were shown as 52.8 ± 8.1 and 0.0360 ± 0.0028 in control rats, 35.8 ± 6.7 and 0.0296 ± 0.0024 in smoking rats and 42.2 ± 5.1 and 0.0324 ± 0.0023 in the simvastatin-treated rats, respectively. In contrast, the values of FVC/FVC0.3 and Cdy in the cigarette smoke-challenged rats were lower than the PBS-challenged rats, whereas the lowering FVC/FVC0.3 and Cdy in the challenged rats may be partly reversed in the rats treated with simvastatin (P < 0.05). However, there was a significant difference in the measurements between control and the simvastatin-treated rats (both P < 0.01).
Figure 3.
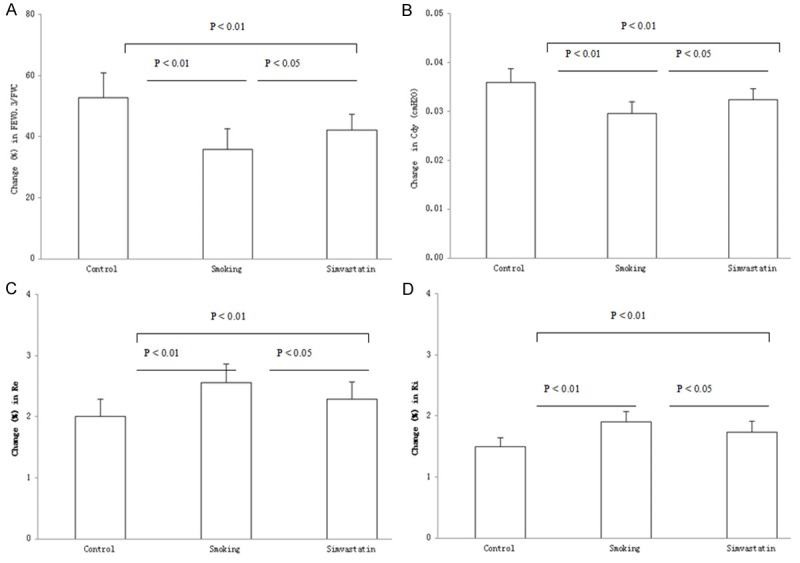
Lung function testing. A ratio of forced expiratory volume at 0.3 s and forced vital capacity (FEV0.3/FVC) (A), dynamic lung compliance (Cdy) (B) and respiratory resistance during expiratory (Re) (C) and inspiratory (Ri) (D) phases of tidal breathing were examined in control) and smoking rats treated with and without simvastatin. Measurements were expressed as Mean ± SD (n = 10).
Ri and Re were synchronously determined with the examinations of FEV0.3/FVC and Cdy in the rats and the results are shown in Figure 3C, 3D. The average values (cm H2O) for Re and Ri were shown as 2.01 ± 0.28 and 1.50 ± 0.14 in control rats, 2.56 ± 0.30 and 1.90 ± 0.17 in smoking rats, and 2.29 ± 0.28 and 1.73 ± 0.18 in the simvastatin-treated rats, respectively. In contrast, Re and Ri in the cigarette smoke-exposed rats were significantly increased over the control rats (both P < 0.01). Treatment with simvastatin resulted in a significant decrease in the airway resistance in the challenged rats (both P < 0.05). However, there was a significant difference in the Re and Ri measurements between control and the simvastatin-treated rats (both P < 0.01).
Changes in pulmonary artery pressure and RVHI
mPAP and RVHI were determined in the experimental rats and the results are shown in Figure 4A, 4B. The values for both mPAP (mmHg) and RVHI were shown as 17.27 ± 01.40 and 0.200 ± 0.007 in control rats, 29.81 ± 1.55 and 0.223 ± 0.011 in the challenged rats, and 20.04 ± 1.50 and 0.208 ± 0.007 in the challenged rats treated with simvastatin, respectively. The values for both mPAP and RVHI were increased in the challenged rats as compared to control and the simvastatin-treated rats (both P < 0.01). Though treatment with simvastatin may significantly lower mPAP and reduce RVHI, there was a statistical difference observed in the measurements between control and the simvastatin-treated rats (P < 0.01 or 0.05).
Figure 4.

Changes in pulmonary artery pressure and ratio of right/left ventricular mass. Mean pulmonary artery pressure (mPAP) was assessed using right heart catheterization in rats exposed to air (control) and cigarette smoke (smoking) in presence and absence of simvastatin (A). Right ventricular hypertrophy index (RVHI) was examined by measuring mass of right and left ventricular wall and ventricle septum (B). It was calculated with a formula shown as RVHI = right ventricular wall/(left ventricular wall + ventricular septal). Measurements were expressed as Mean ± SD (n = 10).
5-HTT expression level in BALF and serum
A 5-HTT expression level in BALF and serum was assessed in the experimental rats and the results are shown in Figure 5A, 5B. The values for 5-HTT contents (pg/ml) in BALF and serum were 46.64 ± 9.36 and 25.93 ± 3.84 in control rats, 108.61 ± 38.35 and 99.38 ± 40.71 in smoking rats, and 78.03 ± 20.08 and 65.96 ± 34.39 in the simvastatin-treated rats, respectively. In contrast, the protein contents in the samples from smoking rats showed a 3.83- and 1.51-fold increase in serum and a 2.33- and 1.39-fold increase in BALF over control and the simvastatin-treated rats, respectively. 5-HTT expression levels in BALF and serum were significantly increased in the challenged rats as compared to the control rats (both P < 0.01). Treatment with simvastatin resulted in a significant decrease in the 5-HTT protein contents in BALF and serum (both P < 0.05). However, there was a statistical difference observed in the protein contents between control and the simvastatin-treated rats (both P < 0.01).
Figure 5.
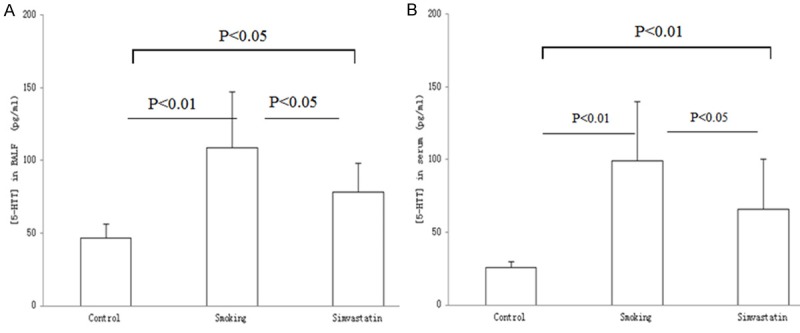
5-HTT content in BALF and serum. A 5-HTT expression level in BALF (A) and serum (B) were determined in rats exposed to air (control) and cigarette smoke (smoking) in presence and absence of simvastatin, respectively. 5-HTT contents in the investigated samples were obviously different from the treated rats. Measurements were expressed as Mean ± SD (n = 10).
Correlation between a 5-HTT expression level and either RVHI or mPAP was examined and the results are shown in Table 1. In association with an increased 5-HTT content in the investigated samples, the values of RVHI or mPAP in the cigarette smoke-exposed rats were significantly enhanced with a positive correlation between the 5-HTT level and either RVHI or mPAP in this model of COPD.
Table 1.
Correlation between 5-HTT level and RVHI or mPAP in the challenged rats
| BALF (r-value) | Serum (r-value) | P value | |
|---|---|---|---|
| 5-HTT vs RVHI | 0.508 | 0.479 | < 0.01 |
| 5-HTT vs mPAP | 0.675 | 0.693 | < 0.01 |
Discussion
The animal model of COPD was first established through a protocol modified by a 4-month challenge for the rats exposed to a high level of smoking (20 cigarettes, twice a day) in a smoking apparatus. Low power photomicrograph of COPD-like lesion in the challenged lung tissues showed obviously peribronchiolar inflammation with a large amount of inflammatory cells infiltration around the small airways, whereas an enlargement of alveolar airspaces, destruction of septal walls of alveoli and pulmonary bullae formed in the lungs were also observed under a microscope as compared to control. These pathologic features of the lungs indicated that the rat model of cigarette smoke-induced COPD was well developed. Treatment with simvastatin alleviated morphological impairments in the challenged lungs, suggesting that this agent may help slow down the progression of the disease. Since simvastatin is of the nature of anti-inflammation which could influence inflammatory airway disease [12], it lead us to speculate that its effect on the pathological changes was produced due to the results of ameliorating inflammatory response in the aggressive lung disease. In clinical practice, weight loss and malnutrition are a very common complication in COPD patients [13], whereas these changes primarily affect the patients’ quality of life and thereby they are also independent prognostic indicators of both morbidity and mortality [14]. In investigating nutritional status, weight gain in smoking rats was significantly decreased as compared to control rats in this model. Treatment with simvastatin showed a slight but significant increase in body weight of the rats, suggesting that the agent had a beneficial effect on nutritional support. In association with the findings in the challenged lungs, it’s likely that simvastatin provided the benefit for the smoking rats probably through alleviating inflammatory response in the lungs to lower resting energy expenditure due to the response.
In support of the above-stated pathological findings, MLI and MAN in the animal model of COPD were also measured in understanding distal air space size and alveolar density in the challenged lungs because the measurements of both morphometric techniques have been involved in the direct and unbiased estimation for the quantitative analysis of lung structure [15,16]. The results showed that enlargement of air spaces was obvious with a significant decrease in alveolar number detected in the smoke-exposed lungs as compared to control. These provided a direct evidence of lung injury, which was completely identical to the microscopic findings in this model. Simvastatin applied for the smoking rats partly reduced the structural damages in the challenged lungs, it led us to conclude that agent attenuated but not terminated lung lesions in its therapeutic benefits. Since MLI and MAN as an accurate and efficient stereological approach are a key structural determinant of the alveolar airspace size and lung architecture, it was reasonable to speculate that simvastatin may protect the rats against smoke-induced lung injury rather than altering the remolded lung structures.
Since the presence of airflow obstruction is key in diagnosing COPD and reduction of FEV3/FVC ratio is an indicator of mild lung injury in obstructive lung disease [17], FEV3/FVC, Cdy, Re and Ri were tested in the experimental animals, respectively. Data showed a significant decrease in FEV3/FVC and Cdy concomitantly with an obvious increase in Re and Ri in the smoking rats as compared to control, demonstrating lung function decline with certain degree of severity having been reached in this model of COPD. Treatment with simvastatin partly but significantly reversed the function decline, suggesting that the drug intervention was effective in ameliorating airflow obstruction of small airway based on the pathological findings of the lung from the smoke-exposed rats treated with this agent. It needs to point out that the parameters of lung function tests were recorded using such a method regarding 6.0 ml air injected into the lungs to expand chest of the animals. The method used in this study offers an advantage over agonist stimulation because the airflow response is more related to the condition of in vivo airway obstruction and/or narrowing.
COPD is the most common cause of PH and as the disease progresses, signs of right ventricular dysfunction may develop (Cor pulmonale) because of a maladaptive response to PH [18]. In this study, mPAP and RVHI were examined based on the consideration that development of PH secondary to COPD and Cor pulmonale adversely affect survival. The data showed a significant increase in mPAP and HVHI in the smoking rats as compared to control. Treatment with simvastatin resulted in a significant decrease in both measurements, indicating that the agent may mitigate remodeling of pulmonary circulation and right ventricular hypertrophy in the major therapeutic effort. It has been reported that simvastatin ameliorated the structural and functional derangement of rat lungs, partly by preventing pulmonary vascular abnormality [19]. Moreover, the simvastatin-treated rats displayed reduced PH and right ventricular pressure [20]. Thus it is conceivable that the effects of this agent on mPAP and RVHI are achieved with a major target lacked on PH that is a common cause of right ventricular hypertrophy.
In association with the increases in mPAP and RVHI, a 5-HTT expression level in serum and BALF was examined since it may be a key determinant of pulmonary vessel remodeling in addition to contributing to the uptake of 5-HT passing through the lung [21]. The results indicated that the 5-HTT levels in serum and BALF from smoking rats were a 2.33- and 3.83-fold increase over control, respectively. Furthermore, the 5-HTT contents in the investigated samples correlated to development of mPAP and RVHI. It has been clear that 5-HTT is a membrane bound protein that controls plasma level of 5-HT which is one of the most potent naturally occurring pulmonary vasoconstrictors [22]. Even though the physiological role of the 5-HTT protein in cardiac cells and pulmonary vascular smooth muscle cells has not yet been completely elucidated, these findings strongly supported such a conclusion that an increase in 5-HTT expression level not only contributed to remodeling of pulmonary circulation and morphological changes of right ventricle but also correlated with their severity in the diseased model. Treatment with simvastatin partly but significantly reduced 5-HTT protein expression in the samples from the smoking rats, strongly supporting the findings regarding its effects on PH and RVHI. Moreover, it was showing a clinical interest that the predominant effect of this agent on PH and RVHI was considered in lowering PH through down-regulating the 5-HTT expression level because the right ventricle gradually underwent hypertrophy in response to the increased PH [23]. Our findings could arise important clues to simvastatin’s role in such an animal model and thus may provide information for a therapeutic opportunity in COPD patients with reduced mortality and medical costs.
In conclusion, a rat model of COPD is well-established with a chronic exposure to a high level of smoking. Simvastatin partly reverses lung function decline and attenuates remodeling of pulmonary circulation and structural changes of lung and right ventricle possibly through reducing 5-HTT contents in the rat model of COPD.
Disclosure of conflict of interest
None.
References
- 1.Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765–773. doi: 10.1016/S0140-6736(07)61380-4. [DOI] [PubMed] [Google Scholar]
- 2.Rabe KF, Beghe B, Luppi F, Fabbri LM. Update in chronic obstructive pulmonary disease 2006. Am J Respir Crit Care Med. 2007;175:1222–1232. doi: 10.1164/rccm.200704-586UP. [DOI] [PubMed] [Google Scholar]
- 3.Bohadana A, Teculescu D, Martinet Y. Mechanisms of chronic airway obstruction in smokers. Respir Med. 2004;98:139–151. doi: 10.1016/j.rmed.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Naeije R, Barberà JA. Pulmonary hypertension associated with COPD. Critical Care. 2001;5:286–289. doi: 10.1186/cc1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lau WK, Chan-Yeung MM, Yip BH, Cheung AH, Ip MS, Mak JCW. The role of circulating serotonin in the development of chronic obstructive pulmonary disease. PLoS One. 2012;7:e31617. doi: 10.1371/journal.pone.0031617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108:1141–1150. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eddahibi S, Chaouat A, Morrell N, Fadel E, Fuhrman C, Bugnet AS, Dartevelle P, Housset B, Hamon M, Weitzenblum E, Adnot S. Polymorphism of the Serotonin Transporter Gene and Pulmonary Hypertension in Chronic Obstructive Pulmonary Disease. Circulation. 2003;108:1839–44. doi: 10.1161/01.CIR.0000091409.53101.E8. [DOI] [PubMed] [Google Scholar]
- 8.Ulrich S, Hersberger M, Fischler M, Nussbaumer-Ochsner Y, Treder U, Russi EW, Speich R. Genetic polymorphisms of the serotonin transporter, but not the 2a receptor or nitric oxide synthetase, are associated with pulmonary hypertension in chronic obstructive pulmonary disease. Respiration. 2010;79:288–295. doi: 10.1159/000226243. [DOI] [PubMed] [Google Scholar]
- 9.Palinski W. New evidence for beneficial effects of statins unrelated to lipid lowering. Arterioscler Thromb Vasc Biol. 2001;21:3–5. doi: 10.1161/01.atv.21.1.3. [DOI] [PubMed] [Google Scholar]
- 10.Davignon J, Leiter LA. Ongoing clinical trials of the pleiotropic effects of statins. Vasc Health Risk Manag. 2005;1:29–40. doi: 10.2147/vhrm.1.1.29.58937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin effects beyond lipid lowering: are they clinically relevant? Eur Heart J. 2003;24:225–248. doi: 10.1016/s0195-668x(02)00419-0. [DOI] [PubMed] [Google Scholar]
- 12.Hothersall E, McSharry C, Thomson NC. Potential therapeutic role for statins in respiratory disease. Thorax. 2006;61:729–734. doi: 10.1136/thx.2005.057976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ezzell L, Jensen GL. Malnutrition in chronic obstructive pulmonary disease. Am J Clin Nutr. 2000;72:1415–6. doi: 10.1093/ajcn/72.6.1415. [DOI] [PubMed] [Google Scholar]
- 14.Schols WJ, Slangen J, Volovics L, Wouters EFM. Weight loss is a reversible factor in the prognosis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:1791–7. doi: 10.1164/ajrccm.157.6.9705017. [DOI] [PubMed] [Google Scholar]
- 15.Andersen MP, Parham AR, Waldrep JC, McKenzie WN, Dhand R. Alveolar fractal box dimension inversely correlates with mean linear intercept in mice with elastase-induced emphysema. Int J Chron Obstruct Pulmon Dis. 2012;7:235–43. doi: 10.2147/COPD.S26493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochs M, Nyengaard JR, Jung A, Knudsen L, Voigt M, Wahlers T, Richter J, Gundersen HJ. The number of alveoli in the human lung. Am J Respir Crit Care Med. 2004;169:120–4. doi: 10.1164/rccm.200308-1107OC. [DOI] [PubMed] [Google Scholar]
- 17.Morris ZQ, Coz A, Starosta D. An isolated reduction of the FEV3/FVC ratio is an indicator of mild lung injury. Chest. 2013;144:1117–23. doi: 10.1378/chest.12-2816. [DOI] [PubMed] [Google Scholar]
- 18.Weitzenblum E, Chaouat A. Cor pulmonale. Chron Respir Dis. 2009;6:177–85. doi: 10.1177/1479972309104664. [DOI] [PubMed] [Google Scholar]
- 19.Lee JH, Lee DS, Kim EK, Choe KH, Oh YM, Shim TS, Kim SE, Lee YS, Lee SD. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. 2005;172:987–993. doi: 10.1164/rccm.200501-041OC. [DOI] [PubMed] [Google Scholar]
- 20.Nishimura T, Faul JL, Berry GJ, Vaszar LT, Qiu D, Pearl RG, Kao PN. Simvastatin Attenuates Smooth Muscle Neointimal Proliferation and Pulmonary Hypertension in Rats. Am J Respir Crit Care Med. 2002;166:1403–1408. doi: 10.1164/rccm.200203-268OC. [DOI] [PubMed] [Google Scholar]
- 21.Eddahibi S, Hanoun N, Lanfumey L, Lesch KP, Raffestin B, Hamon M, Adnot S. Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J Clin Invest. 2000;105:1555–1562. doi: 10.1172/JCI8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacLean MR, Herve P, Eddahibi S, Adnot S. 5-hydroxytryptamine and the pulmonary circulation: receptors, transporters and relevance to pulmonary arterial hypertension. Br J Pharmacol. 2000;131:161–168. doi: 10.1038/sj.bjp.0703570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shujaat A, Minkin R, Eden E. Pulmonary hypertension and chronic cor pulmonale in COPD. Int J Chron Obstruct Pulmon Dis. 2007;2:273–282. [PMC free article] [PubMed] [Google Scholar]