

Abstract
Autophagy is an important constitutive intracellular catalytic process that occurs in basal conditions, as well as during stress in all tissues. It is induced during cellular growth, tissue differentiation and metabolic demands. The regulated expression is cytoprotective while its deregulation leads to varieties of diseases. It plays a vital role in ischemic heart disease, being beneficial and adaptive during ischemia while detrimental and lethal during reperfusion. Reperfusion injury is the consequence of this deregulated autophagy and the motive of its persistence during reperfusion is still obscure. A long standing debate persists as to the dual nature of autophagy and defining its clearer role in cell death as compared to the widely studied process, apoptosis. Despite the progresses in understanding of the process and identification of critical mediators, there is no therapeutic strategy to address its final outcome, the reperfusion injury. This lack of effective therapeutic strategies has even questioned the validity of the process as a single entity. We still continue to witness the devastation with standard cure of reperfusion. In this article, we review the process, highlight reperfusion injury and outline important studies being conducted for the prevention of reperfusion injury and offer cardio-protection.
Keywords: Autophagy, cardiomyocytes, myocardial ischemia, myocardial reperfusion, Ischemia/reperfusion injury
Introduction
Ischemic heart disease (IHD) is the leading cause of death worldwide [1] and continues to rise. The global burden of disease studies, carried out by the World Health Organization and the World Bank projects the number of deaths to reach 11.1 million (from 6.2 million in 1990) by 2020, which can be seen on their website [2]. The subsets of IHD; angina pectoris, unstable angina and myocardial infarction (MI) differ in the severity, frequency and duration of sign and symptoms. These diseases share a common pathology of atherosclerotic plaque buildup in the intima of coronary artery and clinically manifest according to the severity of coronary obstruction. MI, commonly called as ‘heart attack’ is the most severe form of IHD. The standard mode of cure is re-establishment of coronary perfusion (reperfusion) by thrombolytic therapy, primary percutaneous intervention (PPCI) or coronary artery bypass grafting (CABG). This has been a revolutionary strategy that has limited death and morbidity associated with MI, but reperfusion gives rise to a newer form of ailment called as reperfusion injury (RI) that is responsible for post reperfusion cardiac morbidities and heart failure. Moreover, a rough estimate states that there are about 1.6 million patients that undergo some form of reperfusion therapy in hospitals and specialist clinics in the western world alone [3] and the total number worldwide would be alarming. These data cannot be taken as a reference but the severity can be true. Validation of newer therapeutic targets and strategies, which could limit RI associated morbidities, is still an unmet clinical need.
Autophagy is functional from the very embryonic period [4] and plays a fundamental role in growth and differentiation of body tissues. Its dynamic role during muscle development in vital tissues like the heart, diaphragm and alveolar cells makes it a survival ensuring process [5,6]. It is responsible for nutrient homeostasis, energy salvage and degradation of old, malfunctioning organelles within a cell. Additionally, it has a cytoprotective component which fights the invading pathogens and provides immune protection [7]. In contrast to these salutary survival promoting aspects, autophagy is also a type II programmed cell death and can initiate cell death in different circumstances. This delirious autophagic response have been attributed to many diseases and disorders such as –neurodegenerative diseases [8], cancer [9], liver diseases [10], cardiac diseases [11], Metabolic syndromes [12], ageing [13], inflammations [14]. From the very inception, the process of autophagy has faced controversies regarding the cell death process and has been subjected into enormous cross talks with apoptosis, another well studied cell death process type I. Advances in research and medicine have established the process of autophagy, with its dual nature as a separate functioning entity with a purpose. The purpose for survival is well understood but the purpose for death is unknown. Numerous studies have been conducted and are being conducted with drugs targeting different pathways, which will be outline later in the text. Listed below in Table 1 are the overall effects of autophagy in basal conditions, conditions of stress and when the process gets deranged.
Table 1.
Role of autophagy in health and development
| Biological effects | Cytoprotective |
|
| |
| Embryo | Pre-implantation of fertilized oocyte, elimination of maternal mRNAs [15], elimination of paternal mitochondria [16], gastrulation, stem cell differentiation, placental development, organogenesis, differentiation of erythrocytes, adipocytes, lymphocytes [17], neuron [18] |
| Birth | Nutritional deficiency on initiating sucking. Fetus to neonate transition [5] |
| Adult | Adaptations to exogenous stimuli (Hormonal, growth factors, cell density loads, nutrient depletion, oxidative stress) |
| House keeping/quality control | Disposal of misfiled and damaged proteins aggregates [19] and dysfunctional organelles [20] |
| Nutrient Recycling | Organelle degradation end product–glucose, amino acids, fatty acids and nucleic acids recycling. |
| Fighting pathogens | Identification of ligands of different bacteria and viruses and engulfing them for degradation. |
| Immune protective | Thymic selection, Effector of TLR signaling, Effector of Th1/Th2 polarization, Antigen presentation |
|
| |
| Longevity | Anti ageing |
|
| |
| Defective autophagy | Neurodegenerative diseases-Huntington’s disease, Parkinson’s disease, Alzheimer’s Disease, Lafora Disease, Lysosomal Diseases |
| Cancer | |
| Liver disease-Hepatocellular carcinoma, Hepatitis, Fibrosis | |
| Metabolic syndromes-Diabetes, Obesity, pancreatitis | |
| Infectious disease | |
| Cardiac and musculoskeletal: Cardiomyopathy, hypertensive heart failure, Hypertrophy, ischemia/reperfusion injury, Pompe’s Disease | |
| Inflammation-Crohn’s Disease | |
The direct contribution of the process of autophagy in the development of reperfusion injury has been established and numerous researches aimed on minimizing this injury have been conducted and are being conducted.
This article provides an update on the process of autophagy and reperfusion injury as well as outlines significant studies and clinical trials at present.
Literature search methodology
For this review, the PubMed database was searched for articles concerning autophagy and its role in the development of ischemia, reperfusion injury which were published in English before July 2014. We used the search terms “Autophagy” and “Ischemia-Reperfusion Injury”. Clinical studies were considered if they evaluated the association of autophagy with the pathogenesis, pathological features, pharmacological targets in ischemia and reperfusion (I/R) injury, accordance to Author’s judgment.
Characteristics of autophagy
Overview of autophagy
Discovery of the process, word
The phenomenon of ‘self-eating’ was first observed by Ashford and Porter during 1962 in the rat liver cells [21]. The word ‘autophagy’ was coined by Belgian biochemist Christian de Duve in 1963 [22], which is Greek in origin and means “to eat” (phagy) “oneself” (auto). The cell gets rid of cytoplasmic contents, long lived proteins or even organelles by means of lysosomes, also known as-type II programmed cell death. Depending upon the route of cargo transport, autophagy is of three types; macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) [23]. Macroautophagy, hereafter termed as ‘autophagy’ engulfs the cytoplasmic content into autophagosomes while microautophagy utilizes lysosomal membrane which engulfs the cytoplasmic content. During CMA, a chaperone identifies a specific protein peptides or polypeptides in the cytosol. The autophagosomes and the chaperone transports the content for degradation to the lysosomes while microautophagy requires no transportation. Autophagy has been observed in invertebrates as well as in mammals. Many of the genes and proteins of autophagy were first discovered in yeast and have been named as autophagy related genes (Atg) and their respective proteins [24]. By now, more than 30 autophagy related genes (Atg) have been identified and many of them have homologues in mammalian cell [25].
Autophagy components
The process of autophagy comprises of triggers and stimulators, sensors and genetic regulators, autophagy related genes and proteins and the final effectors.
Signaling pathways
Trigger
Almost any signal can be a trigger for autophagy, some activating the pathway and some suppressing the pathway. By far, energy depletion and oxygen deficient environment are the most powerful triggers for stimulating autophagy, while the reverse environment inhibits autophagy. Similarly, the growth factors, hormones, receptors with cytokine activities, receptors with tyrosine kinase activities and receptors that recognize pathogen ligands can also activate autophagy. The low energy sensors activate the adenosine-monophosphate-activated protein kinase (AMPK) pathway or the Beclin-1/class III phophatidylinositol kinase (PI3K) pathway while hypoxia induces transcriptional factors and mediators for autophagy. The high energy sensor is mammalian target of rapamycin (mTOR) which on the other hand is a negative regulator of autophagy. In addition to the nutrient status within the cell, activation of cell surface receptors with tyrosine kinase activities such as endothelial derived growth factor receptor (EGFR), neurotropins (TrkA/B), fibroblast growth factor receptor (FGFR), platelet derived growth factor receptor (PDGFR) through adaptor protein GRB2, leads to series of phosphorylations and activations of Ras proteins and later Raf protein kinases. This Raf further activates mitogen activated-protein kinases (MAPK) like- extracellular signal regulated kinase 1/2 (ERK 1/2), phosphoprotein 38 (p38), c-jun N-terminal kinase (JNK), which is a positive regulator of autophagy process [26,27]. Similarly, invading pathogen express ligands that are recognized by toll like receptor (TRAIL-R), esp. TLR 7 which activate autophagy [28]. In the same manner, tumor necrosis factor receptor (TNF-R), that is activated by various cytokines can either induce survival autophagy via tumor necrosis factor receptor type-1 associated death domain protein (TRADD), ribosome-inactivating protein (RIP), tumor necrosis factor receptor-associated factor 2 (TRAF2), inhibitors of apoptosis (cIAP1) to nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) or can induce death autophagy via fas-associated protein with death domain (FADD) or procaspases 8 and 10 [29]. Additionally, BCL-2/adenovirus E1B 19kDa interacting protein (BNIP), death associated protein kinase (DAPK), dynamin related protein 1 (DRP-1) [30-32] have also been identified to stimulate autophagy in tissues. Here, we discuss autophagy in a cardiomyocyte in different settings.
Inhibitory pathway: Phosphatidylinositol 93.4.5)-triphosphate kinase class I (PI3K-I)-Akt-mTOR axis
With nutrient abundance that is modulated by signals from PI3Ks of cytokine receptors, G protein-coupled (GPC) receptors, growth factor receptors and integrin receptors, the PI3K-Akt-mTOR axis gets activated. The Akt exerts an inhibitory action on the assembly of tuberous sclerosis complex 1 and 2 (TCS1/TCS2), that relieves the inhibition of Rheb on mammalian target of rapamycin kinase 1 (mTORC1) thereby activating it. The mTORC1 [33] negatively regulates autophagy by inactivating Unc-51 like autophagy activating kinase 1 (ULK1 kinase) that consists of ULK1, Atg 13 and Atg 17.
Stimulatory pathways: AMPK axis, Beclin-1/PI3K III axis and the Hypoxia inducible factor(Hif-1)-Protein kinase C delta (PKCδ)–c-Jun N-terminal kinase 1 (JNK1) axis
The status of nutrient and energy depletion is sensed and modulated by liver kinase B1 (LKB1), Ca2+/calmodulin–dependent kinase kinase beta (CaMKKβ) and transforming growth factor β activated kinase-1 (TAK1), resulting in phosphorylation of threonin residue at 172 position and activation of AMPK [34]. AMPK, on one hand initiates autophagy by stimulating p27 gene that starts manufacturing autophagic machineries [35]; On the other hand it inhibits mTORC1 by dissociating TSC1 from TSC2, rendering it inactive. Similarly, hypoxia targets the hypoxia induced hypoxia inducible factor Hif-1- protein kinase C delta PKCδ-JNK1 axis [36]. This axis promotes interaction of Beclin-1 and phophotidylinositol 3 kinase class III (PI3K III), an essential step in the phagophore induction. Additionally, this interactions is also induced or promoted by nutrient depletion, especially amino acid depletion [37] (notably leucine depletion in cardiac tissue [38]) and molecular mediators like Ca2+ and reactive oxygen species (ROS). Moreover, organelle stress itself can directly activate or generate mediators that influence both pathways. For instance, DNA damage and oxidative stress can inactivate mTORC1 through sestrin, a protein induced by p53 gene, which culminates in mTORC1 inhibition [39]. Similarly, endoplasmic reticulum (ER) stress can activate endoplasmic reticulum kinase (PERK), which through a series of activation of eukaryotic translation initiation factor 2α (eIF2α) kinase Fcn2 (another energy sensor), induces its downstream target of Gcn4 [40], and can activate autophagy related genes to initiate the manufacturing of autophagic proteins [41]. Likewise, another protein, regulated in development and DNA damage responses-1 (REDD-1) can be activated by hypoxia and can directly disrupt the assembly of TSC1 and TSC2 [42]. Moreover, oxidative, genotoxic stress and organelle stress can induce nuclear transcription factor p53 and activate autophagy by transactivating AMPK [43], while the cytoplasmic p53 protein can inhibit autophagy by activating mTORC1. The key steps and interactions of pathways of autophagy is outlined in Figure 1.
Figure 1.
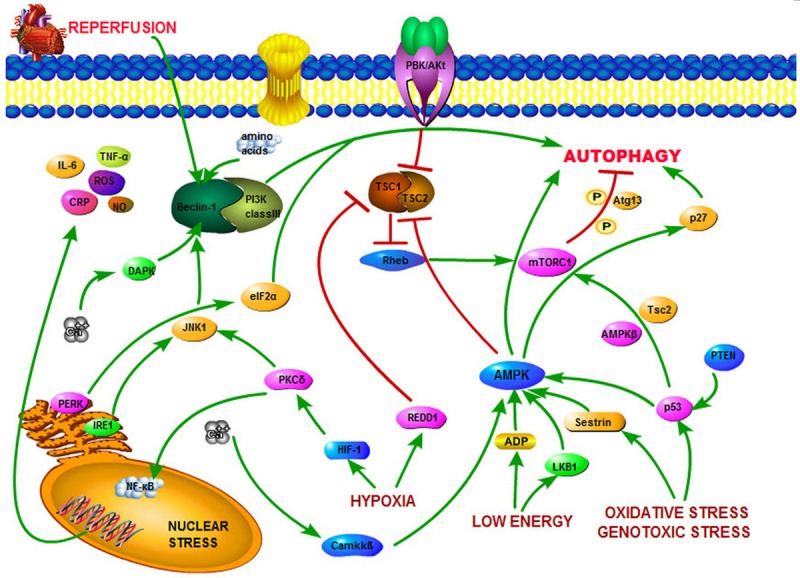
Important autophagic pathway during ischemia and reperfusion: The inhibitory pathway mediated by PI3P-Akt-mTORC1 halts autophagy whiles the stimulatory pathways-AMPK and Beclin-1-PI3K initiates autophagy. Reperfusion is also seen to start the autophagy activation pathway.
Reperfusion
It is logical at this point to state that besides the quality and quantity of previous blood flow, perfusion and reperfusion share the same physiology and should follow the same inhibitory pathway of autophagy. Surprisingly, with unexplained processes and undiscovered molecular mediators, Beclin-1 and microtubule associated protein 1 (MAP 1) light chain 3 (LC3), which is a marker of autophagosome maturation is reported to be elevated, signifying the ongoing autophagy and cellular damage [44]. This form of cellular death goes against the theoretical principles of survival due to nutrient and oxygen abundance and has indeed challenged reperfusion strategy in different fields, notably cardiology.
Key mediator molecules
Reactive oxygen species (ROS) is well known that hypoxia, organelle stress (especially the mitochondrial and endoplasmic reticulum [45]) and dysfunctional enzymes during myocardial ischemia generate tremendous amount of ROS. ROS is a powerful activator of starvation-induced autophagy [46], that causes oxidative damage to the organelles and lipid perioxidation of lipid membranes especially in the mitochondria, which leads to fatal cell death [47]. Generation of ROS continues or increases from the mitochondria even with the commencement of myocardial reperfusion by a mechanism called ROS-induced ROS release when electron transport resumes [48]. Besides cellular damages, ROS is also responsible for post-ischemic contractile dysfunction, called ‘myocardial stunning’ where the heart muscles show reduced contractions after reperfusion [49] although it is reversible.
Calcium is another molecule that gets accumulated and propagates ischemia/reperfusion (I/R) injury in the cell. The lack of ATP with the resultant anaerobic glycolysis generates lactic acid that makes the environment acidic. To neutralize this environment initially sodium comes inside the cell in exchange of hydrogen ion and with time to get rid of excess sodium from the cell, calcium comes in and sodium ion is extruded [50]. This becomes the last step that the cell can offer to maintain homeostasis. With no reversal of ischemia, piling up of calcium ensues. Simultaneously, the ER stress also spills Ca2+ into the cytosol. This calcium then activates ROS and induces opening of the mitochondrial permeability transition pore (mPTP), cessation of electron transport chain, loss of membrane potential, matrix swelling, inner membrane remodeling and finally outer membrane rupture with subsequent release of mitochondrial cytochrome C and other apoptogenic proteins [51]. This leads to intracellular catastrophe. Reperfusion, adds more and further brings in calcium that further induces ROS and exacerbate damage by disrupting the plasma membranes, damage to the sarcoplasmic reticulum and mitochondrial re-energization [52].
Beclin-1 protein plays a crucial role in the induction and maturation of autphagosome. It is a mammalian ortholog of yeast Atg 6. Beclin-1 is normally in an inhibited state being bound to Bcl-2 protein (anti-apoptotic), and localized in the endoplasmic reticulum. This molecule is seen to demonstrate a dual nature of action in experimental models. It is essential for cellular survival in mice L292 fibroblasts [53] and murine embryonic fibroblasts (MEFs) [54]. On the contrary, it is a necessary component of autophagy mediated cell death and this has been documented by genetic suppression of Beclin-1, that resulted in augmentation (rather than impairment) of cellular survival in cardiomyocytes [55]. The exact trigger for Beclin-1 activation is unclear but some studies have identified intracellular amino acids depletion [19], and activated ROS [56] as a strong stimulus. Similarly, nutrient depletion itself weakens the bond between Beclin-1 and Bcl-2 thus liberating Beclin-1(which is its active form). The reason of expression of Beclin-1 during reperfusion is still a mystery but some undiscovered mediators may be activating Beclin-1 directly, may be weakening the Beclin-1-Bcl-2 bond or may be inhibiting the Bcl-2 molecule itself. These hypotheses need more research and validity. Moreover, Beclin-1 inhibition or activation by Bcl-2 protein has been seen an indirect control of autophagy by apoptosis. Thus, Beclin-1 proteins role at the juncture of cellular death or survival and autophagy or apotosis has invited numerous cross talks among scientists. Identification of the ‘switch’ that turns one process on and the other off and the vice versa has been a platform of researches.
The release of Bcl-2 homology domain 3 (BH3) -only proteins occurs in response to hypoxia and release of ROS. It is the downstream target of HIF-1, and can trigger autophagy by controlling the activities of pores and channels in the outer mitochondrial membrane, inducing mPTP opening and mitochondrial damage [57]. Not only during autophagy, but its role on initiating apoptosis mediated cell death in ventricular myocytes [58] has also been documented. Some of the studies have also reported that BNIP protein not only influences but also up regulates autophagy, the purpose being for the removal of damaged mitochondria to confer cardioprotection [59]. These contrasting roles of BNIP protein during autophagy has also been a subject of researches and crosstalks. Till date, 6 BH3 proteins (BNIP3, PUMA, Bid, Bad, HGTD-P, Noxa) have been identified that take part in ischemia-reperfusion death pathways in heart and/or brain [46].
Autophagosome formation and execution
As mentioned earlier, any form of stress triggers autophagosome formation, which fuses with the lysosome with ultimate degradation of the sequestered material through the cytoplasm to vacuole (Cvt) pathway for degradation in autophagy [60]. Here, we outline only the molecules involved in the formation and execution of autophagy machinery, as the entire steps, interactions and the discussion is beyond the scope of this article.
Pre induction and induction
The initiation of autophagosome formation starts with protrusion of a phagophore where a portion of cell membrane cups off from the endoplasmic reticulum [61,62]. It is mediated by different inducers with their own pathways. The identified inducers of autophagy in mammals are-ULK1, Unc-51 like autophagy activating kinase 2 (ULK2), 200KD focal adenosine kinase (FAK) family kinase-interacting protein (FIP200), Atg 13, Atg 101, Atg 9 and WD repeat domain, phosphoinositide interacting 1 protein (WIPI-1) [63,64].
Vesicle nucleation
Manufacturing of a double layered membrane starts at the phagophore which is a complex process and occurs with the interaction of Class III PI3K and beclin-1 along with contributions from beclin-1 regulators such as- Barkor, BAX- interacting factor-1 (Bif-1), activating molecule for BECN 1 regulated autophagy protein 1 (AMBRA 1), ultraviolet-radiation resistance-associated gene (UVRAG) and p150 (mammalian homolog of yeast Vps15) [37,65]. This structure starts engulfing the cytoplasmic content.
Vesicle elongation and maturation
Further elongation of the newly formed membranous vacuolar structure occurs which is aided by the Atg 12 conjugation system, Atg 8 conjugation system, Atg 5, Atg 10, Atg 16L, Atg 4, LC3, Beclin-1, Atg 7, Atg 3, phosphatidylethanolamine (PE) and Atg 9 [66]. A mature autophagosome is then formed.
Vesicle fusion with lysosomes
The newly formed autophagosome fuses with lysosomes by the interactions with lysosomal membrane proteins, lysosomal-associated membrane protein 1 (LAMP-1), lysosomal-associated membrane protein 2 (LAMP-2), the small GTPase Ras related protein 7 (Rab7) and an ATPase called SKD1 [67].
Digestion and Disassembly of Atgs
Finally, the degradation of the inner vesicle along with its contents occur which is dependent on a series of lysosomal/vacuolar acid hydrolases, including Cathepsins B, D and L [68]. The small molecular end-products of degradation, including amino acids, fatty acids, glucose and nucleotides, are released into the cytosol by permeases for reuse. Similarly, disassembly of the Atg proteins occurs that are mediated by Atg 2, Atg 9 and Atg 18 (Figure 2) [69].
Figure 2.

The autophagic (macroautophagy) pathway for cellular degradation. The triggers initiate phagophore which starts sequestering cytoplasmic content. Phagophore then nucleates, elongates and matures. A mature autophagosomes fuses with the lysosomes where degradation of the sequestered cytoplasmic content occurs. The end product of degradation -glucose, amino acids, fatty acids, nucleic acids along with the Atg proteins are released into the cytosol for re-use.
I/R injury
It is clear that autophagic cellular damage during ischemia and reperfusion is the basis of reperfusion injury. This injury was firstly observed by Jenings et al. during 1960 in a canine model, who also noticed that reperfusion accelerated the process of necrosis formation [70]. Since then, numerous studies have been conducted that have broadened our understanding of the micro-process, the mechanical influences and the pharmacological interactions. This has exposed several possibilities in preventing RI. Many animal studies, in vitro studies and trials have been conducted so as to limit RI. So far the human trials and the enthusiastic search for therapeutic targets have been disappointing.
Reperfusion injury in a nutshell
With the commencement of reperfusion in cardiac tissues, there is increased blood flow that brings in nutrition, oxygen, polymorphonucler cells (PMNs) and platelets to a previously ischemic area. The electron transport in the functional mitochondria resumes. ROS production starts immediately within the first few minutes [71] by the activated neutrophils [72] as well as by a process called ROS-induced ROS release, besides the continuous ROS release from previously damaged organelles. Researchers have found that ROS targets and activates Beclin-1, regulatory factors, activating factors and the endothelium by which the neutrophils and platelets are attracted [73], leading to microvascular plugging. In addition to the ill effects on the cellular and organelle morphology, ROS also damages the sarcolemma with liberation of calcium to the pool that is already filled up with calcium due to acidotic environment. The balance of calium is altered and this disturbance in calcium homeostasis also plays an important role in the development of RI [74]. This calcium open the mPTP [75] and release caspases, second mitochondrial activator of caspase (SMAC), inhibitor of apoptosis (IAP) and endonucleases [76]. The calcium dependent protease (calpain I) are also activated that result in proteolysis of troponin I [77] with histological evidences of cell death [78]. The surviving myocardium too, is on the death pathway as demonstrated by the accumulation of autophagosomes and prominent up-regulation of lysosomal enzymes [79]. Besides these, a commonly observed feature of reperfusion is the rapid onset of contraction band necrosis [80] which marks the loss of plasticity and cell architecture. These processes are all interrelated and lead to reversible, treatable conditions such as ventricular arrhythmias [81] and myocardial stunning [82], but also can lead to irreversible condition like microvascular obstruction (MVO). MVO is defined as the “inability to reperfuse a previously ischemic region [83] grossly due to ultra structural damage to the microvasculature [84], endothelial abnormalities, neutrophil occlusion and erythrocyte stasis at the capillary level [75]. The presence of MVO has been demonstrated as sluggish coronary blood flow and impaired myocardial blush grade [85] even after successful reperfusion (PPCI). As a sequel of MI, the myocyte become edematous and necrotic, which can compress the microvasculature and increase distal vascular resistance, arteriolar spasm and progressive endothelial dysfunction [86]. This endothelial dysfunction in a severe case leads to endothelial damage and extravasation of blood into the interstitium. This form of intramyocardial hemorrhaging within the area of infarction has been demonstrated by cardiac MRI [87] after reperfusion. Additionally, if there were prior exposures of sublethal ischemia, the myoctes and tissue in course time develop adaptations, a form of cardioprotection called the ischemic preconditioning [88], which with reperfusion gets abolished and the drive for survival in a compromised environment is lost. Besides these, the myocardial tissues are also unable to mount a reactive hyperemic response leading to increased vascular resistance in a previously ischemic and damaged tissue, due to loss of endothelial nitric oxide synthase (eNOS) that cannot generate Nitric oxide (NO), a physiological vasculature modulator. Another commonly noticed feature of MI is hyperglycemia (even in the absence of diabetes mellitus). It has been implicated with further deterioration of left ventricular functioning after reperfusion [89] and increased mortality after MI [90]. All these microscopic changes collectively give rise to ‘reperfusion injury’ or in severe cases the ‘lethal reperfusion injury.’ The process of superimposed cell death by reperfusion injury is outlined in Figure 3.
Figure 3.
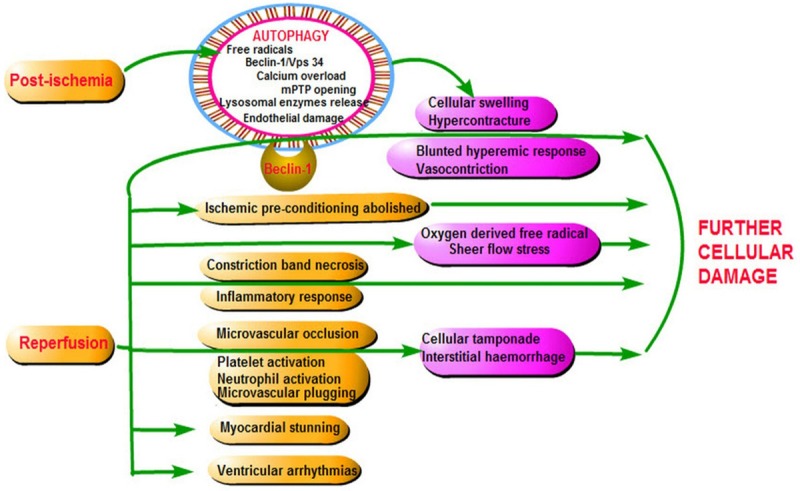
The overall effects of reperfusion on an ischemic cell. Reperfusion causes further cellular damage.
Autophagy and I/R injury in myocardium
Autopahgy starts in the ischemic period and continues or even increases during the later stages of reperfusion [82]. It is an attempt to survive the severely limiting conditions during ischemia, while it is a quest for survival following the overwhelming activation of cellular processes during reperfusion. The importance of I/R injury lies in the fact that each functional myocyte is vital for the normal cardiac functioning, as the turnover/regeneration is very slow; only about 1% of cardiomyocytes are renewed annually at the age of 25 which declines to 0.45%, at the age of 75 [91]. With this low and insufficient pool of myocytes at hand, reperfusion adds more insult as indirect evidences have demonstrated RI to contribute 40-50% of the final MI size [52]. The process of RI is not static, it is continuous as presence of MVO for up to 48 hours [92] and autophagy for up to 3 days in a previous ischemic area [93], have been documented. Opening an artery does not ensure perfusion as a phenomenon of vascular stasis called the ‘no reflow’ can occur which may last from 2 minutes to 3.5 hours [94]. The perfused myocardium can remain unresponsive (myocardial stunning) or even show irregular rhythm, contributed by persistant ROS activation, though reversible. RI can be perceived here, as a window that exposes the world of intracellular devastations of autophagy. The final outcomes of RI on the heart can be larger infarct size, lower left ventricular ejection fraction, adverse left ventricular remodeling and worse clinical outcomes [52].
Cross talk and the complex inter-relationship: autophagy and apoptosis
Apoptosis and autophagy as well as necrosis are the major processes of mammalian cell death. The former two differ from the latter by the feature of tissue inflammation. Apoptosis and autophagy both do not exhibit tissue inflammation and share many same death signals like ceramide, TNF-related apoptosis inducing ligand, FADD, DAPK, DAPK-1 [30-32,95]. The interactions between the two processes are complex demonstrating agonism, antagonism or synergism. There are numerous literatures highlighting individual interactions; here we outline some important ones.
Autophagic regulation of apoptosis
The proteins involved during autophagy such as Atg5 and Atg 12 [96], Beclin-1 take part in the regulation of apoptosis. Studies have also demonstrated a mutual co-reguation between the two where depletion of Atg 5 and Atg 3 was associated with suppression of caspase-8 activation and apoptosis [97]. Moreover, studies have also demonstrated that autophagy can regulate, sequester and eliminate caspase 8 [98], an effector of apoptosis.
Apoptotic regulation of autophagy
The apoptotic proteins such as Bcl-2, BCL-xl, and most of pro-apoptotic BH3 only proteins can initiate the process of autophagy. Similarly the negative regulation have also been observed in studies where the pro-apoptotic protein Bax [99] and Ste20-like kinase 1 (Mst1) [100] suppressed autophagy while the effectors of apoptosis, the caspases were also seen to inhibit autophagic proteins- Beclin-1, vps34, Atg3, Atg4D, AMBRA1 and p62 [101]. Studies have demonstrated cell death by apoptosis when autophagy is suppressed [79]. To add more, the inhibitor of apoptosis- the flice inhibitory protein (FLIP), has been found to be a negative regulator of autophagy [102].
With this diversity of interactions, some scholars have felt that the contributions of apoptosis on cell death far outweighs to that of autophagy, and have even suggested autophagy as being a smaller effector of the death process. Nonetheless, it stands out as a different entity with the hallmark of early organelle damage and late cytoskeleton derangement as compared to apoptosis where the reverse happens. There are distinctions in the histology and the roles played. Autophagy plays an added role of cellular renovation and sustenance [103] by means of ‘self eating’ as compared to the apoptotic role of ‘self killing’. Further researches are warranted to answer the controversies related to autophagy mediated cell death, death signals generation, death signal interaction and its pharmacological and genetic regulation.
Experimental studies within the last 5 years
There are numerous studies that have been conducted all over the world in light to autophagy and reperfusion injury. Researches, literatures and reviews have highlighted the findings of effective pharmaceutical agents and the list seems encouraging. However, most of the experimental studies face a setback while relating into larger trials or human studies due to discrepancies in reliability and reproducibility. The coronary artery and the biological environment differ significantly to that of experimental models. The presence of concomitant co-morbid conditions, latent preconditioned myocardium and the changes associated with medications makes the results variable. Besides, there are inherent complexities and dangers associated with conducting larger human trials which has given rise to numerous experimental studies. We would like to focus on the studies that have been conducted during the last five years.
Studies aimed at suppressing autophagy at the cellular level
Most of the experimental studies that have been conducted are seen to target the PI3K and the Akt pathway to inhibit autophagy. These molecules interact from the cell membranes and have been an easy target to reach. Newer drugs or drugs that have been beneficial have been tried. The final effect is observed as beneficial and cardioprotective or detrimental with elevated levels of autophagosomes, elevated levels of Beclin-1 or the autophagic markers LC3B-II. Besides the aforementioned target, AMPK, Beclin-1 and the paracrine pathways have also been targeted. These studies demand a broader version and translations into human studies. Most of these studies have utilized histological as well as radiological evidence to relate to cardioprotective effect such as decreased in infarct size, improved or good left vetricular functioning. Some of the recently studies autophagic pathways that showed cardioprotection with their respective drugs are outlined in Table 2.
Table 2.
Cardio protection delivering drugs, their interacting pathways on I/R injury: experimental/animal study during the last 5 years
| Target pathway | Pharmaceutical agents |
|---|---|
| PI3K/Akt | Salidroside (LY294002) decrease Bax and Caspase-3(2013) [104], Luteolin (polyphenol falvonoid) inhibit Bax and apoptosis (2011) [105], PPAR-alpha activation as a preconditioning like intervention (2012) [106]/PTCA induced ischemic postconditioning (2012) [107], Ligustrazine (TMP) by itself and by phosphorylating eNOS to produce NO(2012) [108], Sitagliptin via GLP-1/GLP-1 receptor (2013) [109] |
| Globular adiponectin modulate SERCA and suppress ER stress (2013) cardiomyocyte [110], Anandamide induction of HSP72 mediated by CB (2) and CB (1) receptors (2013) [111], Remote ischemic postconditioning by upregulating ALDH2 expressions (2014) [112] | |
| PI3K/ Akt and ERK1/2 (RISK pathway) | Epigallocatechin-3 gallate [113] |
| PI3K/AKT and ERK1/2- MEK1/2 | A3 adenosine receptor agonist, 2-CL-IB-MECA, inhibit caspase-3 (2014) [114] |
| PI3K/Akt and ERK1/2 | Berbamine (from Barberry) maintains intracellular calcium and prevents calpain activation (2012) cardiomyocytes [115] |
| PI3K/Akt and PKC pathway | Urantide (2012) [116] |
| PI3K and p38MAPK | Dobutamin-mediated heme oxygenase-1 induction (2013) [117], BNP post conditioning inhibit HMGB1, TNF-α and IL-6. (2014) [118] |
| PI3K/Akt/ERK | Intralipid (GSK-3β) inhibit of mPTP (2011) [119] |
| Achyranthes bidentata (herb) (2013) [120] | |
| PI3K/Akt and ERK1/ERK2 | Urocortin 2 autocrine/paracrine and pharmacological effects (2013) mouse ex vivo [121], GHRH through activation of RISK and SAFE pathway (2013) [122] |
| PI3K, Akt/PKCε/MitoKATP/ROS signaling | Catestatin (2013) [123] |
| PI3K/Akt/mTOR/SVV | Survivin potentiates the anti apoptotic effects of inslin. (2011) [124] |
| Arachidonic acid Akt/PKCα/β | t-AUBC inhibition of sHE enhances the activity of EETs (2010) [125] |
| Aldolase reductase modulates GSK-3β | |
| Phosphorylation (2012) [126] | |
| PI3K/PKG/GSK3β | ANP prevents mPTP opening by inactivating GSK3β (2012) [127], Sufentanil postconditioning (2012) [128] |
| Activation of RISK/GSK-3β and inhibition of p38 MAPK | Curcumin (turmeric) (2012) [129] |
| PI3K/Akt/GSK3β | Dansen (salvia miltiorrhiza) (2013) [130] |
| PI3K/Akt and JAK2/STAT3 pathways | Fasudil inhibits ROCK, attenuates ER stress and modulates SERCA activity (2012) [131] |
| PI3K and NO | Therapeutic hypothermia at 34°C (2012) [132] |
| PI3K/Akt/NDRG2 | Potentiates the anti-apoptotic effect of insulin (2013) [133] |
| GSK3β activation | Sevoflurane (2013) [134] |
| ROS signalling | Mitochondrial BK(Ca) (2013) [135] |
| Akt/PTEN pathway | Ischemic postconditioning-mediated miRNA-21 (2013) [136] |
| PI3K/Akt/FOXO3A/Bim pathway | Sodium tanshinone IIA sulfonate (2013) [137] |
| Paracrine pathway TIMP-1 | Cardiac fibroblasts (2014) [138] |
| AMPK/Akt/JNK signaling activation | Labdane diterpenes (2011) [139] |
| Rosiglitazone (2011) [140] | |
| AMPK pathway | CTRP9 protein (2012) [141], Berberine (2012) [142], Macrophage migration inhibition factor, MIF (2013) [143], Omentin (2014) [144] |
| AMPK/Akt | D-dopachrome tautomerase enzyme (2014) mice [145], Antithrombin (2014) [146] |
| AMPK | Propofol inhibition, phosphorylation of mTOR (2010) [147] |
| Beclin-1 | Ginsenoside Rg1 (2012) [148] |
| Inhibition of activation of AMPKα, Beclin-1, activation of mTOR, decreased level of LC3B-2 | Trimetazidime (2010) [149] |
| K+ channel opener | Nikorandil (2011) [150] |
Source: PubMed. Abbreviation: Akt/PKB: Ak strain transforming/protein kinase B, ALDH2: aldehyde dehydrogenase 2, ANP: atrial natriuretic peptide, BNP: brain natriuretic peptide, CB: cannabinoid receptor, CTRP9: C1q/TNF-related protein 9, EETs: epoxyeicosatrienoic acids, ERK: extracellular signal-regulated kinases, GHRH: growth hormone releasing hormone, GLP-1: glucagon-like-peptide-1, GSK-3β: glycogen synthase kinase3 beta, HMGB1: high-mobility group protein B1, HSP72: heat shock rotein72, IL-6: interleukin-6, JNK: c-jun N-terminal kinase, MAPK: mitogen-activated protein kinase, MEK: methyl ethyl ketone (butanone), NDRG: N-myc downstream-regulated gene, PI3K: phophatidylinositol kinase, PKC: protein kinase C, PPAR-α: perioxisome proliferator-activated receptor-alpha, RISK: reperfusion injury salvage kinases, RISK: reperfusion injury salvage kinase, ROCK: rho-kinase, sEH: soluble epoxide hydrolase, SERCA: sarco/endoplasmic reticulum Ca2+-ATPase, SAFE: survivor activating factor enhancement, tAUBC: trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexylo-benzoic acid, TIMP-1: Tissue inhibitor of metalloproteinase-1, TMP: 2,3,5,6-tetramethylpyrazine, TNF-α: tumor necrosis factor alpha.
Results of previous trials
It is worthy to mention here that the clinical studies and in vivo studies are far less in cardiac tissues as compared to non cardiac tissues and animal models. Some of the compounds have gained access to human trials in other diseases like neurodegenerative diseases and cancer. Newer compounds like reservatrol, ginesonide, Chinese herbal medicine have been impressive in suppressing autophagy in non cardiac tissues and demand further study. Due to the beneficial effects in experimental models, selected drugs have been tried on clinical trials. Some of the important targets have been ROS [151], calcium [152], or pharmaceutical agent that inhibit these or their detrimental effects like–magnesium sulphate [153], sodium-hydrogen exchange inhibitor, adenosine [154], ANP [155], GIK [156], GLP-1 [157], exetanide [158], anti-inflammatory mediators [159-161]. Most of them have demonstrated a positive cardioprotective effects but in variations and have not been able to reach 100% of the set target. The concept of preconditioning and post conditioning in limiting reperfusion injury has been impressive too but it is too early to advocate its application. A clearer, broader role of these pharmaceutical agents and maneuvers is demanded, which are being searched by wider trials of today.
Future trends
The search for therapeutic target and effective strategy is ongoing and to our dismay, till date, no effective therapeutic strategy exists. Despite these unrewarding searches, newer drugs and their positive interactions continue to be discovered out of these trials. So, human trials are underway, the results of many trials are awaited. Listed in Table 3 are some newer therapeutic agents and the status of the trials (as the entire exhausting list is not the aim of the article) aimed at restricting reperfusion injury.
Table 3.
Ongoing studies aimed towards limiting reperfusion injury. Note: The trials that cover the cardiac surgical aspects and the maneuvers are excluded
| Drug | Sponsor | Target |
|---|---|---|
| Name of study (NCT) | Status | |
| Bendavia | Stealth Peptide Inc. Boston, USA | Mitochondrial electron transport chain |
| EMBRACE (NCT01572909) | Ongoing | |
| Cyclosporin A | Mario Negri Institute for Pharmacological Research, Italy | mPTP inhibitor |
| CYCLE (NCT01650662) | Ongoing | |
| Cyclosporin A | Hospices Civils de Lyon, France | Clincal outcomes |
| CIRCUS (NCT01502774) | Ongoing | |
| Nitric Oxide | Barts & The London NHS Trust, London, UK | Microvasculatu-re |
| NITRITE-AMI (NCT01584453) | Ongoing | |
| NIAMI (NCT01388504) | ||
| NOMI trial (NCT01398384) | ||
| Endovascular catheter cooling | Philips Helthcare, Lund University, Uppsala University | Infarct size reduction |
| CHILL-MI (NCT01379261) | Ongoing | |
| Melatonin | Herlev Hospital, Aalborg, Denmark | Antioxidant |
| (NCT01172171) | Phase II | |
| TR040303 | Trophus, European commission | mPTP antagonist |
| MITOCARE (NCT001374321) | Completed | |
| Atorvastatin | R&D Cardiologie, Netherlands | Left ventricular remodelling |
| The REPERATOR study (NCT00286312) | Completed | |
| POST AMI | University of Padua, Padua, Italy | Post conditioning |
| (NCT01004289) | Completed | |
| PRIME | Hospices Civils de Lyon, France | Delayed post conditioning |
| (NCT01483755) | Completed | |
| Post Cond No Reflow | Hospices Civils de Lyon, France | Microvascular obstruction lesions |
| (NCT01208727) | Completed | |
| Streptokinase | Istanbul University, Turkey | Microvasculatu-re |
| (NCT00627809) | Completed | |
| Endothelin | Mayo Clinic, Minnesota, USA | Microvasculature |
| (NCT00586820) | Completed | |
| Edaravone | Kumamoto University, Kumamoto, Japan | Antioxidant |
| (NCT00265239) | Completed | |
| Adenosine | Xijing Hospital, Shaanxi, China | Preconditionin-g |
| (NCT00881686) | Completed | |
| FX06, | Fibrex Medical Research & Development GmbH, | Fibrin derived peptide B beta 15-42 |
| The “F.I.R.E.” study (NCT 003269760). | Completed | |
| Adiponectin | Universiteit Antwerpen, Antwerp, Belgium | Obesrvational study |
| R2ACE (NCT01414452) | Completed | |
| Rosiglitazone | Radboud University, Nijmegen, Netherlands | Interventional study |
| (NCT00405015) | Completed | |
| Short term Statin | Radboud University, Nijmegen, Netherlands | Endothelial dysfunction |
| (NCT00987974) | Completed | |
| Black tea (Camellia sinensis) | Radboud University, Nijmegen, Netherlands | Endothelial function |
| (NCT01660516) | Completed | |
| Metformin | Radboud University, Nijmegen, Netherlands | Endothelial damage |
| MetFMD (NCT01610401) | Completed | |
| Role of Glucose and Statins | Zablocki VA Medical Center, Milwaukee, USA. | Anaesthetic preconditionin-g |
| (NCT00995670) | Completed | |
| Aspirin, Clopidogrel and Statins | Gyeongsang National University Hospital, Gyeonsangnam-do, Korea | Platelet activation, inflammation and myonecrosis |
| ACCEL-LOADING-ACS-Study (NCT01354808) | Completed | |
| Remote Post conditioning | IRCCS Policlinico S. Matteo, Italy | Post conditioning |
| RemPostCon (NCT00865722) | Completed | |
| Erythropoetin | University Hospital, Montmellier, | Infact size reduction |
| Intra-CO-EpoMI (NCT01043991) | Completed | |
| Milrinone and Esmolol | Ming-He Huang University of Texas, USA | Infarct size reduction |
| COMET-AMI (NCT 02098629) | Recruiting | |
| Remote ischemic conditioning | Arhus University Hospital Denmark, Serbia, Spain, UK | Clincal outcomes |
| CONDI2 (NCT01857414) | Recruiting | |
| Endothelial arginase inhibitor | Karolinska Institute, Stockholm, Sweden | Vasodilation by Nitric Oxide |
| (NCT02009527) | Recruiting | |
| Vit C | University of Soma La Sapienza, Rome, Italy | Antioxidant |
| TREVI (NCT01090895) | Recruiting | |
| Spironolactone | University College, London, UK | Infarct size reduction Mineralo-corticoid receptor antagonist |
| MINIMISE STEMI (NCT01882179) | Recruiting | |
| Melatonin | Herlev Hospital, Aalborg, Denmark | Infact size reduction |
| (NCT0117217) | Recruiting | |
| Glucagonlike Peptide-1 | Chinese PLA General Hsopital, Beijing, China | Myocardial salvage |
| (NCT02001363) | Recuiting | |
| DREAM | University of Leicester, Leicestershire, UK | Remote ischemic conditioning |
| (NCT01664611) | Recruiting | |
| Cooling | Medical University of Vienna, Austria | Infarct size reduction |
| STATIM (NCT01777750) | Recruiting |
Source: www.clinicaltrial.gov/CT2/show (accessed on September 10, 2014).
Conclusion
Autophagy is integral for cardiac cell survival. Its uncontrolled continuum during reperfusion leads to fatal outcomes which has been the target of many researches. Rigorous search for an effective therapeutic strategy has been unsuccessful so far. A noteworthy point here is manipulating the autophagy pathways and mediators have been impressive in most experimental studies but not suitable for clinical trials due to various reasons. The failed attempts needs to translate into studies centered around the autophagy related genes and autophagy related proteins. Reperfusion injury cannot be attended without taking care of autophagy process as reperfusing a cardiac tissue cures the MI like the warm tip of an iceberg; the submerged injuries are vast, rigid and detrimental. However the concern that this process has received is indeed encouraging and we hope that the results of many molecular targets, which are awaited, initiated or planned would surely relate to effective cardio protection in the future.
Disclosure of conflict of interest
None.
References
- 1.Finegold JA, Asaria P, Francis DP. Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. Int J Cardiol. 2013;168:934–45. doi: 10.1016/j.ijcard.2012.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zalesak M, Styk J, Ledvenyiova V, Carnicka S, Nemcekova M, Ravingerova T. Presence of cardiovascular risk factors in secondary school students in two Slovak regions. Cesk Fysiol. 2012;61:36–40. [PubMed] [Google Scholar]
- 3.Steffenino G, Chinaglia A, Millesimo G, Gnavi R, Picariello R, Orlando A PRIMA Investigators. Management of acute ST-elevation myocardial infarction in the coronary care units of Piedmont in 2005: results from the PRIMA regionwide survey. J Cardiovasc Med (Hagerstown) 2008;9:169–77. doi: 10.2459/JCM.0b013e3281ac210c. [DOI] [PubMed] [Google Scholar]
- 4.Tra T, Gong L, Kao LP, Li XL, Grandela C, Devenish RJ, Wolvetang E, Prescott M. Autophagy in human embryonic stem cells. PLoS One. 2011;6:e27485. doi: 10.1371/journal.pone.0027485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 6.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nedjic J, Aichinger M, Mizushima N, Klein L. Macroautophagy, endogenous MHC II loading and T cell selection: the benefits of breaking the rules. Curr Opin Immunol. 2009;21:92–7. doi: 10.1016/j.coi.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–58. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, Perlmutter DH. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–32. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 11.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 12.Codogno P, Meijer AJ. Autophagy: a potential link between obesity and insulin resistance. Cell Metab. 2010;11:449–51. doi: 10.1016/j.cmet.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 14.Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol. 2009;10:461–70. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117–20. doi: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- 16.Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–4. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- 17.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 18.Vazquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, de Pablo F. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy. 2012;8:187–99. doi: 10.4161/auto.8.2.18535. [DOI] [PubMed] [Google Scholar]
- 19.Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–7. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- 20.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashford TP, Porter KR. Cytoplasmic components in hepatic cell lysosomes. J Cell Biol. 1962;12:198–202. doi: 10.1083/jcb.12.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–3. doi: 10.4161/auto.6398. [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–45. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 25.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 26.Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–5. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- 27.Webber JL, Tooze SA. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 2010;29:27–40. doi: 10.1038/emboj.2009.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc Natl Acad Sci U S A. 2004;101:3438–43. doi: 10.1073/pnas.0400443101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorburn J, Moore F, Rao A, Barclay WW, Thomas LR, Grant KW, Cramer SD, Thorburn A. Selective inactivation of a Fas-associated death domain protein (FADD)-dependent apoptosis and autophagy pathway in immortal epithelial cells. Mol Biol Cell. 2005;16:1189–99. doi: 10.1091/mbc.E04-10-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, Huang WL, Zeng YX, Zhu XF. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene. 2009;28:886–98. doi: 10.1038/onc.2008.441. [DOI] [PubMed] [Google Scholar]
- 31.Jia L, Dourmashkin RR, Allen PD, Gray AB, Newland AC, Kelsey SM. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br J Haematol. 1997;98:673–85. doi: 10.1046/j.1365-2141.1997.2623081.x. [DOI] [PubMed] [Google Scholar]
- 32.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–68. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 35.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 36.Decuypere JP, Parys JB, Bultynck G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells. 2012;1:284–312. doi: 10.3390/cells1030284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mortimore GE, Poso AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7:539–64. doi: 10.1146/annurev.nu.07.070187.002543. [DOI] [PubMed] [Google Scholar]
- 39.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talloczy Z, Jiang W, Virgin HW 4th, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–5. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–8. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834–45. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–53. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 44.Meyer G, Czompa A, Reboul C, Csepanyi E, Czegledi A, Bak I, Balla G, Balla J, Tosaki A, Lekli I. The cellular autophagy markers Beclin-1 and LC3B-II are increased during reperfusion in fibrillated mouse hearts. Curr Pharm Des. 2013;19:6912–8. doi: 10.2174/138161281939131127122510. [DOI] [PubMed] [Google Scholar]
- 45.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–33. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 46.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergamini CM, Gambetti S, Dondi A, Cervellati C. Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des. 2004;10:1611–26. doi: 10.2174/1381612043384664. [DOI] [PubMed] [Google Scholar]
- 48.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolli R. Mechanism of myocardial “stunning”. Circulation. 1990;82:723–38. doi: 10.1161/01.cir.82.3.723. [DOI] [PubMed] [Google Scholar]
- 50.Avkiran M, Marber MS. Na(+)/H(+) exchange inhibitors for cardioprotective therapy: progress, problems and prospects. J Am Coll Cardiol. 2002;39:747–53. doi: 10.1016/s0735-1097(02)01693-5. [DOI] [PubMed] [Google Scholar]
- 51.Bernardi P, Rasola A. Calcium and cell death: the mitochondrial connection. Subcell Biochem. 2007;45:481–506. doi: 10.1007/978-1-4020-6191-2_18. [DOI] [PubMed] [Google Scholar]
- 52.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen SY, Chiu LY, Maa MC, Wang JS, Chien CL, Lin WW. zVAD-induced autophagic cell death requires c-Src-dependent ERK and JNK activation and reactive oxygen species generation. Autophagy. 2011;7:217–28. doi: 10.4161/auto.7.2.14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 55.Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS, Stephanou A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006;40:846–52. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- 56.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–17. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 57.Webster KA, Graham RM, Thompson JW, Spiga MG, Frazier DP, Wilson A, Bishopric NH. Redox stress and the contributions of BH3-only proteins to infarction. Antioxid Redox Signal. 2006;8:1667–76. doi: 10.1089/ars.2006.8.1667. [DOI] [PubMed] [Google Scholar]
- 58.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–31. doi: 10.1161/01.res.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- 59.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–57. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 60.Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell. 2005;16:2544–53. doi: 10.1091/mbc.E04-08-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–7. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 63.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun Q, Fan W, Zhong Q. Regulation of Beclin 1 in autophagy. Autophagy. 2009;5:713–6. doi: 10.4161/auto.5.5.8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F. Structure of Atg5. Atg16, a complex essential for autophagy. J Biol Chem. 2007;282:6763–72. doi: 10.1074/jbc.M609876200. [DOI] [PubMed] [Google Scholar]
- 67.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 68.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jennings RB, Sommers HM, Smyth GA, Flack HA, Linn H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch Pathol. 1960;70:68–78. [PubMed] [Google Scholar]
- 71.Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R, McCay PB. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci U S A. 1989;86:4695–9. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res. 1999;43:860–78. doi: 10.1016/s0008-6363(99)00187-x. [DOI] [PubMed] [Google Scholar]
- 73.Xu Y, Huo Y, Toufektsian MC, Ramos SI, Ma Y, Tejani AD, French BA, Yang Z. Activated platelets contribute importantly to myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;290:H692–9. doi: 10.1152/ajpheart.00634.2005. [DOI] [PubMed] [Google Scholar]
- 74.Gross GJ, Kersten JR, Warltier DC. Mechanisms of postischemic contractile dysfunction. Ann Thorac Surg. 1999;68:1898–904. doi: 10.1016/s0003-4975(99)01035-8. [DOI] [PubMed] [Google Scholar]
- 75.Oerlemans MI, Koudstaal S, Chamuleau SA, de Kleijn DP, Doevendans PA, Sluijter JP. Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int J Cardiol. 2013;165:410–22. doi: 10.1016/j.ijcard.2012.03.055. [DOI] [PubMed] [Google Scholar]
- 76.Webster KA. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiol. 2012;8:863–84. doi: 10.2217/fca.12.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res. 1997;80:393–9. [PubMed] [Google Scholar]
- 78.Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98:425–44. [PMC free article] [PubMed] [Google Scholar]
- 79.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 80.Rodriguez-Sinovas A, Abdallah Y, Piper HM, Garcia-Dorado D. Reperfusion injury as a therapeutic challenge in patients with acute myocardial infarction. Heart Fail Rev. 2007;12:207–16. doi: 10.1007/s10741-007-9039-9. [DOI] [PubMed] [Google Scholar]
- 81.Hearse DJ, Tosaki A. Free radicals and reperfusion-induced arrhythmias: protection by spin trap agent PBN in the rat heart. Circ Res. 1987;60:375–83. doi: 10.1161/01.res.60.3.375. [DOI] [PubMed] [Google Scholar]
- 82.Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation. 2001;104:2981–9. doi: 10.1161/hc4801.100038. [DOI] [PubMed] [Google Scholar]
- 83.Krug A, Du Mesnil de R, Korb G. Blood supply of the myocardium after temporary coronary occlusion. Circ Res. 1966;19:57–62. doi: 10.1161/01.res.19.1.57. [DOI] [PubMed] [Google Scholar]
- 84.Kloner RA, Rude RE, Carlson N, Maroko PR, DeBoer LW, Braunwald E. Ultrastructural evidence of microvascular damage and myocardial cell injury after coronary artery occlusion: which comes first? Circulation. 1980;62:945–52. doi: 10.1161/01.cir.62.5.945. [DOI] [PubMed] [Google Scholar]
- 85.Iwakura K, Ito H, Takiuchi S, Taniyama Y, Nakatsuchi Y, Negoro S, Higashino Y, Okamura A, Masuyama T, Hori M, Fujii K, Minamino T. Alternation in the coronary blood flow velocity pattern in patients with no reflow and reperfused acute myocardial infarction. Circulation. 1996;94:1269–75. doi: 10.1161/01.cir.94.6.1269. [DOI] [PubMed] [Google Scholar]
- 86.Luo AK, Wu KC. Imaging microvascular obstruction and its clinical significance following acute myocardial infarction. Heart Fail Rev. 2006;11:305–12. doi: 10.1007/s10741-006-0231-0. [DOI] [PubMed] [Google Scholar]
- 87.Ganame J, Messalli G, Dymarkowski S, Rademakers FE, Desmet W, Van de Werf F, Bogaert J. Impact of myocardial haemorrhage on left ventricular function and remodelling in patients with reperfused acute myocardial infarction. Eur Heart J. 2009;30:1440–9. doi: 10.1093/eurheartj/ehp093. [DOI] [PubMed] [Google Scholar]
- 88.Yellon DM, Alkhulaifi AM, Pugsley WB. Preconditioning the human myocardium. Lancet. 1993;342:276–7. doi: 10.1016/0140-6736(93)91819-8. [DOI] [PubMed] [Google Scholar]
- 89.Ishihara M. Acute hyperglycemia in patients with acute myocardial infarction. Circ J. 2012;76:563–71. doi: 10.1253/circj.cj-11-1376. [DOI] [PubMed] [Google Scholar]
- 90.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123:1262–74. doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rochitte CE, Lima JA, Bluemke DA, Reeder SB, McVeigh ER, Furuta T, Becker LC, Melin JA. Magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation. 1998;98:1006–14. doi: 10.1161/01.cir.98.10.1006. [DOI] [PubMed] [Google Scholar]
- 93.Baines CP. How and when do myocytes die during ischemia and reperfusion: the late phase. J Cardiovasc Pharmacol Ther. 2011;16:239–43. doi: 10.1177/1074248411407769. [DOI] [PubMed] [Google Scholar]
- 94.Ambrosio G, Weisman HF, Mannisi JA, Becker LC. Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation. 1989;80:1846–61. doi: 10.1161/01.cir.80.6.1846. [DOI] [PubMed] [Google Scholar]
- 95.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–5. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 96.Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. 2010;142:590–600. doi: 10.1016/j.cell.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, Kester M, Wang HG. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–68. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–77. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M, Sadoshima J. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–88. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Norman JM, Cohen GM, Bampton ET. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2010;6:1042–56. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- 102.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11:1355–62. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 104.Xu MC, Shi HM, Gao XF, Wang H. Salidroside attenuates myocardial ischemia-reperfusion injury via PI3K/Akt signaling pathway. J Asian Nat Prod Res. 2013;15:244–52. doi: 10.1080/10286020.2012.762358. [DOI] [PubMed] [Google Scholar]
- 105.Fang F, Li D, Pan H, Chen D, Qi L, Zhang R, Sun H. Luteolin inhibits apoptosis and improves cardiomyocyte contractile function through the PI3K/Akt pathway in simulated ischemia/reperfusion. Pharmacology. 2011;88:149–58. doi: 10.1159/000330068. [DOI] [PubMed] [Google Scholar]
- 106.Ravingerova T, Carnicka S, Nemcekova M, Ledvenyiova V, Adameova A, Kelly T, Barlaka E, Galatou E, Khandelwal VK, Lazou A. PPAR-alpha activation as a preconditioning-like intervention in rats in vivo confers myocardial protection against acute ischaemia-reperfusion injury: involvement of PI3K-Akt. Can J Physiol Pharmacol. 2012;90:1135–44. doi: 10.1139/y2012-052. [DOI] [PubMed] [Google Scholar]
- 107.Ma XJ, Yin HJ, Guo CY, Jiang YR, Wang JS, Shi DZ. Ischemic postconditioning through percutaneous transluminal coronary angioplasty in pigs: roles of PI3K activation. Coron Artery Dis. 2012;23:245–50. doi: 10.1097/MCA.0b013e3283526a7d. [DOI] [PubMed] [Google Scholar]
- 108.Lv L, Jiang SS, Xu J, Gong JB, Cheng Y. Protective effect of ligustrazine against myocardial ischaemia reperfusion in rats: the role of endothelial nitric oxide synthase. Clin Exp Pharmacol Physiol. 2012;39:20–7. doi: 10.1111/j.1440-1681.2011.05628.x. [DOI] [PubMed] [Google Scholar]
- 109.Chang G, Zhang P, Ye L, Lu K, Wang Y, Duan Q, Zheng A, Qin S, Zhang D. Protective effects of sitagliptin on myocardial injury and cardiac function in an ischemia/reperfusion rat model. Eur J Pharmacol. 2013;718:105–13. doi: 10.1016/j.ejphar.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 110.Guo J, Bian Y, Bai R, Li H, Fu M, Xiao C. Globular adiponectin attenuates myocardial ischemia/reperfusion injury by upregulating endoplasmic reticulum Ca(2)(+)-ATPase activity and inhibiting endoplasmic reticulum stress. J Cardiovasc Pharmacol. 2013;62:143–53. doi: 10.1097/FJC.0b013e31829521af. [DOI] [PubMed] [Google Scholar]
- 111.Li Q, Shi M, Li B. Anandamide enhances expression of heat shock protein 72 to protect against ischemia-reperfusion injury in rat heart. J Physiol Sci. 2013;63:47–53. doi: 10.1007/s12576-012-0228-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu Y, Jia XJ, Zong QF, Zhang GJ, Ye HW, Hu J, Gao Q, Guan SD. Remote ischemic postconditioning protects the heart by upregulating ALDH2 expression levels through the PI3K/Akt signaling pathway. Mol Med Rep. 2014;10:536–42. doi: 10.3892/mmr.2014.2156. [DOI] [PubMed] [Google Scholar]
- 113.Kim SJ, Li M, Jeong CW, Bae HB, Kwak SH, Lee SH, Lee HJ, Heo BH, Yook KB, Yoo KY. Epigallocatechin-3-gallate, a green tea catechin, protects the heart against regional ischemia-reperfusion injuries through activation of RISK survival pathways in rats. Arch Pharm Res. 2014;37:1079–85. doi: 10.1007/s12272-013-0309-x. [DOI] [PubMed] [Google Scholar]
- 114.Hussain A, Gharanei AM, Nagra AS, Maddock HL. Caspase inhibition via A3 adenosine receptors: a new cardioprotective mechanism against myocardial infarction. Cardiovasc Drugs Ther. 2014;28:19–32. doi: 10.1007/s10557-013-6500-y. [DOI] [PubMed] [Google Scholar]
- 115.Zhang CM, Gao L, Zheng YJ, Yang HT. Berbamine protects the heart from ischemia/reperfusion injury by maintaining cytosolic Ca(2+) homeostasis and preventing calpain activation. Circ J. 2012;76:1993–2002. doi: 10.1253/circj.cj-11-1431. [DOI] [PubMed] [Google Scholar]
- 116.Zhang JY, Chen ZW, Yao H. Protective effect of urantide against ischemia-reperfusion injury via protein kinase C and phosphtidylinositol 3'-kinase-Akt pathway. Can J Physiol Pharmacol. 2012;90:637–45. doi: 10.1139/y2012-048. [DOI] [PubMed] [Google Scholar]
- 117.Wang J, Yang H, Hu X, Fu W, Xie J, Zhou X, Xu W, Jiang H. Dobutamine-mediated heme oxygenase-1 induction via PI3K and p38 MAPK inhibits high mobility group box 1 protein release and attenuates rat myocardial ischemia/reperfusion injury in vivo. J Surg Res. 2013;183:509–16. doi: 10.1016/j.jss.2013.02.051. [DOI] [PubMed] [Google Scholar]
- 118.Hu G, Huang X, Zhang K, Jiang H, Hu X. Anti-inflammatory effect of B-Type natriuretic peptide postconditioning during myocardial ischemia-reperfusion: involvement of PI3K/Akt signaling pathway. Inflammation. 2014;37:1669–74. doi: 10.1007/s10753-014-9895-0. [DOI] [PubMed] [Google Scholar]
- 119.Rahman S, Li J, Bopassa JC, Umar S, Iorga A, Partownavid P, Eghbali M. Phosphorylation of GSK-3beta mediates intralipid-induced cardioprotection against ischemia/reperfusion injury. Anesthesiology. 2011;115:242–53. doi: 10.1097/ALN.0b013e318223b8b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tie R, Ji L, Nan Y, Wang W, Liang X, Tian F, Xing W, Zhu M, Li R, Zhang H. Achyranthes bidentata polypeptides reduces oxidative stress and exerts protective effects against myocardial ischemic/reperfusion injury in rats. Int J Mol Sci. 2013;14:19792–804. doi: 10.3390/ijms141019792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li J, Qi D, Cheng H, Hu X, Miller EJ, Wu X, Russell KS, Mikush N, Zhang J, Xiao L, Sherwin RS, Young LH. Urocortin 2 autocrine/paracrine and pharmacologic effects to activate AMP-activated protein kinase in the heart. Proc Natl Acad Sci U S A. 2013;110:16133–8. doi: 10.1073/pnas.1312775110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Penna C, Settanni F, Tullio F, Trovato L, Pagliaro P, Alloatti G, Ghigo E, Granata R. GH-releasing hormone induces cardioprotection in isolated male rat heart via activation of RISK and SAFE pathways. Endocrinology. 2013;154:1624–35. doi: 10.1210/en.2012-2064. [DOI] [PubMed] [Google Scholar]
- 123.Perrelli MG, Tullio F, Angotti C, Cerra MC, Angelone T, Tota B, Alloatti G, Penna C, Pagliaro P. Catestatin reduces myocardial ischaemia/reperfusion injury: involvement of PI3K/Akt, PKCs, mitochondrial KATP channels and ROS signalling. Pflugers Arch. 2013;465:1031–40. doi: 10.1007/s00424-013-1217-0. [DOI] [PubMed] [Google Scholar]
- 124.Si R, Tao L, Zhang HF, Yu QJ, Zhang R, Lv AL, Zhou N, Cao F, Guo WY, Ren J, Wang HC, Gao F. Survivin: a novel player in insulin cardioprotection against myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2011;50:16–24. doi: 10.1016/j.yjmcc.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 125.Chaudhary KR, Abukhashim M, Hwang SH, Hammock BD, Seubert JM. Inhibition of soluble epoxide hydrolase by trans-4- [4-(3-adamantan-1-yl-ureido)-cyclohexyloxy] -benzoic acid is protective against ischemia-reperfusion injury. J Cardiovasc Pharmacol. 2010;55:67–73. doi: 10.1097/FJC.0b013e3181c37d69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Abdillahi M, Ananthakrishnan R, Vedantham S, Shang L, Zhu Z, Rosario R, Zirpoli H, Bohren KM, Gabbay KH, Ramasamy R. Aldose reductase modulates cardiac glycogen synthase kinase-3beta phosphorylation during ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2012;303:H297–308. doi: 10.1152/ajpheart.00999.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Laurence J, Cooke H, Sikder SK. Effect of tamoxifen on regulation of viral replication and human immunodeficiency virus (HIV) long terminal repeat-directed transcription in cells chronically infected with HIV-1. Blood. 1990;75:696–703. [PubMed] [Google Scholar]
- 128.Wu QL, Shen T, Ma H, Wang JK. Sufentanil postconditioning protects the myocardium from ischemia-reperfusion via PI3K/Akt-GSK-3beta pathway. J Surg Res. 2012;178:563–70. doi: 10.1016/j.jss.2012.05.081. [DOI] [PubMed] [Google Scholar]
- 129.Jeong CW, Yoo KY, Lee SH, Jeong HJ, Lee CS, Kim SJ. Curcumin protects against regional myocardial ischemia/reperfusion injury through activation of RISK/GSK-3beta and inhibition of p38 MAPK and JNK. J Cardiovasc Pharmacol Ther. 2012;17:387–94. doi: 10.1177/1074248412438102. [DOI] [PubMed] [Google Scholar]
- 130.Yin Y, Guan Y, Duan J, Wei G, Zhu Y, Quan W, Guo C, Zhou D, Wang Y, Xi M, Wen A. Cardioprotective effect of Danshensu against myocardial ischemia/reperfusion injury and inhibits apoptosis of H9c2 cardiomyocytes via Akt and ERK1/2 phosphorylation. Eur J Pharmacol. 2013;699:219–26. doi: 10.1016/j.ejphar.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 131.Li Y, Zhu W, Tao J, Xin P, Liu M, Li J, Wei M. Fasudil protects the heart against ischemia-reperfusion injury by attenuating endoplasmic reticulum stress and modulating SERCA activity: the differential role for PI3K/Akt and JAK2/STAT3 signaling pathways. PLoS One. 2012;7:e48115. doi: 10.1371/journal.pone.0048115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mochizuki T, Yu S, Katoh T, Aoki K, Sato S. Cardioprotective effect of therapeutic hypothermia at 34 degrees C against ischaemia/reperfusion injury mediated by PI3K and nitric oxide in a rat isolated heart model. Resuscitation. 2012;83:238–42. doi: 10.1016/j.resuscitation.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 133.Sun Z, Tong G, Ma N, Li J, Li X, Li S, Zhou J, Xiong L, Cao F, Yao L, Wang H, Shen L. NDRG2: a newly identified mediator of insulin cardioprotection against myocardial ischemia-reperfusion injury. Basic Res Cardiol. 2013;108:341. doi: 10.1007/s00395-013-0341-5. [DOI] [PubMed] [Google Scholar]
- 134.Ma LL, Zhang FJ, Qian LB, Kong FJ, Sun JF, Zhou C, Peng YN, Xu HJ, Wang WN, Wen CY, Zhu MH, Chen G, Yu LN, Liu XB, Wang JA, Yan M. Hypercholesterolemia blocked sevoflurane-induced cardioprotection against ischemia-reperfusion injury by alteration of the MG53/RISK/GSK3beta signaling. Int J Cardiol. 2013;168:3671–8. doi: 10.1016/j.ijcard.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 135.Borchert GH, Hlavackova M, Kolar F. Pharmacological activation of mitochondrial BK(Ca) channels protects isolated cardiomyocytes against simulated reperfusion-induced injury. Exp Biol Med (Maywood) 2013;238:233–41. doi: 10.1177/1535370212474596. [DOI] [PubMed] [Google Scholar]
- 136.Tu Y, Wan L, Fan Y, Wang K, Bu L, Huang T, Cheng Z, Shen B. Ischemic postconditioning-mediated miRNA-21 protects against cardiac ischemia/reperfusion injury via PTEN/Akt pathway. PLoS One. 2013;8:e75872. doi: 10.1371/journal.pone.0075872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang MQ, Zheng YL, Chen H, Tu JF, Shen Y, Guo JP, Yang XH, Yuan SR, Chen LZ, Chai JJ, Lu JH, Zhai CL. Sodium tanshinone IIA sulfonate protects rat myocardium against ischemia-reperfusion injury via activation of PI3K/Akt/FOXO3A/Bim pathway. Acta Pharmacol Sin. 2013;34:1386–96. doi: 10.1038/aps.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Abrial M, Da Silva CC, Pillot B, Augeul L, Ivanes F, Teixeira G, Cartier R, Angoulvant D, Ovize M, Ferrera R. Cardiac fibroblasts protect cardiomyocytes against lethal ischemia-reperfusion injury. J Mol Cell Cardiol. 2014;68:56–65. doi: 10.1016/j.yjmcc.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 139.Cuadrado I, Fernandez-Velasco M, Bosca L, de Las Heras B. Labdane diterpenes protect against anoxia/reperfusion injury in cardiomyocytes: involvement of AKT activation. Cell Death Dis. 2011;2:e229. doi: 10.1038/cddis.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Morrison A, Yan X, Tong C, Li J. Acute rosiglitazone treatment is cardioprotective against ischemia-reperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice. Am J Physiol Heart Circ Physiol. 2011;301:H895–902. doi: 10.1152/ajpheart.00137.2011. [DOI] [PubMed] [Google Scholar]
- 141.Kambara T, Ohashi K, Shibata R, Ogura Y, Maruyama S, Enomoto T, Uemura Y, Shimizu Y, Yuasa D, Matsuo K, Miyabe M, Kataoka Y, Murohara T, Ouchi N. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J Biol Chem. 2012;287:18965–73. doi: 10.1074/jbc.M112.357939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chang W, Zhang M, Li J, Meng Z, Xiao D, Wei S, Chen L, Wang C, Hatch GM. Berberine attenuates ischemia-reperfusion injury via regulation of adenosine-5'-monophosphate kinase activity in both non-ischemic and ischemic areas of the rat heart. Cardiovasc Drugs Ther. 2012;26:467–78. doi: 10.1007/s10557-012-6422-0. [DOI] [PubMed] [Google Scholar]
- 143.Wang J, Tong C, Yan X, Yeung E, Gandavadi S, Hare AA, Du X, Chen Y, Xiong H, Ma C, Leng L, Young LH, Jorgensen WL, Li J, Bucala R. Limiting cardiac ischemic injury by pharmacological augmentation of macrophage migration inhibitory factor-AMP-activated protein kinase signal transduction. Circulation. 2013;128:225–36. doi: 10.1161/CIRCULATIONAHA.112.000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kataoka Y, Shibata R, Ohashi K, Kambara T, Enomoto T, Uemura Y, Ogura Y, Yuasa D, Matsuo K, Nagata T, Oba T, Yasukawa H, Numaguchi Y, Sone T, Murohara T, Ouchi N. Omentin prevents myocardial ischemic injury through AMP-activated protein kinase- and Akt-dependent mechanisms. J Am Coll Cardiol. 2014;63:2722–33. doi: 10.1016/j.jacc.2014.03.032. [DOI] [PubMed] [Google Scholar]
- 145.Qi D, Atsina K, Qu L, Hu X, Wu X, Xu B, Piecychna M, Leng L, Fingerle-Rowson G, Zhang J, Bucala R, Young LH. The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J Clin Invest. 2014;124:3540–50. doi: 10.1172/JCI73061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ma Y, Wang J, Gao J, Yang H, Wang Y, Manithody C, Li J, Rezaie AR. Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury. Thromb Haemost. 2015;113:338–49. doi: 10.1160/TH14-04-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Noh HS, Shin IW, Ha JH, Hah YS, Baek SM, Kim DR. Propofol protects the autophagic cell death induced by the ischemia/reperfusion injury in rats. Mol Cells. 2010;30:455–60. doi: 10.1007/s10059-010-0130-z. [DOI] [PubMed] [Google Scholar]
- 148.Zhang ZL, Fan Y, Liu ML. Ginsenoside Rg1 inhibits autophagy in H9c2 cardiomyocytes exposed to hypoxia/reoxygenation. Mol Cell Biochem. 2012;365:243–50. doi: 10.1007/s11010-012-1265-3. [DOI] [PubMed] [Google Scholar]
- 149.Khan M, Meduru S, Mostafa M, Khan S, Hideg K, Kuppusamy P. Trimetazidine, administered at the onset of reperfusion, ameliorates myocardial dysfunction and injury by activation of p38 mitogen-activated protein kinase and Akt signaling. J Pharmacol Exp Ther. 2010;333:421–9. doi: 10.1124/jpet.109.165175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ahmed LA, Salem HA, Attia AS, Agha AM. Pharmacological preconditioning with nicorandil and pioglitazone attenuates myocardial ischemia/reperfusion injury in rats. Eur J Pharmacol. 2011;663:51–8. doi: 10.1016/j.ejphar.2011.04.038. [DOI] [PubMed] [Google Scholar]
- 151.Mehta JL, Nichols WW, Saldeen TG, Chandna VK, Nicolini FA, Lawson DL, ter Riet MF. Superoxide dismutase decreases reperfusion arrhythmias and preserves myocardial function during thrombolysis with tissue plasminogen activator. J Cardiovasc Pharmacol. 1990;16:112–20. doi: 10.1097/00005344-199007000-00016. [DOI] [PubMed] [Google Scholar]
- 152.Nicolau JC, Ramires JA, Maggioni AP, Garzon SA, Pinto MA, Silva DG, Nogueira PR, Maia LF, Vendramini P, Bassi I. Diltiazem improves left ventricular systolic function following acute myocardial infarction treated with streptokinase. The Calcium Antagonist in Reperfusion Study (CARES) Group. Am J Cardiol. 1996;78:1049–52. doi: 10.1016/s0002-9149(96)00535-8. [DOI] [PubMed] [Google Scholar]
- 153.Woods KL, Fletcher S, Roffe C, Haider Y. Intravenous magnesium sulphate in suspected acute myocardial infarction: results of the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2) Lancet. 1992;339:1553–8. doi: 10.1016/0140-6736(92)91828-v. [DOI] [PubMed] [Google Scholar]
- 154.Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW AMISTAD-II Investigators. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) J Am Coll Cardiol. 2005;45:1775–80. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 155.Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, Seguchi O, Myoishi M, Minamino T, Ohara T, Nagai Y, Nanto S, Watanabe K, Fukuzawa S, Hirayama A, Nakamura N, Kimura K, Fujii K, Ishihara M, Saito Y, Tomoike H, Kitamura S, investigators JW. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370:1483–93. doi: 10.1016/S0140-6736(07)61634-1. [DOI] [PubMed] [Google Scholar]
- 156.Selker HP, Beshansky JR, Sheehan PR, Massaro JM, Griffith JL, D'Agostino RB, Ruthazer R, Atkins JM, Sayah AJ, Levy MK, Richards ME, Aufderheide TP, Braude DA, Pirrallo RG, Doyle DD, Frascone RJ, Kosiak DJ, Leaming JM, Van Gelder CM, Walter GP, Wayne MA, Woolard RH, Opie LH, Rackley CE, Apstein CS, Udelson JE. Out-of-hospital administration of intravenous glucose-insulin-potassium in patients with suspected acute coronary syndromes: the IMMEDIATE randomized controlled trial. JAMA. 2012;307:1925–33. doi: 10.1001/jama.2012.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Read PA, Hoole SP, White PA, Khan FZ, O'Sullivan M, West NE, Dutka DP. A pilot study to assess whether glucagon-like peptide-1 protects the heart from ischemic dysfunction and attenuates stunning after coronary balloon occlusion in humans. Circ Cardiovasc Interv. 2011;4:266–72. doi: 10.1161/CIRCINTERVENTIONS.110.960476. [DOI] [PubMed] [Google Scholar]
- 158.Woo JS, Kim W, Ha SJ, Kim JB, Kim SJ, Kim WS, Seon HJ, Kim KS. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol. 2013;33:2252–60. doi: 10.1161/ATVBAHA.113.301586. [DOI] [PubMed] [Google Scholar]
- 159.Rusnak JM, Kopecky SL, Clements IP, Gibbons RJ, Holland AE, Peterman HS, Martin JS, Saoud JB, Feldman RL, Breisblatt WM, Simons M, Gessler CJ Jr, Yu AS. An anti-CD11/CD18 monoclonal antibody in patients with acute myocardial infarction having percutaneous transluminal coronary angioplasty (the FESTIVAL study) Am J Cardiol. 2001;88:482–7. doi: 10.1016/s0002-9149(01)01723-4. [DOI] [PubMed] [Google Scholar]
- 160.Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F HALT-MI Investigators. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol. 2002;40:1199–204. doi: 10.1016/s0735-1097(02)02136-8. [DOI] [PubMed] [Google Scholar]
- 161.Lin GM, Li YH, Jaiteh LE, Han CL. Pexelizumab fails to inhibit assembly of the terminal complement complex in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention. Insight from a substudy of the Assessment of Pexelizumab in Acute Myocardial Infarction (APEX-AMI) trial. Am Heart J. 2012;164:e19. doi: 10.1016/j.ahj.2012.09.012. author reply e21. [DOI] [PubMed] [Google Scholar]