

Abstract
Lung cancer is the most common cause of cancer-related death in the world. The main types of lung cancer are small cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC); non small cell lung carcinoma (NSCLC) includes squamous cell carcinoma (SCC), adenocarcinoma and large cell carcinoma, Non small cell lung carcinoma accounts for about 80% of the total lung cancer cases. Dihydroartemisinin (DHA) inhibits cell proliferation and induces apoptosis in several cancer cell lines. The effects of DHA on cell growth and proliferation in lung cancer cells remain to be elucidated. Here, we demonstrate that DHA inhibited cell proliferation in the A549 lung cancer cell line through suppression of the AKT/Gsk-3β/cyclin D1 signaling pathway. DHA significantly inhibited cell proliferation of A549 cells in a concentration and time dependent manner as determined by MTS assay. Flow cytometry analysis demonstrated that DHA treatment of A549 cells resulted in cell cycle arrest at the G1 phase, which correlated with apparent downregulation of both mRNA and protein levels of both PCNA and cyclin D1. These results suggest that DHA is a potential natural product for the treatment of lung cancer.
Keywords: Dihydroartemisinin, cell proliferation, cell cycle, cyclinD1
Introduction
Lung cancer is the most common cause of cancer-related death in men and women, and is responsible for 1.38 million deaths annually [1,2]. In China, lung cancer has the highest morbidity and mortality among malignant tumors around the country, which leads to 600 thousands deaths annually. Lung cancer can be divided into two major types: non-small-cell lung cancer (NSCLC) and small cell lung cancer (SCLC) [1,3,4]. Non-small-cell lung carcinoma (NSCLC) is subdivided into three broad categories: pulmonary squamous cell carcinoma (SCC), pulmonary adenocarcinoma (AC) and large cell carcinoma [5]. Prolonged cigarette smoking is the most common cause of lung cancer [6], especially in China which has the largest number of smokers (300 million) in the world. Non-small cell lung cancer accounts for about 80% of the total lung cancer cases in clinic. Nearly 40% of lung cancers are adenocarcinoma, which usually originate in peripheral lung tissue. Squamous cell carcinoma accounts for about 30% of lung cancers. A hollow cavity and associated cell death are commonly found at the center of the tumor [7,8].
Recently, it was found that DHA inhibited both cell proliferation and cell cycle progression of rat aortic vascular smooth muscle cells by inhibiting the activation of ERK1/2 and expression of c-fos [9-11]. These data suggest that DHA could be a potential natural product for the treatment of lung cancer. In this study, we demonstrated the anti-proliferative effects of DHA on lung cancer cells, and found that DHA inhibited cell proliferation and induced cell apoptosis via the AKT/GSK3β/cyclinD1 pathway.
Materials and methods
Cells and cell culture
The A549 cell line was obtained from the cell bank of the Committee on Type Culture Collection of the Chinese Academic of Science (CCTCC, Shanghai, China). Cells were seeded at a density of 1 × 104 cells/cm2 and maintained in DMEM growth media supplemented with 10% fetal bovine serum (FBS, Gibco), 100 U/ml penicillin and 100 mg/ml streptomycin, in a humidified atmosphere of 5% CO2 at 37°C.
Cell proliferation assay
Cell proliferation was determined using the MTS assay (Promega). Cells were seeded at a concentration of 5000 cells/well in 24-well plates and incubated for 1, 2, 3, 4 and 5 days. At each time point, 100 μl media and 20 μl MTS was added to each well. Following incubation at 37°C for 2 hours, absorbance at 490 nm was detected using the spectrometer.
Flow cytometry
A549 cells (2 × 106) were plated in 100-mm plates with 15 ml of media, with or without DHA. After 2 days, the cells were resuspended in PBS containing 1% Triton X, 0.1 mg/ml RNase A, and 0.05 mg/ml propidium iodide. The cells were subjected to FACS caliber flow cytometry, and the percentage of cells in each phase of the cell cycle was obtained using Modfit software.
Cell cycle assay
The cell cycle was analyzed by using flow cytometry (FCM) with propidium iodide staining. Both floating and attached cells were collected by trypsin digestion and low-speed centrifugation, washed with cold PBS, and then fixed in ice-cold 70% ethanol overnight. The fixed cells were collected by brief centrifugation and resuspended in PBS, after which the cells were treated with RNaseA and stained with propidium iodide for 1 hour at room temperature, and finally analyzed by FCM.
Apoptosis assay
Apoptosis was assayed using the Annexin V-FITC Apoptosis Kit (Keygen, China) according to the manufacturer’s instructions. Briefly, the cells were harvested and washed twice with PBS, followed by resuspension in Annexin-V binding buffer, and then FITC-conjugated Annexin V and PI were added. After incubation for 10 min at room temperature in the dark, another binding buffer was added, and the samples were immediately analyzed using FCM.
Quantitative RT-PCR
Total RNA was isolated from the cultured cells using Trizol reagent (Invitrogen) according to the manufacturer’s instruction. Reverse transcription of total RNA was carried out at 42°C for 50 min and then at 70°C for 15 min using the SuperScriptTM first strand synthesis system (Invitrogen). The resulting single-stranded cDNA was amplified by PCR using primers specific for proliferating cell nuclear antigen (PCNA) (forward primer, 5’-GAAGCCACCCACACCATCAC-3’;reverse primer, 5’-TTCTTCAAAAATCTGACCATTCCAA-3’), CyclinD1 (forward primer, 5’-GAGGAGCAGCTCGCCAA-3’; reverse primer, 5’-CTGTCAAGGTCCGGCCAGCG-3’), and beta-actin (forward primer, 5’-TGGTCCTCTGGGCATCTCAGGC-3’; reverse primer, 5’-GGTGAACCTGCTGTTGCCCTCA-3’). Quantitative real-time PCR was performed using the SYBR Premix ExTaq II (TLiRNaseH Plus) (TaKaRa, Otsu, Japan) with a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Three independent experiments were performed. The resulting data were expressed as mean ± SE.
Immunoblotting analysis
Cells were lysed with RIPA buffer (Beyotime, China) and boiled for 5 minutes. The protein concentration of each lysate was measured using the BCA method (Beyotime, China). Equal quantities of protein from each cell lysate were separated on SDS-polyacrylamide electrophoresis gels and transferred to PVDF membranes (Millipore, Billerica, MA). The membranes were blocked with 5% skim milk, incubated with each primary antibody overnight at 4°C, washed with TBST buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.05% Tween 20) and incubated with secondary antibodies. The proteins were visualized using enhanced chemiluminescence (GE Healthcare Biosciences).
Statistical analysis
All experiments were performed at least three times and statistical analysis was done using the SPSS17.0 package (SPSS Inc., Chicago, USA). The values were expressed as mean with SD. The ANOVA test was used whenever more than two groups were compared, and the significance level was set at P < 0.05. P values of less than 0.05 (*P < 0.05) were considered to be statistically significant.
Results
DHA inhibits cell proliferation of A549 cells
To determine the effects of DHA on cell proliferation, we treated A549 cells with 0 μM, 10 μM, 20 μM and 30 μM DHA for 1 day to 5 days, and determined cell proliferation by MTS assay. DHA did not affect cell viability at day one (Figure 1). From the second to fifth day, DHA inhibited cell proliferation in a concentration and time dependent manner. Treatment of A549 cells with 10 μM DHA for 5 days and 30 μM DHA for 4 days resulted in 75% of loss of cell viability.
Figure 1.
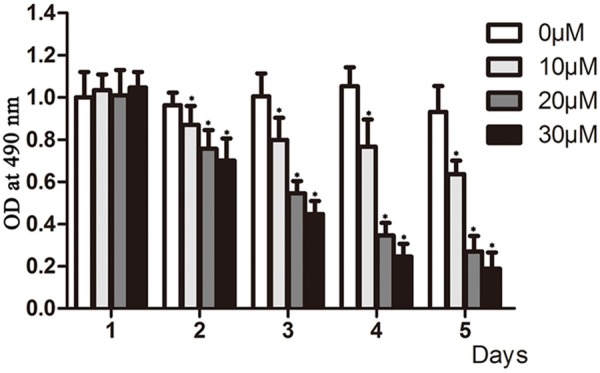
DHA inhibited the proliferation of A549 Cells. A. MTS detection of the proliferation of A549 cells. A549 cells were seeded at 5000 cells/well in 96-well plates and treated with different concentrations of DHA (0 μM, 10 μM, 20 μM and 30 μM) for the indicated days. Cell proliferation was determined by the MTS assay, the y axis represent cell proliferation percent of control. *P < 0.05.
DHA induces cell cycle arrest
To identify the mechanism by which DHA inhibits cell proliferation, we treated A549 cells with 0 μM, 10 μM, 20 μM and 30 μM DHA for 48 hour and analyzed the cell cycle distribution by flow cytometry. DHA significantly arrested cells at G1 phase of the cell cycle and induced a sub-G1 peak, indicating cell apoptosis (Figure 2A). Compared with the control, treatment of cells with 30 μM DHA resulted in 89.54% of cells arrested at G1 phase and 11.62% of cells in sub-G1 (Figure 2B). Thus, DHA prevented cell cycle progression from G1 to S phase transition and induced apoptosis in A549 cells.
Figure 2.
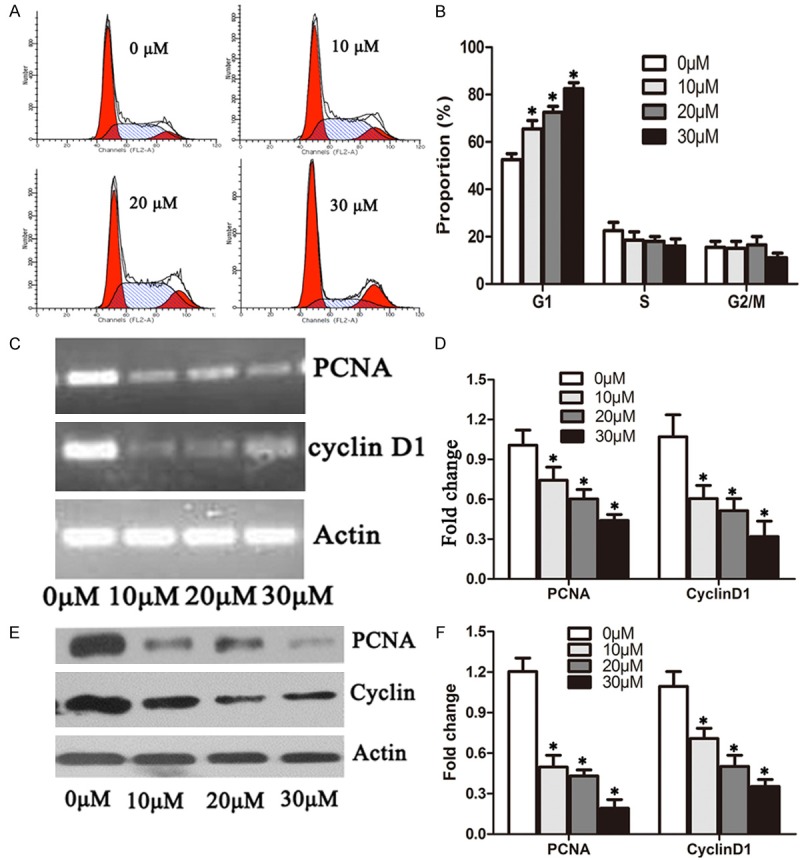
DHA induced cell cycle arrest at G1 phase in A549 cells. A. A549 cells treated with 0 μM, 10 μM, 20 μM and 30 μM DHA for 48 hours were subjected to flow cytometry analysis of the cell cycle distribution. B. Quantification of the dataset from A. C. A549 cells were treated with 0 μM, 10 Μm, 20 μM and 30 μM DHA 48 hours, respectively. RT-PCR analysis was done to determine the mRNA levels of PCNA, cyclin D1 and actin. D. Quantification of PCNA and Cyclin D1 mRNA levels from RT-PCR as in C. The mRNA levels of PCNA and Cyclin D1 were normalized to beta-actin. E. Total proteins were extracted for the immunoblotting of PCNA, cyclin D1 and actin. F. Quantification of PCNA and Cyclin D1 levels from immunblots as in E. The protein levels of PCNA and Cyclin D1 were normalized to beta-actin. *P < 0.05.
G1 phase progression is controlled by cyclin D dependent kinases 4, 6 (CDK4/6) and PCNA. We asked whether DHA inhibits the activity of CDK4/6. We determined the gene expression of PCNA and cyclin D1, the major regulator of CDK4/6, by examining the mRNA levels of PCNA and cyclin D1 in A549 cells, treated with 0 μM, 10 μM, 20 μM and 30 μM DHA for 48 hours. The mRNA levels of both PCNA and cyclin D1 were significantly reduced by 10 μM DHA and almost ablated by 30 μM DHA (Figure 2C, 2D). Consistent with these findings, we showed that DHA significantly reduced the protein levels of both PCNA and cyclin D1 (Figure 2E, 2F). To gain further insight into the molecular mechanisms by which DHA arrested cell cycle at the G1 phase, we detected p21 protein levels and found that DHA increased p21 in a concentration-dependent way (Figure 3B, 3C). The results indicated that DHA inhibited cell proliferation by down-regulating PCNA gene expression and prevented G1 to S phase progression through blocking cyclin D1 expression.
Figure 3.
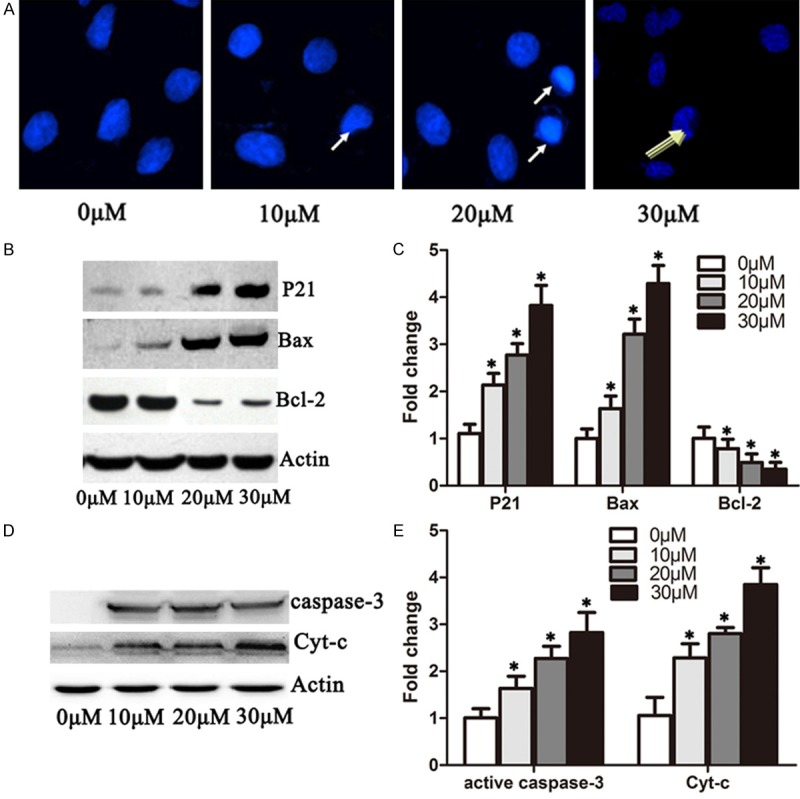
DHA induced apoptosis in A549 cells by increasing p21, Bax, cytochrome-c and active caspase 3 protein level and inhibiting Bcl-2. A. Immufluorescence microscopy analysis of 549 cells treated with 0 μM, 10 μM, 20 μM and 30 μM DHA for 72 hours. Cells were stained with propidium iodide. The condensed nucleus and cleaved nucleus were enhanced by 20 μM and 20 μM DHA as the arrow indicates. B. DHA cells were treated with 0 μM, 10 μM, 20 μM and 30 μM of DHA 48 hours, respectively. Western blots were performed to detect the protein levels of p21, Bax, Bcl-2 and actin. C. Quantification of p21, Bax and Bcl-2 protein levels from immunblots in B. The protein levels of p21, Bax and Bcl-2 were normalized to beta-actin. D. A549 cells were treated as in B. Western blots were performed to detect the protein levels of cytochrome-c and active-Caspase 3. E. Quantification of cytochrome-c and active-Caspase 3 levels from immunoblots as in D. The protein levels of cytochrome-c and active-Caspase3 were normalized to beta-actin. *P < 0.05.
DHA increases the ratio of Bax/Bcl-2 and the activation of caspase 3 and cytochrome-c
To determine the induction of apoptosis by DHA (Figure 2B), we treated A549 cells with 0 μM, 10 μM, 20 μM and 30 μM DHA for 72 hours, stained the cells with PI and observed cell morphology under microscope. The cell nucleus became condensed when treated with 5 μM DHA for 72 hours. Moreover, 10 μM and 30 μM DHA for 72 hours induced apparent apoptotic bodies and nuclear cleavage (Figure 3A). To confirm our findings, we determined the protein levels of Bax and Bcl-2 by immunoblotting. Bax protein was gradually induced with the increase in dosage of DHA. In contrast, Bcl-2 was significantly down-regulated as the concentration of DHA increased (Figure 3B), indicating that DHA induces cell apoptosis by increasing the ratio of Bax/Bcl-2 (Figure 3C). To corroborate these observations, we further detected activated Caspase-3 and cytochrome-c in A549 cells treat with DHA. Interestingly, DHA significantly increased the activated-Caspase3 and cytochrome-c protein levels in a dose dependent manner (Figure 3D, 3E).
DHA inhibits activation of the AKT/GSK-3β pathway
Arrest of cells in the G1 phase of the cell cycle provides a window for the induction of cell apoptosis. To determine whether DHA inhibits cell proliferation via down-regulation of the AKT/GSK pathway, we examined the expression of AKT, GSK-3β, p-AKT and p-GSK-3β in A549 cells treated with DHA. DHA did not affect the total protein levels of AKT and GSK-3β, but did dramatically decrease p-AKT or p-GSK-3β levels (Figure 4A). The p-AKT and p-GSK-3β signals were 8 times and 13 times lower in cells treated with 30 μM DHA than levels observed in the control cells (Figure 4B), indicating that DHA blocks cell proliferation via inhibition of the AKT/GSK-3β pathway.
Figure 4.
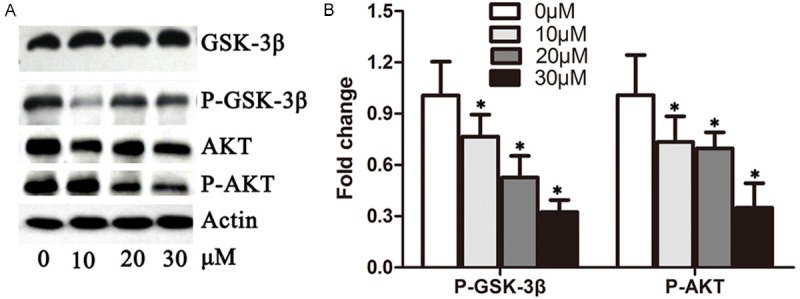
DHA reduced the activity of the AKT/GSK-3β pathway. A. A549 cells were treated with 0 μM, 10 μM, 20 μM and 30 μM DHA for 48 hours, respectively. Total proteins were extracted for the immunoblotting of AKT, p-AKT, GSK-3beta, p-GSK-3beta and actin. B. Quantification of p-AKT and p-GSK-3beta levels from immunoblots in A. The protein levels of p-AKT and p-GSK-3beta were normalized to beta-actin. *P < 0.05.
Discussion
In this study, we believe that this is the first time that it has been shown that DHA inhibited cell proliferation and induced cell apoptosis in the lung cancer A549 cell line. This was accompanied by downregulation of AKT/GSK-3β signaling. Many new therapeutic strategies have been explored for the treatment of lung cancer. For example, the semi-synthetic epothilone B derivative, ixabepilone, has been demonstrated as having potent anti-tumor activity against lung cancer cells [12,13]. Targeting of Notch signaling pathway with drugs such as the γ-secretase inhibitors also showed a promising future development for the treatment of lung cancer [14]. In this study, we found that DHA down-regulated the AKT pathway, one of the key cancer cell growth-promoting signaling pathways. Moreover, DHA induced A549 cell apoptosis by increasing the ratio of Bax/Bcl-2 and the levels of active caspase-3 and cytochrome-c. Thus, we propose that DHA is a promising therapeutic agent for the treatment of lung cancer. Our data demonstrated that DHA inhibits the proliferation of lung cancer A549 cells. Self-sufficiency in cell growth and resistance to anti-proliferative signaling are hallmarks of cancer cells. To ensure homeostasis of the cell number and maintenance of normal tissue architecture and function, normal tissues tightly control the production and release of growth-promoting signals, which instruct entry to and progression through the cell growth and division cycle. Cancer cell has obtained the capability of deregulating these signal pathways that regulate progression through the cell cycle as well as cell growth. In our experiment, DHA inhibited the growth of A549 cells in a concentration and time dependent manner. FCM analysis demonstrated that DHA arrested A549 cell cycle at G1 phase. PCNA is one of the most important factors governing cell proliferation. DHA significantly reduced both the mRNA and protein levels of PCNA. Cyclin D1 is the major G1 phase cyclin and over-expressed in most cancer cells. We found that cyclin D1 mRNA and protein levels were significantly down-regulated by DHA. AKT-Gsk-3β signaling promotes cell cycle progression by upregulating the positive regulators of cell growth including PCNA and cyclin D1 [15-17]. In agreement with the reduction of PCNA and cyclin D1, DHA dramatically inhibited the activation of the AKT/GSK-3β signaling pathway in A549 cells. Our data suggest that DHA inhibits A549 cell proliferation via the AKT/GSK-3β-cyclinD1 signal pathway.
In addition, we found that DHA induced cell apoptosis in A549 cells as evidenced by nuclear condensation. The ratio of Bax/Bcl-2, active caspase-3 and cytochrome-c protein are critical factors for the induction of apoptosis [18-21]. Consistent with these findings, DHA increased the ratio of Bax/Bcl-2 and up-regulated the protein levels of active caspase-3 and cytochrome-c.
In conclusion, our study demonstrated that DHA inhibited cell proliferation in A549 cells through suppression of the AKT/Gsk-3β/cyclinD1 signaling pathway. Furthermore, DHA induced apoptosis by increasing the ratio of Bax/Bcl-2 and active caspase-3 and cytochrome-c, suggesting that DHA is a potential drug for the therapy of lung cancer.
Acknowledgements
This work was supported by the Technology Plan Project of Chongqing (No. 20130141).
Disclosure of conflict of interest
No potential conflicts of interest were disclosed.
References
- 1.Fossella F, Pereira JR, von Pawel J, Pluzanska A, Gorbounova V, Kaukel E, Mattson KV, Ramlau R, Szczesna A, Fidias P, Millward M, Belani CP. Randomized, multinational, phase III study of docetaxel plus platinum combinations versus vinorelbine plus cisplatin for advanced non-small-cell lung cancer: the TAX 326 study group. J. Clin. Oncol. 2003;21:3016–3024. doi: 10.1200/JCO.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 2.Biesalski HK, Bueno de Mesquita B, Chesson A, Chytil F, Grimble R, Hermus RJ, Kohrle J, Lotan R, Norpoth K, Pastorino U, Thurnham D. European Consensus Statement on Lung Cancer: risk factors and prevention. Lung Cancer Panel. CA Cancer J Clin. 1998;48:167–176. doi: 10.3322/canjclin.48.3.167. [DOI] [PubMed] [Google Scholar]
- 3.Shen YC, Chou CJ, Chiou WF, Chen CF. Anti-inflammatory effects of the partially purified extract of radix Stephaniae tetrandrae: comparative studies of its active principles tetrandrine and fangchinoline on human polymorphonuclear leukocyte functions. Mol Pharmacol. 2001;60:1083–1090. [PubMed] [Google Scholar]
- 4.Choi HS, Kim HS, Min KR, Kim Y, Lim HK, Chang YK, Chung MW. Anti-inflammatory effects of fangchinoline and tetrandrine. J Ethnopharmacol. 2000;69:173–179. doi: 10.1016/s0378-8741(99)00141-5. [DOI] [PubMed] [Google Scholar]
- 5.Gulcin I, Elias R, Gepdiremen A, Chea A, Topal F. Antioxidant activity of bisbenzylisoquinoline alkaloids from Stephania rotunda: cepharanthine and fangchinoline. J Enzyme Inhib Med Chem. 2010;25:44–53. doi: 10.3109/14756360902932792. [DOI] [PubMed] [Google Scholar]
- 6.Hsu JH, Wu YC, Liou SS, Liu IM, Huang LW, Cheng JT. Mediation of Endogenous beta-endorphin by Tetrandrine to Lower Plasma Glucose in Streptozotocin-induced Diabetic Rats. Evid Based Complement Alternat Med. 2004;1:193–201. doi: 10.1093/ecam/neh027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King VF, Garcia ML, Himmel D, Reuben JP, Lam YK, Pan JX, Han GQ, Kaczorowski GJ. Interaction of tetrandrine with slowly inactivating calcium channels. Characterization of calcium channel modulation by an alkaloid of Chinese medicinal herb origin. J Biol Chem. 1988;263:2238–2244. [PubMed] [Google Scholar]
- 8.Lin TY, Lu CW, Tien LT, Chuang SH, Wang YR, Chang WH, Wang SJ. Fangchinoline inhibits glutamate release from rat cerebral cortex nerve terminals (synaptosomes) Neurochem Int. 2009;54:506–512. doi: 10.1016/j.neuint.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Mantena SK, Sharma SD, Katiyar SK. Berberine, a natural product, induces G1-phase cell cycle arrest and caspase-3-dependent apoptosis in human prostate carcinoma cells. Mol Cancer Ther. 2006;5:296–308. doi: 10.1158/1535-7163.MCT-05-0448. [DOI] [PubMed] [Google Scholar]
- 10.Maffucci T, Falasca M. Phosphoinositide 3-kinase-dependent regulation of phospholipase Cgamma. Biochem Soc Trans. 2007;35:229–230. doi: 10.1042/BST0350229. [DOI] [PubMed] [Google Scholar]
- 11.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lonning PE, Brown PO, Borresen-Dale AL, Botstein D. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Song F, Chen X, Li Y, Fan J, Wu X. Bmi-1 regulates epithelial-to-mesenchymal transition to promote migration and invasion of breast cancer cells. Int J Clin Exp Pathol. 2014;7:3057–3064. [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richardson A, Sledge GW, Carey LA. Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res. 2008;14:8010–8018. doi: 10.1158/1078-0432.CCR-08-1208. [DOI] [PubMed] [Google Scholar]
- 15.Wikman H, Kettunen E. Regulation of the G1/S phase of the cell cycle and alterations in the RB pathway in human lung cancer. Expert Rev Anticancer Ther. 2006;6:515–530. doi: 10.1586/14737140.6.4.515. [DOI] [PubMed] [Google Scholar]
- 16.Vincenzi B, Schiavon G, Silletta M, Santini D, Perrone G, Di Marino M, Angeletti S, Baldi A, Tonini G. Cell cycle alterations and lung cancer. Histol Histopathol. 2006;21:423–435. doi: 10.14670/HH-21.423. [DOI] [PubMed] [Google Scholar]
- 17.Gugger M, Kappeler A, Vonlanthen S, Altermatt HJ, Ris HB, Lardinois D, Borner MM, Heighway J, Betticher DC. Alterations of cell cycle regulators are less frequent in advanced non-small cell lung cancer than in resectable tumours. Lung Cancer. 2001;33:229–239. doi: 10.1016/s0169-5002(01)00196-9. [DOI] [PubMed] [Google Scholar]
- 18.Soumaoro LT, Uetake H, Higuchi T, Takagi Y, Enomoto M, Sugihara K. Cyclooxygenase-2 expression: a significant prognostic indicator for patients with colorectal cancer. Clin Cancer Res. 2004;10:8465–8471. doi: 10.1158/1078-0432.CCR-04-0653. [DOI] [PubMed] [Google Scholar]
- 19.Dai L, Wang L, Deng L, Liu J, Lei J, Li D, He J. Novel multiarm polyethylene glycol-dihydro artemisinin conjugates enhancing therapeutic efficacy in non-small-cell lung cancer. Sci Rep. 2014;4:5871. doi: 10.1038/srep05871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu LK, Wu HF, Guo ZR, Chen XJ, Yang D, Shu YQ, Zhang JN. Targeted efficacy of dihydroartemisinin for translationally controlled protein expression in a lung cancer model. Asian Pac J Cancer Prev. 2014;15:2511–2515. doi: 10.7314/apjcp.2014.15.6.2511. [DOI] [PubMed] [Google Scholar]
- 21.Sun Q, Teong B, Chen IF, Chang SJ, Gao J, Kuo SM. Enhanced apoptotic effects of dihydroartemisinin-aggregated gelatin and hyaluronan nanoparticles on human lung cancer cells. J Biomed Mater Res B Appl Biomater. 2014;102:455–462. doi: 10.1002/jbm.b.33023. [DOI] [PubMed] [Google Scholar]